
Abstract
The HD superfamily has been studied in detail for several decades. The plant-specific HD-Zip I subfamily attracts the most attention because of its involvement in plant development and stress responses. In this review, we provide a comprehensive insight into the evolutionary events responsible for the functional redundancy and diversification of the HD-Zip I genes in regulating various biological processes. We summarized the evolutionary history of the HD-Zip family, highlighting the important role of WGDs in its expansion and divergence of retained duplicates in the genome. To determine the relationship between the evolutionary origin and functional conservation of HD-Zip I in different species, we performed a phylogenetic analysis, compared their expression profiles in different tissues and under stress and traced the role of orthologs and paralogs in regulating developmental processes. We found that HD-Zip I from different species have similar gene structures with a highly conserved HD and Zip, bind to the same DNA sequences and are involved in similar biological processes. However, they exhibit a functional diversity, which is manifested in altered expression patterns. Some of them are involved in the regulation of species-specific leaf morphology and phenotypes. Here, we discuss the role of changes in functional domains involved in DNA binding and protein interaction of HD-Zip I and in cis-regulated regions of its target genes in promoting adaptive innovations through the formation of de novo regulatory systems. Understanding the role of the HD-Zip I subfamily in organism-environment interactions remains a challenge for evolutionary developmental biology (evo-devo).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regulation of gene transcription is a complex process in which specific proteins act as commutators within regulatory cascades controlling the functions of genes. Transcription-associated proteins are divided into three functional groups: DNA-binding transcription factors (TFs), transcription cofactors and proteins associated with chromatin structure. TFs are protein-coding genes that recognize and bind to specific DNA sequences in promoters of target genes to control biological processes in all living organisms. Their activity can be modulated by mechanisms involving their synthesis, subcellular localization, interaction with other proteins and posttranslational modifications.
In plants, the evolutionary history of TFs is clearly associated with dramatic geological and climatic changes during land colonization that required the developing of new regulatory mechanisms. The rapid adaptation of plants to unfavorable environmental conditions such as increased CO2 concentration, light intensity, desiccation and limited nutrient availability is attributed to the increase in the number of genes that resulted from whole genome duplications (WGDs) and then their gradual sub- or neofunctionalization, leading to morphological, physiological and regulatory changes in organisms (Rensing 2014). Most copies of TFs were retained in genomes after the α-WGD event because regulatory proteins are protected from mutations due to their pleiotropic effects (Prud’homme et al. 2007). However, a gene duplicate is often released from negative selective pressure and even single-nucleotide mutations in the DNA-binding or interaction domains can alter its binding preferences and promote gene regulatory network (GRN) evolution (Paris et al. 2013; Rosas et al. 2014). In particular, the retention and subsequent diversification of duplicated TFs involved in stress responses and affecting to changes in stress signaling pathways in plants have been documented (Mizoi et al. 2012; Yang et al. 2014). Some studies suggest that phenotypic diversity is due to mutations in the cis-regulatory motif rather than in the coding sequence of TF (Wray 2007). A genome-wide analysis of cis-regulatory sites in paralogs of A. thaliana confirmed that changes in their architecture are reflected in changes in the regulatory network (Arsovski et al. 2015). In contrast, regions outside DNA-binding domains involved in protein–protein interactions are conserved, especially for hormone-related TFs such as ARF, PIF, MYC2, FHY1 and ABI3A (Romani, Moreno 2021). Therefore, further studies are needed to determine the mechanisms underlying the conservatism or functional divergence of DNA binding preference between WGD-derived members of TF families and their target genes.
