
Abstract
The reaction of a square-planar platinum(II) complex containing two bis(2-diphenylphosphinoethyl)phenylphosphine (triphos), [Pt(triphos)2](NO3)2, with [Au(tu)2]Cl (tu = thiourea) gave a new trinuclear AuI2PtII complex, [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2, through Au-P coordination. While the [Pt(triphos)2]2+ unit in [Pt(triphos)2](NO3)2 adopted the trans-meso configuration, only the cis-racemic isomer was observed for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. 31P NMR spectroscopy indicated rapid equilibrium among the possible isomers of [Pt(triphos)2]2+, which facilitated the trans-to-cis transformation at the PtII center in this system. Additionally, we observed that this structural transformation led to an increase in the emission intensity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Square-planar platinum(II) complexes are kinetically inert, which often enables the formation of geometric isomers, namely, cis/trans or E/Z isomers [1]. These isomers play crucial roles in influencing the biological activity [2], optical/electronic properties [3], and chemical reactivity [4, 5]. Consequently, controlled syntheses of specific isomers are needed to develop functional platinum(II) coordination compounds. Structural manipulations of platinum(II) complexes, especially those with phosphine ligands, have garnered increasing interest from coordination chemists. This interest is primarily due to the fascinating isomerization behavior and photoluminescent features of these complexes [6]. In 1970, Mastin and Haake reported that a square-planar platinum(II) complex with two triphenylphosphines and two chloride ligands, [Pt(PPh3)2Cl2], showed cis-trans equilibrium in solution and was directed to the trans-isomer by irradiation with UV light [7]. After this report, cis-trans isomerism of platinum(II) complexes with monophosphine or diphosphine ligands was widely studied and utilized for stimuli-responsive systems, such as molecular gears and anion receptors [8,9,10,11]. Typically, these compounds featured a platinum(II) center surrounded by two phosphine groups and two heteroatoms, as in [PtII(P)2(X)2]. This category of compounds exhibited distinct electronic states, with the total energies differing for the cis and trans isomers [12]. This variability facilitates preferential formation of one isomer or enables isomerization with chemical/physical stimuli. When modified asymmetrically by substituent groups (i.e., [PtII(PA)2(PB)2]), platinum(II) complexes bound by four phosphine groups can also form cis-trans isomers [13, 14]. These isomers tend to have nearly equal energy levels for the cis and trans isomers, so it is more challenging to regulate isomerism. While several studies have reported crystallization of these isomers [15,16,17,18], structural control of the geometric isomers through chemical reactions remains elusive.
In this context, we investigated the stereochemistry of a square-planar platinum(II) complex, [Pt(triphos)2]2+ (triphos = bis(2-diphenylphosphinoethyl)phenylphosphine). This complex was prepared and isolated as an organotin(IV) chloride salt by Garcı́a-Fernández and coworkers in 2001, who proposed based on 31P and 195Pt NMR spectroscopic results that two of the three phosphorous (P) atoms were bound to PtII and one terminal P atom remained uncoordinated [19]. Although both cis and trans isomers are theoretically possible for the structure of [Pt(triphos)2]2+, its stereostructure has remained ambiguous due to the absence of crystallographic data. In this study, we crystallized the nitrate salt [Pt(triphos)2](NO3)2 and determined the structure. X-ray crystallography revealed that the complex [Pt(triphos)2](NO3)2 formed the trans isomer, with two uncoordinated phosphorous atoms oriented on opposite sides. Furthermore, the reaction of [Pt(triphos)2](NO3)2 with the gold(I) complex [Au(tu)2]Cl (tu = thiourea) resulted in the formation of a heterometallic PtIIAuI2 complex, [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. In this complex, the two uncoordinated P atoms in [Pt(triphos)2]2+ were bound by {AuI(tu)}+ units through Au–P bonds (Scheme 1). This meant that [Pt(triphos)2](NO3)2 functioned as a ditopic P-donating metalloligand [20–23]. Notably, while the initial [Pt(triphos)2](NO3)2 adopted the trans isomer, the [Pt(triphos)2]2+ unit observed in [Pt(triphos)2{Au(tu)}2]4+ adopted the cis configuration. To our knowledge, trans-to-cis isomerization of a square-planar [PtII(PA)2(PB)2] unit via coordination with metal ions is unprecedented. An increase in the photoluminescence was also observed after the transformation from [Pt(triphos)2]2+ to [Pt(triphos)2{Au(tu)}2]4+. The present study demonstrated that combining square-planar PtII complexes with two linear triphosphine ligands produced a structurally flexible ditopic P-donating metalloligand. Furthermore, the spectroscopic and structural data for the two complexes provide an essential dataset for understanding the cis/trans isomers of [PtII(PA)2(PB)2]-type square-planar complexes.

Synthetic routes to [Pt(triphos)2](NO3)2 and [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2
Experimental section
Materials
The starting complexes [Pt(triphos)Cl]Cl [24] and [Au(tu)2]Cl [25] were prepared according to methods described in the literature. Other chemicals were purchased and used without further purification.