The abundance of TFs has been confirmed in many plants whose genomes have been sequenced and they account for between 0.5 and 13% of all protein-coding genes (PlantTFDB v5.0 http://planttfdb.gao-lab.org/; Table S1). These regulatory genes were classified according to the specificity of their DNA-binding domain, from 46 families in Trifolium pratense and Helianthus annuus to 71 families in Kalanchoe laxiflora (Luscombe et al. 2000; Table S1). The observed differences in gene abundance among plant species are due to their polyploidy status or genome duplication. The A. thaliana genome encodes 2296 TFs classified into 58 families according to specific DNA-binding domains, structural features and functions (PlantTFDB v5.0; http://planttfdb.gao-lab.org/, Table S1). Approximately 60% of them are from three rounds of WGD: γ (1R), β (2R), and α (3R) (Maere et al. 2005). Thus, this number is 1.3 and 1.7 times higher than in Drosophila and Caenorhabditis elegans, respectively, and is consistent with the eukaryotic evolutionary model, which indicates a higher rate of gene expansion in land plants than in animals (Shiu et al. 2005). About 40% of A. thaliana TFs such as HD-Zip, AP2/EREBP, ARF, NAC, SBP, GARP, MADS-box, ABI3/VP1 and WRKY are plant-specific TFs. This confirms an independent evolution of lineage-specific TF families that began as early as the Precambrian (Catarino et al. 2016; Bowman et al. 2017; Wilhelmsson et al. 2017; Rensing 2014; Johnson 2017).
The homeobox proteins are transcription factors found in all living organisms that contain the highly conserved homeodomain (HD) involved in DNA-binding. The first plant HD-containing gene was discovered in maize by Vollbrecht and coworkers (1991). The KNOTTED1 gene was identified by transposon tagging and its constitutive expression in transgenic maize plants showed knot-like structures in the leaves. In plants, the superfamily of HD genes is divided into 11 families: BEL, DDT, HD-Zip, KNOX, LD, NDX, PHD, PINTOX, PLINC, SAWADEE and WOX (Mukherjee et al. 2009). The plant-specific HD-Zip gene family is characterized by the presence of the highly conserved 60 amino acid HD adjacent to the less conserved leucine zipper motif (LZ) (Ariel et al. 2007). It is divided into four subfamilies: HD-Zip I, HD-Zip II, HD-Zip III and HD-Zip IV, depending on DNA-binding specificity, gene structure, number of heptad repeats in LZ, presence of additional motifs and biological functions. The HD is crucial for recognition and binding of these transcription factors to the cis-regulatory element in target genes, whereas LZ has some heptad repeats with a leucine residues in the center position and functions as a dimerization motif that is critical for efficient DNA binding (Tron et al. 2004). The HD-Zip proteins interact with DNA only as dimers. The three-dimensional structure of HD contains three characteristic α-helices, of which the second and third helices form a helix-turn-helix DNA-binding motif (Desplan et al. 1988). The HD-Zip I and HD-Zip II genes recognize the pseudopalindromic sequence CAATNATTG with the central nucleotides A/T and C/G, respectively, in the regulatory regions of target genes (Sessa et al. 1993; Palena et al. 1999). Whereas, the HD-Zip III and HD-Zip IV genes recognize GTAAT(G/C)ATTAC and CATT(A/T)AATG sequences, respectively, in their target genes (Sessa et al. 1998; Tron et al. 2001).
In this review, we provide new insights into the evolution and evolutionary processes that led to the functional redundancy and/or diversification of the HD-Zip I transcription factors in plants. We summarized the evolutionary history of the HD-Zip family, highlighting the important role of WGDs in the separation of four subfamilies in flowering plants and the functional diversity of the retained duplicates in the genome. To correlate the common evolutionary origin and functional conservation of orthologous and paralogous genes, we defined the phylogenetic relationships of the 483 HD-Zip I proteins from different species (including monocots, dicots, ferns and mosses) based on amino acid sequence similarity. We then compared their expression profiles in different tissues and under stress conditions. We also tracked the role of orthologous genes from different species and paralogous genes from the same species in regulating growth and development and in responding to stress. These analyzes confirmed the functional complexity of the HD-Zip I subfamily, resulting from its evolution in the context of WGD and the adaptation of different species to a rapidly changing environment. Many of these genes have similar structures, bind to the same DNA sequences in target genes and exhibit similar expression profiles during developmental processes and under stress, confirming their partial functional redundancy. However, some of them exhibit a functional diversity, manifested in altered expression patterns and related to the site-specificity of their activity (organs or tissues) or to external stimuli such as water availability and light. In addition, some members of HD-Zip I are involved in the regulation of species-specific leaf morphology and phenotypes. We discuss the role of changes in functional domains involved in DNA binding and protein interaction of HD-Zip I proteins and in cis-regulated regions of their target genes, which are responsible for the evolution of morphological diversity in plants and promote adaptive innovations through the formation of de novo regulatory systems to control various biological processes. Understanding the role of the HD-Zip I subfamily in organism-environment interactions remains a challenge for evolutionary developmental biology (evo-devo).