Preparation of the complexes
[Pt(triphos)2](NO3)2·CH2Cl2
To a colorless solution containing [Pt(triphos)Cl]Cl (81 mg, 0.10 mmol) in 30 mL of methanol was added 54 mg (0.099 mmol) of triphos and 30 mL of methanol, which yielded a yellow suspension. After stirring for 2 h, 1.0 mL of a 1 M NaNO3 aqueous solution and 15 mL of water were added to the resulting clear yellow solution, which was then slowly evaporated for 3 days. The crude [Pt(triphos)2](NO3)2 was collected by filtration and washed with water. The crude product was purified by recrystallization from CH2Cl2-n-hexane. Yield: 86 mg (58%). Anal. calcd. for [Pt(triphos)2](NO3)2∙CH2Cl2 = C69H68Cl2N2O6P6Pt: C, 56.26; H, 4.65; N, 1.90%. Found: C, 56.45; H, 4.94; N, 2.01%. IR spectrum (cm–1, ATR): 1433, 745 (νP−CH2−), 1101, 696 (νP−Ph), 1333 (νNO3−). 1H NMR spectrum (methanol-d4, 500 MHz), δ: 7.64–6.82 (50 H, m), 2.56–2.16 (15 H, m). 31P{1H} NMR spectrum (methanol-d4, 202 MHz), δ: 45.2 (t, 1JP−Pt = 1085 Hz) and 17.9 (t, 1JP−Pt = 624 Hz).
[Pt(triphos)2{Au(Tu)}2]Cl2(NO3)2·5H2O
To a solution containing [Pt(triphos)2](NO3)2·CH2Cl2 (51 mg, 0.036 mmol) in 3 mL of methanol was added a solution containing [Au(tu)2]Cl (31 mg, 0.081 mmol) in 2 mL of methanol. After stirring for 1.5 h at room temperature in the dark, 0.150 mL of an aqueous solution of 1 M NaCl was added to the resulting colorless solution, and Et2O was diffused in for 7 days. The resulting colorless needle-like crystals were collected by filtration. Yield: 25 mg (33%). Anal. Calcd. for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2∙5H2O = C70H84Au2Cl2N6O11P6PtS2: C, 40.13; H, 4.04; N, 4.01%. Found: C, 40.21; H, 3.88; N, 4.33%. IR spectrum (cm–1, ATR): 3052 (νN−H), 1435, 753 (νP−CH2−), 1103, 694 (νP−Ph), 1330 (νNO3−), 1635 (νC=S). 1H NMR spectrum (methanol-d4, 500 MHz), δ: 7.91–6.63 (50 H, m), 3.63–3.37 (3 H, m), 3.17n2.58 (11 H, m). 31P{1H} NMR spectrum (methanol-d4, 202 MHz), δ: 92.2 (t. 1JP–Pt = 1342 Hz), 43.4 (t, 1JP–Pt = 1221 Hz), 34.6 (s), 33.2 (s).
Physical measurements
IR spectra were recorded with a JASCO FT/IR-4100 infrared spectrophotometer using the ATR method at room temperature. Elemental analyses (C, H, N) were performed at Osaka University with a YANACO CHN Corder MT-5. X-ray fluorescence spectrometry was performed with a SHIMADZU EDX-7000 spectrometer. The TG and DTA measurements were performed with a SHIMADZU DTG-60 system. The PXRD patterns were recorded with a BRUKER D2 PHASER diffractometer at room temperature. 1H and 31P NMR spectra were recorded with a JEOL ECS400 (400 MHz) spectrometer in methanol-d4 with tetramethylsilane (TMS) as an internal standard for 1H spectra and triphenylphosphine as an external standard for 31P spectra. Powder X-ray diffraction (PXRD) measurements were performed under ambient conditions with a BRUKER D2 PHASR diffractometer. The simulated powder patterns were generated from the single-crystal X-ray structures with Mercury 2023.2 [26]. The diffuse solid-state reflection spectra were measured with a JASCO V-670 UV/VIS spectrometer at room temperature using MgSO4. The photoluminescence spectra were recorded with a JASCO FP-8500 spectrometer. The internal emission quantum yields (Φ) were obtained via absolute measurements with an integrating sphere (JASCO ILFC-847); the internal surface was coated with highly reflective Spectralon. An ESC-842 calibrated light source (WI) and an ESC-843 calibrated light source (D2) were used to calibrate the emission intensities and measure the absolute quantum yields.
X-ray structural determinations
The single-crystal X-ray diffraction dataset for [Pt(triphos)2](NO3)2·CH2Cl2 was collected at 100 K with a Synergy Custom X-ray diffractometer equipped with a Hypix-6000HE hybrid photon counting detector and a Rigaku VariMax rotating-anode X-ray source with a Mo target (λ = 0.71073 Å). The dataset for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 was collected with a PILATUS3 X CdTe 1 M detector with a synchrotron X-ray source at the BL02B1 beamline at Spring-8. The intensity data were collected via the ω-scan technique and empirically corrected for absorption. All structures were solved by the intrinsic phasing method within the SHELXT program [27] and were refined on F2 by the full-matrix least-squares technique using the SHELXL program [28] via the Olex2 interface [29]. The hydrogen atoms, with the exception of those on the water molecules, were calculated and placed with riding models. All nonhydrogen atoms were refined anisotropically, while the H atoms were refined isotropically. Nitrate anions and water molecules could not be modeled and were, therefore, removed from the electron density map with the Olex2 solvent mask command [29]. ISOR instructions were applied for C35 in [Pt(triphos)2](NO3)2·CH2Cl2 and C35 and N2 in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2.
The crystallographic data are summarized in Table 1. Selected bond distances and angles are summarized in Tables 2 and 3.
Results and discussion
Synthesis and structural characterization of [Pt(triphos)2](NO3)2
The reaction of [Pt(triphos)Cl]Cl with one equivalent of triphos in methanol triggered a color change from colorless to yellow. Subsequently, an excess of NaNO3 was added to isolate the nitrate salt of [Pt(triphos)2]2+ as a crude powder. Slow diffusion of n-hexane into a solution of the crude powder in CH2Cl2 yielded colorless block crystals ([Pt(triphos)2](NO3)2) in moderate yield (58%). The IR spectrum of [Pt(triphos)2](NO3)2 exhibited an intense band at 1343 cm–1 attributed to the νN−O stretch of the NO3– ion, along with sharp signals corresponding to νP−Ph (1103, 694 cm–1) and νP−C (1435, 753 cm–1) vibrations of the triphos ligands (Fig. 1a) [30]. The CHN elemental and fluorescence X-ray analyses were consistent with the CH2Cl2 adduct [Pt(triphos)2](NO3)2·CH2Cl2 (Fig. S1). Thermogravimetric (TG) analyses indicated a weight loss of 3.0% at 92 °C, corresponding to loss of the CH2Cl2 molecule of solvation (Fig. S2). This value, which was lower than the theoretical value of 5.6% for the loss of one CH2Cl2, suggested the efflorescent nature of [Pt(triphos)2](NO3)2.

IR spectra of a [Pt(triphos)2](NO3)2 and b [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2
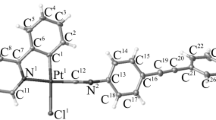
A single-crystal X-ray analysis of [Pt(triphos)2](NO3)2 revealed that the asymmetric unit comprised half of a mononuclear platinum(II) complex cation with two triphos ligands, [Pt(triphos)2]2+, situated at the crystallographic inversion center, and half of a solvated CH2Cl2 molecule. Although the NO3– ions could not be modeled, they were presumed to occupy the crystal void spaces and were disordered. As illustrated in Fig. 2, each triphos ligand was bonded to the square-planar PtII center in a bidentate-P,P′ fashion, engaging one of the two terminal PPh2 groups and the central PPh group. The PtII center was coordinated by two PPh2 and PPh groups from the triphos ligands, forming a [Pt(PA)2(PB)2]-type chromophore with a trans configuration, thereby positioning the uncoordinated PPh2 groups on opposite sides. In addition to cis-trans isomerism around the PtII center, the coordinated PPh group, which was a stereogenic phosphorous atom, exhibited an R–S configuration, leading to meso(RS)-racemic (RR/SS) isomerism in [Pt(triphos)2]2+. Among the four possible isomers illustrated in Chart 1, [Pt(triphos)2](NO3)2 was found to adopt the trans-meso isomer in the crystals. The Pt–PPh2 (2.3190(9) Å) and Pt–PPh (2.3268(9) Å) bond distances were longer than those in a related AuIPtII complex with a similar asymmetric triphos coordination mode, [AuPt(triphos)Cl3] (Pt–PPh2 = 2.23(1) Å, Pt–PPh = 2.23(1) Å) [24]. Elongation of the Pt–P bonds in [Pt(triphos)2](NO3)2 resulted because the trans influence of phosphine groups was larger than that of Cl– [31, 32], which is why [Pt(triphos)2](NO3)2 reached solution equilibrium on the NMR timescale (vide infra).

Perspective view of [Pt(triphos)2](NO3)2. The thermal ellipsoids are illustrated at 50% probability. Color code: Pt: white, P: orange, Cl: green, C: gray, H: pale blue. (Color figure online)

Four possible isomers of [Pt(triphos)2]2+; a Trans-meso, b trans-racemic, c cis-meso, and d cis-racemic
It should be noted that the powder X-ray diffraction pattern of [Pt(triphos)2](NO3)2 was consistent with the simulated pattern for the crystal structure, which indicated that only the trans-meso isomer was formed in the bulk sample of [Pt(triphos)2](NO3)2 (Fig. 3a). In the packing diagram for [Pt(triphos)2](NO3)2, the complex cations formed two intermolecular C–H···π interactions between the phenyl groups of the uncoordinated PPh2 and coordinated PPh2 groups (CH···Cg = 2.53 Å and 3.07 Å), creating a 2D sheet supramolecular structure in the crystallographic ab plane (Fig. 4a). The 2D structures were stacked along the c axis and accommodated the CH2Cl2 molecule in the interstitial space (Fig. 4b). We assumed that the adjusted crystal packing to accommodate the CH2Cl2 was the key to the selective crystallization of the trans-meso isomer of [Pt(triphos)2](NO3)2. Similar solvent-directed crystallization of one of four isomers was observed for a PtII complex with chlorophosphineamides [16].