Evolution of the Homeodomain Leucine Zipper (HD-Zip) Gene Family in Plants
Together with the availability of fully sequenced genomes, our knowledge of the evolution of the HD superfamily has increased significantly. Initial alignments of HD sequences from different taxa showed a closer evolutionary relationship between plants and animals than between different plant families (Bharathan et al. 1997). This suggests that the divergence of HDs began before the separation of plants, animals and fungi, and only later within the plant kingdom. In plants, almost all HD families except LD and NDX have already been identified in chlorophytes (Romani et al. 2018). Members of the LD family were found in charophytes and the NDX family only in land plants, suggesting that these families were lost in chlorophytes due to gene loss or arose later in evolution. Among the HD superfamily, the HD-Zip family is one of the best characterized gene groups in plants. The numerous phylogenetic studies have shed light on the complex evolution of the four classes in this family (Prigge and Clark 2006; Mukherjee et al. 2009; Romani et al. 2018). However, little is known about its origin, especially in the early stages of plant differentiation. Therefore, it is controversial whether the HD-Zip family arose as a new lineage within the HD gene superfamily or whether it was formed by fusion of protein domains (Schena and Davis 1992). The evolutionary model of the HD-Zip gene family assumes that a single HD-Zip-like protein arose during early chlorophyte evolution and then gradually diverged into the four classes in charophytes (Fig. 1). The HD-Zip II-like protein first evolved in chlorophytes and was later inherited by streptophytes. Phylogenetic analysis of these proteins in different plant taxa revealed two clades with presence or absence of the specific CPSCE sequence in their C-terminal part (Romani et al. 2018). These two clades have a common origin and underwent the duplication event before the divergence of Klebsormidiales. However, the clade with the CPSCE-less sequence is conserved only in charophytes and mosses and has been lost in vascular plants. This evolutionary fate of two lineages of HD-Zip II was further supported by differences in the intron–exon structure of genes with CPSCE-less and CPSCE motifs. In addition, the ZIBEL-like motif was detected at the N-terminus of HD-Zip II proteins in Klebsormidiales and is also conserved in all land plants. According to the phylogenetic tree, HD-Zip I, HD-Zip III and HD-Zip IV are monophyletic groups in land plants (Romani et al. 2018). The HD-Zip III- and HD-Zip IV-like proteins evolved from a common ancestor during the early evolution of streptophytes but before the divergence of Klebsormidium. In parallel, the STeroidogenic Acute Regulatory protein-related lipid Transfer/START-Associated Domain (START/SAD) motifs were incorporated into the C-terminal end of the HD-Zip III and HD-Zip IV proteins. The methionine-glutamine-lysine-histidine-leucine-alanine (MEKHLA) motif was found only in the HD-Zip III proteins, appeared in the Klebsormidium species and was retained in later stages of evolution. Although the HD-Zip III and HD-Zip IV genes have a common ancestor, they showed different evolution of exon–intron structure. The gene structure of the members of HD-Zip IV with about 10 exons is conserved among plant taxa, while HD-Zip III underwent modifications that resulted in changes in the number of their exons increasing from 13 in Klebsormidium nitens to 18 in Marchantia and Arabidopsis. In contrast, the phylogenetic analyzes of HD-Zip I-like proteins in different plant taxa were unable to clearly determine their evolutionary fate because of divergent sequences. This is probably due to fact that they evolved faster than the HD-Zip II proteins in both monocotyledons and dicotyledons (Sakakibara et al. 2001). The HD-Zip I genes eventually evolved in Klebsormidiophyceae, which is confirmed by the specific gene structure in Klebsormidiales with an intron within the HD domain that was not found in Marchantia and other land plants. Some HD-Zip I proteins have the AHA motif at the C-terminus, which probably occurred before the divergence of Charales and is conserved in other charophytes. In land plants, this AHA motif is conserved in bryophytes, but in some angiosperm lineages it degenerated later. The HD-Zip I proteins of the mosses also share other regulatory motifs in their N-and C-terminal regions. All this suggests a low level of diversification of HD-Zip I in Physcomitrella patens with functional redundancy.

The evolutionary model of the HD-Zip gene family in plants. Major events including duplications (marked by asterisks) or gene loss (marked by lightning), which took place at different stages of the plant evolution, have been shown. The additional domains are colored in red (Color figure online)
In the evolution of the HD-Zip family, at least some duplication events have been confirmed to have had an impact on its expansion and divergence (Romani et al. 2018). Currently, the number of HD-Zip genes ranges from 15 in H. annuus to 180 in Glycine max (Table S2). The number of each HD-Zip subfamily also varies widely among plant species. The fewest genes were detected in subfamily I, II, III and IV in Selaginella moellendorffii (4), Spinacia oleracea (3), Amborella trichopoda (7), and P. patens (4), respectively (Table S3). In turn, the most genes in subfamilies I, II, III and IV were identified in Gossypium hirsutum (57), Triticum aestivum (39), Gossypium hirsutum (28) and Solanum lycopersicum (45), respectively. These differences are due to the polyploidy status of the plant and evolutionary events, including WGDs.
Phylogeny and Molecular Characteristics of the HD-Zip Subfamily I
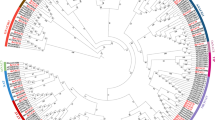
The HD-Zip I subfamily is one of the most abundant groups of homeoboxes, whose members play key roles in plant development and response to environmental conditions (Vlad et al. 2014). Phylogenetic analysis of 483 HD-Zip I proteins of several monocotyledonous and dicotyledonous species confirmed their evolutionary relationships (Fig. 2; Table S4). They are clearly clustered into ten early identified clades: α, β1, β2, γ, δ, ε, φ1, φ2, ζ and η (Henriksson et al. 2005; Li et al. 2019a, b). Similar to previous studies, the clade ζ includes only HD-Zip I proteins from monocotyledons and the clades β2, η and φ2 represent only the HD-Zip I proteins from dicotyledons. The expansion of the HD-Zip I subfamily by WGD was observed in all species studied, including the Brassica, Oryza, Gossypium, Zea, Populus and Nicotiana genera. The HD-Zip I subfamily was the best characterized in the model plant A. thaliana. Therefore, in this review, all data on the structure and function of these genes are presented for A. thaliana and then related to other species. In Arabidopsis, the HD-Zip I subfamily includes 17 members divided into eight clades: α-AtHB3, AtHB20, AtHB13 and AtHB23; β1-AtHB1; β2-AtHB5, AtHB6 and AtHB16; ε-AtHB22 and AtHB51; δ-AtHB21, AtHB40 and AtHB53; γ-AtHB7 and AtHB12; φ1-AtHB52; and φ2-AtHB54 (Fig. S1A). These clades, with the exception of β1, φ1 and φ2, include pairs of paralogous genes located in the WGD-derived duplicated chromosomal regions (Henriksson et al. 2005). Based on the duplication history of the A. thaliana genome, the origin of several pairs of paralogous genes HD-Zip I has been linked to the major gene duplication events that occurred 20 to 60 million years ago (Blanc et al. 2003). Among them, AtHB13 and AtHB23, AtHB7 and AtHB12, AtHB6 and AtHB16, AtHB21 and AtHB40, and AtHB3 and AtHB20 evolved from the recent segmental genome duplication. In contrast, the paralogous genes AtHB21 and AtHB53 likely evolved from ancient segmental duplication (Blanc et al. 2003). The phylogenetically related genes show high sequence similarity with the same exon-intron organization and domain topology (Figs. S1B and S1C; Table S5). In particular, the regions corresponding to HD are highly conserved, whereas LZ shows greater sequence variation. Because LZ is a dimerization motif that is a prerequisite for DNA binding, differences within this motif may affect the efficiency of this process. To form dimers required for recognition of the pseudopalindromic sequence CAAT(A/T)ATTG, Zip folds into an α-helical conformation with a leucine residue at every seventh position on the same side of the helix. This structure allows dimer formation through hydrophobic interactions (Landschulz et al. 1988; Chan et al. 1998; Tron et al. 2004). However, other differences in gene structure and functional domains have been observed between distantly related species. As mentioned previously, the HD-Zip I genes of Kleobsormidiales