Observed (top) and simulated (bottom) powder X-ray diffraction patterns for a [Pt(triphos)2](NO3)2 and b [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2

Perspective views of [Pt(triphos)2](NO3)2; a 2D sheet supramolecular structure viewed from the c axis and b packing structure viewed from the a axis. The blue dotted lines indicate the CH···π interactions. Color code: Pt: white, P: orange, Cl: green, C: gray, H: pale blue. (Color figure online)
Synthesis and structural characterization of [Pt(triphos)2{Au(Tu)}2]Cl2(NO3)2
The [Pt(triphos)2](NO3)2 complex contained two unbound PPh2 groups. Consequently, these groups were anticipated to function as coordination sites for other metal ions, similar to the role of P-donating metalloligands with free phosphine groups, e.g., 1,1’-bis(diphenylphosphino)ferrocene (dppf) [33], (-PhPC5H4FeC5H4-)3 [21], and [Pt(Ar)phenylpyridinates (κ1-dppm)] [22, 23]. To probe this ability, we examined the reaction of [Pt(triphos)2](NO3)2 with two equivalents of [Au(tu)2]Cl, which is a well-known AuI source [34], in methanol. With addition of excess NaCl to the colorless reaction mixture and subsequent diffusion of diethyl ether vapor, we isolated colorless plate-like crystals of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 in satisfactory yield (33%). An X-ray fluorescence analysis revealed that [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 contained Pt and Au as the metallic elements in a 1:2 ratio, in addition to S, P, and Cl (Fig. S1). The IR spectrum revealed bands due to NO3– ions at 1330 cm–1, νP−Ph (1103, 694 cm–1) and νP−C (1435, 753 cm–1) bands for the triphos ligands, and νN−H (3052 cm–1) and νC=S (1635 cm–1) bands for the tu ligands (Fig. 1b) [30, 38]. These results supported the formation of a 1:2 adduct by the [Pt(triphos)2]2+ and [Au(tu)]+ units. Moreover, the elemental analyses indicated the formula of a double salt, [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2·5H2O. The presence of water molecules was supported by the TG analysis (Fig. S2).
The [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 crystals showed weak diffraction with the laboratory instrument, so we conducted single-crystal X-ray analyses with the synchrotron faculty at SPring-8. The asymmetric unit contained half of the AuI2PtII complex cation, [Pt(triphos)2{Au(tu)}2]4+ (Fig. 5), which was situated on the crystallographic C2 axis, and one chloride ion. As found for [Pt(triphos)2](NO3)2], two triphos ligands in [Pt(triphos)2{Au(tu)}2]4+ chelated the square-planar PtII center via bidentate-P,P’ binding. The bond distances around the PtII center (Pt–PPh2 = 2.316(3) Å, Pt–PPh = 2.311(3) Å) were similar to those in [Pt(triphos)2](NO3)2. However, the PtII unit had unexpectedly adopted the cis configuration, and the two stereogenic P atoms had the same chirality. That is, the cis-racemic isomer of [Pt(triphos)2]2+ was contained in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. The two {Au(tu-S)}+ moieties were bound to the free PPh2 sites of the [Pt(triphos)2]2+ unit (Au–P = 2.265(4) Å, Au–S = 2.314(5) Å, S–Au–P = 174.07(15)°) to form a heterometallic trinuclear structure. The powder X-ray diffraction pattern for the bulk sample of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 matched the simulated pattern from the crystal data well (Fig. 3b), which confirmed the phase purity of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2.

A perspective view of the complex cation accommodating the Cl– anion in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. The thermal ellipsoids are illustrated at 50% probability. Color code: Pt: white, Au: pink, P: orange, Cl: green, N: blue, C: gray, H: pale blue. The red dotted lines indicate hydrogen bonds. (Color figure online)
In the crystal packing structure, two Cl– anions were wrapped by the NH2 groups of the tu ligands and the ethylene and phenyl groups of the triphos ligands via NH···Cl (2.25 Å) and CH···Cl (2.64, 2.62, and 2.94 Å) hydrogen bonds. These interactions resulted in formation of the discrete supramolecule {Cl2@[Pt(triphos)2{Au(tu)}2]}2+ (Fig. 5). We assumed that the interaction between [Pt(triphos)2{Au(tu)}2]4+ and Cl– led to preferable formation of the cis-racemic isomer of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. Moreover, these supramolecules were interconnected by additional CH···Cl hydrogen bonds (2.63 Å) between the phenyl groups and Cl– anions of adjacent supramolecules, forming a 1D chain structure (Fig. 6a). The 1D chains also interacted with neighboring chains via CH···π interactions (CH···Cg = 3.21 and 2.71 Å), culminating in a 3D supramolecular structure with large void spaces (34.2%, 3102.3 Å3 per unit cell) (Fig. 6b). We could not model the nitrate anion and solvated water molecules in the crystal structure because they were severely disordered in the void space.