show a different exon–intron structure than Marchantia and other land plants, including the additional intron within the HD domain (Romani et al. 2018). Also, the fern Ceratopteris richardii HD-Zip I genes have seven additional amino acids in half of LZ (Aso et al. 1999). In addition, differences were observed in two amino acid residues in LZ between ferns and angiosperms, which are important for dimerization (Sakakibara et al. 2001). This suggests that DNA binding preference and regulatory capability of HD-Zip I members are different in these plant lineage. Recent reports confirmed the important role of additional motifs in the carboxy- and amino- terminal regions (CTR and NTR) that may be involved in protein interaction (Arce et al. 2011; Capella et al. 2014; Fig. S1B). The variability of these motifs may account for the fact that related HD-Zip I proteins, although recognizing and binding to the same pseudopalindromic sequence CAAT(A/T)ATTG, interact with different specific partner proteins to regulate distinct signaling pathways. Among these additional motifs, the AHA motif downstream of LZ form an amphipathic and negatively charged helix contacting components of the basal transcription complex (Döring et al. 2000). Transactivation of CTR was experimentally confirmed for the Arabidopsis AtHB1, AtHB7, AtHB12 and AtHB13 proteins (Arce et al. 2011; Capella et al. 2014). Also, in the barley HvHox2 protein, 14 additional amino acids were discovered within CTR that are characteristic for an AHA motif (Sakuma et al. 2010). In contrast, in sunflower HD-Zip I proteins defined the role of the N-terminal flexible arm in DNA–protein interaction (Palena et al. 2001).

The phylogenetic relationship of the HD-Zip I proteins in plant species based on data collected in PhyloGenes relase (version 3.0) (http://www.phylogenes.org/). Amino acid sequences of the HD-Zip I subfamily were selected from 18 species representing the major plant taxa: Arabidopsis thaliana, Brassica rapa, Brassica napus, Oryza sativa, Hordeum vulgare, Solanum lycopersicum, Solanum tuberosum, Glycine max, Nicotiana tabacum, Medicago truncatula, Triticum aestivum, Zea mays, Junglas regia, Cucumis sativa, Helianthus annuus, Populus trichocarpa, Gossypium hirsutum and Capsicum annuum. The name of genes have been denoted according to the published data: B. rapa (Khan et al. 2018), O. sativa (Agalou et al. 2008), H. vulgare (Li et al. 2019a, b), S. lycopersicum (Zhang et al. 2014), S. tuberosum (Li et al. 2019a, b), G. max (Chen et al. 2014), N. tabacum (Li et al. 2019a, b), M. truncatula (Li et al. 2022), T. aestivum (Yue et al. 2018), Z. mays (Mao et al. 2016), C. sativa (Liu et al. 2013), P. trichocarpa (Hu et al. 2012), G. hirsutum (Zhang et al. 2016) and C. annuum (Zhang et al. 2021). The phylogenetic tree was constructed using MEGA 11 program with the NJ method and 1000 bootstrap replicates. The analyzed proteins were divided into twelve clades designated α, β1, β2, γ, δ, ε, φ1, φ2, ξ and η, and indicated by a specific color (Henriksson et al. 2005; Li et al. 2019a, b) (Color figure online)
Biological Role of the HD-Zip I Transcription Factors
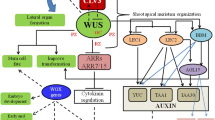
The first HD-containing gene discovered in plants was the KNOTTED1 gene of maize, which is involved in leaf differentiation (Vollbrecht et al. 1991). Since then, entire complements of HD genes have been identified and functionally characterized in many species, demonstrating their diverse biological functions. The HD-Zip I genes are key regulators of plant developmental processes and stress responses (Capella et al. 2015). Transcriptomic studies have shown that they are expressed at different developmental stages and in different organs such as leaves, roots, stems, flowers and fruits, and that they are associated with different external stimuli such as light, phytohormones and stress conditions (Figs. S2 and S3). Some reports confirm that members of HD-Zip I can also regulate developmental processes independent of stress conditions (Komatsuda et al. 2007; Gong et al 2019; Sharif et al. 2021). The summary model of the functions of HD-Zip I genes is shown in Fig. 3.