Perspective views of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. a The 2D chain structure viewed from the b axis. b The packing structure viewed from the a axis. The red and blue dotted lines indicate the hydrogen bonds and CH···π interactions, respectively. Color code: Pt: white, Au: pink, P: orange, Cl: green, N: blue, C: gray, H: pale blue. (Color figure online)
NMR spectral study
X-ray diffraction experiments revealed that in the solid samples of [Pt(triphos)2](NO3)2 and [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2, the [Pt(triphos)2]2+ units selectively formed the trans-meso- and cis-racemic isomers, respectively. To investigate the molecular structure in solution, we performed NMR spectroscopic measurements of these compounds in methanol-d4. The 1H NMR spectra were complicated (Fig. S3); therefore, our analysis was primarily focused on the 31P NMR spectra. The 31P NMR spectrum of [Pt(triphos)2](NO3)2 determined in methanol-d4 at 25 °C exhibited two singlet signals with platinum satellites at δ 46.28 (signal A) and 19.02 ppm (signal B) with an integration intensity ratio of 1:2 (Fig. 7a). The coupling constant JP−Pt of signal A (1087 Hz) was approximately twice that of signal B (622.6 Hz). These spectral features were inconsistent with the Cs symmetric mononuclear structure observed in the single-crystal X-ray study, which should display two singlets with similar JP−Pt values alongside one singlet without platinum satellites. We tentatively assigned the 31P signals A and B to the central PPh and terminal PPh2 groups of triphos ligands, respectively, and we presumed that the coordinated and uncoordinated PPh2 groups rapidly exchanged on the NMR timescale. To verify this dynamic behavior, we measured the variable-temperature (VT) NMR spectra at temperatures from 25 °C to − 80 °C. Upon cooling, signals A and B became broader at -40 °C and disappeared at lower temperatures. At − 80 °C, a new singlet without platinum satellites appeared at − 15.23 ppm (signal C), which indicated uncoordinated PPh2 groups, as typically reported in the literature [36]. Additionally, complex, broad signals appeared between δ 15–50 ppm. We hypothesized that all or some of the four possible stereoisomers of [Pt(triphos)2]2+ (cis-meso, cis-racemic, trans-meso, and trans-racemic) were in equilibrium in solution, and an average structure was observed at 25 °C. Exchange among the isomers required Pt–P bond cleavage. The Pt–P bonds were elongated, which was attributed to the substantial trans influence of the phosphines, may have facilitate bond cleavage within the system.

VT 31P NMR spectra of a [Pt(triphos)2](NO3)2 and b [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 determined in methanol-d4
The 31P NMR spectrum of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 determined in methanol-d4 at 25 °C showed two singlets with platinum satellites at 92.21 (JP−Pt = 1342 Hz) and 43.37 (JP−Pt = 1211 Hz) ppm ascribed to the Pt-PPh2 and Pt-PPh groups, respectively. In addition, the spectrum showed two singlets without platinum satellites at 34.6 and 33.2 ppm due to the Au-PPh2 group (Fig. 7b). The overall spectrum was consistent with the C2 symmetric trinuclear AuI2PtII structure observed in the single-crystal X-ray analysis, except for the appearance of two signals due to the Au-PPh2 groups. The signal splitting presumably resulted from partial exchange of the tu ligands with the solvent in solution. Note that the VT NMR spectra of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 from 25 to − 80 °C showed no drastic spectral changes, unlike that of [Pt(triphos)2](NO3)2. Au-P bond formation may have suppressed Pt-P bond exchange, which maintained the cis-racemic isomer of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 even in solution.
Photophysical properties
The absorption spectra of the [Pt(triphos)2](NO3)2 and [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 complexes in methanol exhibited π-π* transition bands below 270 nm due to the Ph groups of the triphos ligands and a 1MLCT transition from PtII to the phosphine groups at approximately 320 nm (Fig. S4) [37]. In addition, [Pt(triphos)2](NO3)2 showed a weak visible band at approximately 400 nm due to the 3MLCT transition.
The solid samples of [Pt(triphos)2](NO3)2 and [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 showed emission at 77 K. The excitation and emission spectra are illustrated in Fig. 8. The excitation spectra of the two complexes showed π–π* bands at approximately 280 nm and 1MLCT bands at approximately 330 nm. The similar excitation spectra indicated that cis-trans isomerism around the PtII center and the presence/absence of the {Au(tu)}+ moieties did not affect the absorption spectrum. However, the emission band for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 (λem = 474 nm) occurred at a longer wavelength than that of [Pt(triphos)2](NO3)2 (λem = 438 nm). Moreover, the emission band for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 revealed a structured pattern with a separation of ca. 1600 cm–1. This value was similar to the C=S stretching energy (1635 cm–1) of the tu ligand in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2, which was observed in the IR spectra (vide supra). Therefore, we propose that the emission of [Pt(triphos)2](NO3)2 involved charge transfer from PtII to a phosphine (3MLCT) of the [PtP4] moiety, while the emission of [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 was ascribed to charge transfer from S to P (3LLCT) or S to Au (3LMCT) [38]. The large Stokes shift for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 was explained by intramolecular energy transfer from the [PtP4] to [AuPS] moieties and structural distortion of the AuI ion in the triplet state. The quantum yields (Φ) of the two complexes were 6.9% for [Pt(triphos)2](NO3)2 and 16% for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. Multiple H bonds between the complex cationa and Cl– may have reduced the molecular vibrations, which may have contributed to the strong photoluminescence from the AuI2PtII complex [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2.

Excitation (ex) and emission (em) spectra of a [Pt(triphos)2](NO3)2 and b [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 in the solid-state at 77 K
Concluding remarks
We crystallized and elucidated the structure of [Pt(triphos)2](NO3)2, in which two triphos ligands were chelated to the square-planar PtII center with two of three P atoms. We also showed that the [Pt(triphos)2]2+ complex acted as P-donating metalloligands for AuI ions by utilizing the two remaining P atoms. This was evidenced by formation of the trinuclear AuI2PtII complex [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2, which was synthesized by reacting [Pt(triphos)2](NO3)2 with [Au(tu)2]Cl. Notably, we observed a structural transformation within the [Pt(triphos)2]2+ unit; the trans-meso isomer present in [Pt(triphos)2](NO3)2 underwent conversion to the cis-racemic isomer in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. An NMR spectral analysis suggested that this unique trans-to-cis isomerization of the PtII center was triggered by the reaction with AuI and occurred through rapid equilibrium among the possible isomers in solution. The complexes [Pt(triphos)2](NO3)2 and [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 showed photoluminescence at 77 K. The higher luminescence quantum yield for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2 relative to that for [Pt(triphos)2](NO3)2 was tentatively explained by the presence of multiple hydrogen bonds with Cl–, which may have suppressed vibrational quenching of the photoluminescence in [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. Finally, the present study demonstrated that square-planar PtII species with two triphosphine ligands can serve as P-donating metalloligands with dynamic geometric isomerism for the future design and creation of switchable luminescent materials.
Data availability
Crystallographic data (including structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. CCDC numbers: 2327654 for [Pt(triphos)2](NO3)2 and 2327655 for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. Copies of the data can be obtained free of charge on application to CCDC, https://www.ccdc.cam.ac.uk/structures/.
References
Gispert, J.R.: Coordination Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany (2008)
Kishimoto, T., Yoshikawa, Y., Yoshikawa, K., Komeda, S.: Different effects of Cisplatin and Transplatin on the higher-order structure of DNA and gene expression. Int. J. Mol. Sci. 21, 34 (2020). https://doi.org/10.3390%2Fijms21010034
Patterson, H.H., Tewksbury, J.C., Martin, M., Krogh-Jespersen, M.-B., Lomenzo, J.A., Hooper, H.O., Viswanath, A.K.: Luminescence, absorption, MCD, and NQR Study of the Cis and Trans Isomers of Dichlorodiammineplatinum(II). Inorg. Chem. 20, 2297–2301 (1981). https://doi.org/10.1021/ic50221a071
Konno, T., Yoshinari, N., Taguchi, M., Igashira-Kamiyama, A.: Drastic change in Dimensional structures of d-Penicillaminato (AuI2PtII2ZnII)n coordination polymers by Moderate Change in Solution pH. Chem. Lett. 38, 526–527 (2009). https://doi.org/10.1021/ic50221a071
Yoshinari, N., Hashimoto, Y., Igashira-Kamiyama, A., Konno, T.: Synthesis, characterization, and Crystal Structures of Cis and trans isomers of a platinate(II) complex with d-Penicillaminate. Bull. Chem. Soc. Jpn. 84, 623–625 (2011). https://doi.org/10.1246/bcsj.20110036
Geist, F., Jackel, A., Winter, R.F.: Synthesis, Ligand Based Dual Fluorescence and Phosphorescence Emission from BODIPY Platinum Complexes and its application to Ratiometric Singlet Oxygen Detection. Inorg. Chem. 54, 10946–10957 (2015). https://doi.org/10.1021/acs.inorgchem.5b01969
Mastin, S.H., Haake, P.: Synthesis of trans-platinum(II) complexes by photochemical isomerization. J. Chem. Soc. D. 6, 202 (1970). https://doi.org/10.1039/C29700000202
Ube, H., Yasuda, Y., Sato, H., Shionoya, M.: Metal-centred azaphosphatriptycene gear with a photo- and thermally driven mechanical switching function based on coordination isomerism. Nat. Commun. 8, 14296 (2017). https://doi.org/10.1038/ncomms14296
Pascui, A.E., Rees, K.V., Zant, D.W., Broere, D.L.J., Siegler, M.A., Vlugt, J.I.V.D.: Macrocyclic platinum(II) complexes with a bifunctional Diphosphine Ligand. Eur. J. Inorg. Chem. 34, 5578–5714 (2015). https://doi.org/10.1002/ejic.201501055
Melník, M., Mikuš, P.: Organodiphosphines in Pt{η2-P(X)nP}Cl2 (n = 9–15, 17, 18) derivatives – structural aspects. Rev. Inorg. Chem. 41, 41–48 (2021). https://doi.org/10.1515/revic-2020-0010
Owens, S.B. Jr., Smith, D.C. Jr., Lake, C.H., Gray, F.M.: Synthesis, characterization, and cis-trans isomerization studies of cis-[PdCl2{Ph2P(CH2CH2O)3CH2CH2PPh2-P,P’}] and trans-[PtCl2{Ph2P(CH2CH2O)3CH2CH2PPh2-P,P’}] Metallacrown Ethers. Eur. J. Inorg. Chem. 30, 4710–4718 (2008). https://doi.org/10.1515/revic-2020-0010
Romeo, R., Minniti, C., Trozzi, M.: Uncatalyzed Cis-trans isomerization and methanol solvolysis of Arylbromobis(triethylphosphine)platinum(II) complexes. A different role for Steric Hindrance in Dissociative and associative mechanisms. Inorg. Chem. 15, 1134–1138 (1976). https://doi.org/10.1515/revic-2020-0010
Zhang, S., Pattacini, R., Braunstein, P., Cola, L.D., Plummer, E., Mauro, M., Doulaouen, C., Daniel, C.: Synthesis, structure, and Optical properties of Pt(II) and pd(II) complexes with oxazolyl- and pyridyl-functionalized DPPM-Type ligands: A combined experimental and theoretical study. Inorg. Chem. 53, 12739–12756 (2014). https://doi.org/10.1021/ic501566u
Zhang, S., Pattacini, R., Braunstein, P.: Unexpected Metal-Induced isomerisms and Phosphoryl migrations in Pt(II) and pd(II) complexes of the functional phosphine 2-(Bis(diphenylphosphino)methyl)-oxazoline. Inorg. Chem. 50, 3511–3522 (2011). https://doi.org/10.1021/ic1024422
Redwine, K.D., Wilson, W.L., Moses, D.G., Catalano, V.J., Nelson, J.H.: Cooperative Diastereoselectivity of Palladium- and platinum-promoted intramolecular [4 + 2] diels – alder cycloaddition reactions of 3,4-Dimethyl-1-phenylphosphole. Inorg. Chem. 39, 3392–3402 (2000). https://doi.org/10.1021/ic990885j
Lungu, D., Daniliuc, C., Jones, P.G., Nyulászi, L., Benkõ, Z., Bartsch, R., Mont, W.-W.D.: Exceptional Coordination Mode of Unsaturated PNP ligands (Me3Si)2C=PN(R)PPh2 with palladium and platinum dichlorides: Insertion of Phosphaalkene Phosphorus atoms into metal–chlorine bonds. Eur. J. Inorg. Chem. 20, 2901–2905 (2009). https://doi.org/10.1002/ejic.200900236
Deeming, A.J., Cockerton, B.R., Doherty, S.: Substitution of chloride in [PtCl2(PEt3)2] by the chiral anionic ligand [Mo(CO)5(PPhH)]– to give mixed platinum-molybdenum compounds and a 31P{1H} NMR analysis of their fluxionality. X-ray crystal structure of [PtCl(PEt3)2(µ-PPhH){Mo(CO)5}] and trans-[Pt(PEt3)2(µ-PPH)2{Mo(CO)5}2]. Polyhedron. 16, 1945–1956 (1997). https://doi.org/10.1016/S0277-5387(96)00403-2
Barkley, J., Higgins, S.J., McCart, M.K.: Complexes of 2,3-bis(diphenylphosphino)propene with PtII, PdII and RuII: Synthesis, characterisation and rearrangements to complexes of cis-1,2-bis(diphenylphosphino)propene. J. Chem. Soc. Dalton Trans. 16, 1787–1792 (1998). https://doi.org/10.1016/S0277-5387(96)00403-2
Garcı́a-Seijo, M.I., Castiñeiras, A., Mahieu, B., Jánosi, L., Berente, Z., Kollár, L., Garcı́a-Fernández, M.E.: Crystal structure and reactivity of mononuclear cationic palladium(II) and platinum(II) triphos complexes with phenyltin(IV) anions. The formation of polynuclear platinum–triphos ionic and covalent complexes. Polyhedron. 20, 855–868 (2001). https://doi.org/10.1016/S0277-5387(96)00403-2
Balch, A.L., Catalano, V.J.: Formation of luminescent, metal-metal bonded, 3:5 complexes using palladium(II) or platinum(II) and iridium(I) through chelate ring openings. Inorg. Chem. 31, 2569–2575 (1992). https://doi.org/10.1021/ic00038a048
Mizuta, T., Aotani, T., Imamura, Y., Kubo, K., Miyoshi, K.: Structure and Properties of the Macrocyclic Tridentate Ferrocenylphosphine Ligand (-PhPC5H4FeC5H4-)3. Organometallics. 27, 2457–2463 (2008). https://doi.org/10.1021/om800057w
Dolatyari, V., Shahsavari, H.R., Fereidoonnezhad, M., Farhadi, F., Akhlaghi, S., Latouche, C., Latouche, C., Sakamaki, Y., Beyzavi, H.: Luminescent heterobimetallic PtII–AuI complexes bearing N-Heterocyclic carbenes (NHCs) as potent Anticancer agents. Eur. J. Inorg. Chem. 10, 1360–1373 (2019). https://doi.org/10.1021/acs.inorgchem.3c01504
Shahsavari, H.R., Giménez, N., Lalinde, E., Moreno, M.T., Fereidoonnezhad, M., Aghakhanpour, R.B., Khatami, M., Kalantari, F., Jamshidi, Z., Mohammadpour, M.: Luminescent heterobimetallic PtII–AuI complexes bearing N-Heterocyclic carbenes (NHCs) as potent Anticancer agents. Inorg. Chem. 62, 13241–13252 (2019). https://doi.org/10.1002/ejic.201801297
Sevillano, P., Habtemariam, A., Parsons, S., Castiñeiras, A., García, M.E., Sadler, P.J.: Gold(I)-induced chelate ring-opening of palladium(II) and platinum(II) triphos complexes. J. Chem. Soc. Dalton Trans. 16, 2861–2870 (1999). https://doi.org/10.1002/ejic.201801297
Piro, O.E., Castellano, E.E., Piatti, R.C.V., Bolzán, A.E., Arvia, A.J.: Two thiourea-containing gold(I) complexes. Acta Cryst. C58, m252–m255 (2002). https://doi.org/10.1002/ejic.201801297
Macrae, C.F., Sovago, I., Cottrell, S.J., Galek, P.T.A., McCabe, P., Pidcock, E., Platings, M., Shields, G.P., Stevens, J.S., Towler, M., Wood, P.A.: Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 53, 226–235 (2020). https://doi.org/10.1107/S1600576719014092
Sheldrick, G.M.: A short history of SHELX. Acta Cryst. A64, 112–122 (2008). https://doi.org/10.1107/S1600576719014092
Sheldrick, G.M.: Crystal structure refinement with SHELXL. Acta Cryst. C. 71, 3–8 (2015). https://doi.org/10.1107/S2053229614024218
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., Howard, J.A.K., Puschmann, H.J.: OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 42, 339–341 (2009). https://doi.org/10.1107/S0021889808042726
Socrates, G.: Infrared Characteristic Group Frequencies, 3rd edn. Wiley, Chichester, UK (2001)
Kapoor, P., Kukushkin, V.Y., Lövqvist, K., Oskarsson, Å.: Trans influence in platinum(II) complexes. Phenylation of pt(II) dialkylsulfide complexes by BPh4– and SnPh3H. Crystal structures of two polymorphs of trans-chlorobis(dimethylsulfide)(phenyl)platinum(II). J. Organomet. Chem. 517, 71–79 (1996). https://doi.org/10.1016/0022-328X(95)06103-4
Chval, Z., Sip, M., Burda, J.V.: The trans effect in square-planar platinum(II) complexes—A density functional study. J. Comput. Chem. 29, 2370–2381 (2008). https://doi.org/10.1002/jcc.20980
Hrubý, J., Dvořák, D., Squillantini, L., Mannini, M., Slageren, J.V., Herchel, R., Nemec, I., Neugebauer, P.: Co(II)-Based single-ion magnets with 1,1’-ferrocenediyl-bis(diphenylphosphine) metalloligands. Dalton Trans. 49, 11697–11707 (2020). https://doi.org/10.1039/D0DT01512A
Isab, A.A., Fettouhi, M., Ahmad, S., Ouahab, L.: Mixed ligand gold(I) complexes of phosphines and thiourea and X-ray structure of (thiourea-κS)(tricyclohexylphosphine)gold(I)chloride. Polyhedron. 22, 1349–1354 (2003). https://doi.org/10.1016/S0277-5387(03)00129-3
Jensen, K.A., Nielsen, P.H.: Infrared Spectra of thioamides and Selenoamides. Acta Chem. Scand. 20, 597–629 (1966). https://doi.org/10.3891/acta.chem.scand.20-0597
Mason, L.J., Moore, A.J., Carr, A., Helm, M.L.: Lithium bis(2-phenylphosphidoethyl)phenyl-phosphine: A reactive phosphorus intermediate. Heteroat. Chem. 18, 675–678 (2007). https://doi.org/10.1002/hc.20351
Solar, J.M., Ozkan, M.A., Isci, H., Mason, W.R.: Electronic absorption and magnetic circular dichroism spectra of some planar platinum(II), palladium(II), and nickel(II) complexes with phosphorus-donor ligands. Inorg. Chem. 23, 758–764 (1984). https://doi.org/10.1021/ic00174a024
Hashimoto, Y., Yoshinari, N., Naruse, D., Nozaki, K., Konno, T.: Synthesis, structures, and Luminescence properties of Interconvertible AuI2ZnII and AuI3ZnII complexes with mixed bis(diphenylphosphino)methane and d-Penicillaminate. Inorg. Chem. 52, 14368–14375 (2013). https://doi.org/10.1021/ic4024629
Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. 19K05496). The synchrotron radiation experiments were performed at the BL02B1 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Nos. 2022A1573 and 2022B1659). This work is the result of using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0441200023.
Funding
Open Access funding provided by Osaka University.
Author information
Authors and Affiliations
Contributions
KN and HS prepared compounds; KN and TB conducted compound characterization; TB and NY wrote the draft; NY edited the manuscript, and conceived the project. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supporting information
The fluorescence X-ray spectra (Fig. S1), TG profiles (Fig. S2), 1H NMR spectra (Fig. S3), and absorption spectra (Fig. S4) are included in the supplementary material in PDF format. The crystallographic data (including the structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. CCDC numbers: 2327654 for [Pt(triphos)2](NO3)2 and 2327655 for [Pt(triphos)2{Au(tu)}2]Cl2(NO3)2. Copies of the data can be obtained free of charge on application to the CCDC, https://www.ccdc.cam.ac.uk/structures/.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagasato, K., Baba, T., Soma, H. et al. Trans-to-cis isomerization of a platinum(II) complex with two triphosphine ligands via coordination with gold(I) ions. J Incl Phenom Macrocycl Chem 104, 257–268 (2024). https://doi.org/10.1007/s10847-024-01228-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-024-01228-2