The transcriptional activity of the HD-Zip I genes in different biological processes in plants. Positive and negative regulatory actions are indicated by red arrows and blue lines with bars, respectively. Two-letter prefixes for sequence identifiers indicate species of origin: Le-Lycopersicon esculentum, At-Arabidopsis thaliana, Mt-Medicago truncatula (Color figure online)
Regulation of Plant Growth and Development Under Different External Stimuli
The HD-Zip I genes are key regulators of plant growth and development and control various signaling pathways involved in these processes through interactions with other genes. The mechanisms regulating these processes gradually evolved after the divergence of mosses and vascular plants, i.e. around 430 Mya and contributed to their phenotypic diversity and adaptation to different environmental niches. In the moss P. patens, the identified Pphb7 gene is involved in epidermal cell differentiation and regulates various features of rhizoids under auxin induction (Sakakibara et al. 2003). This function of the HD-Zip I gene was not confirmed in flowering plants, so Sakakibara and co-workers proposed that Pphb7 belongs to a new class of regulators that negatively control total chloroplast mass per cell. In A. thaliana, most of 17 HD-Zip I genes that arose by WGD events show only partial functional redundancy in plant growth and development (Fig. S2). The comprehensive transcriptional studies confirmed that 4 and 11 members of the HD-Zip I subfamily are involved in flower and root development, respectively (Henriksson et al. 2005). AtHB1, AtHB7, AtHB12 and AtHB40 are expressed in flowers. Whereas, AtHB1, AtHB3, AtHB5, AtHB6, AtHB7, AtHB12, AtHB16, AtHB20, AtHB21, AtHB23 and AtHB40 are expressed in roots of seedlings growing under well-watered conditions. However, some genes show specific expression patterns and root tissue- or cell-specificity (Perotti et al. 2021, for review). AtHB3 and AtHB20, belonging to the phylogenetic clade α, are up-regulated in roots, and AtHB5 and ATHB6 from the phylogenetic clade β2 are down-regulated in roots but up-regulated in the hypocotyl. The paralogous genes AtHB13 and AtHB23 are induced in the same root cell types and AtHB23 is also transcriptionally activated in stem cells and in the early stages of the lateral root primordium (Perotti et al. 2019). Several members of HD-Zip I with the highest expression level of AtHB20 were detected in both root hair and non-hair epidermal cells. In contrast, AtHB1, AtHB5, AtHB6, AtHB12 and AtHB16 were expressed in the xylem and AtHB1, AtHB16, AtHB51 and AtHB53 were expressed in the procambium. The paralogous genes AtHB7 and AtHB12 also show functional complexity in developmental processes, reflected in different expression patterns and their root tissue specificity. Only AtHB12 was expressed in lateral root primordia, young leaves and inflorescence stems (Hur et al. 2015; Perotti et al. 2021, for review). These genes are also upregulated in flowers and siliques but expressed at different developmental stages (Olsson et al. 2004; Ré et al. 2014; Ribone et al. 2015; Figs. S2 and S4). Transcriptome analyzes showed that HD-Zip I genes in other species are also expressed in different organs and developmental stages, but the evolutionarily related genes show only partial functional similarity (Agalou et al. 2008; Fig. S3). In rice, the OsHOX6, OsHOX8, OsHOX22 and OsHOX23 genes are highly expressed in seedlings. OsHOX3, OsHOX12 and OsHOX24 are expressed during grain ripening and development, whereas OsHOX8, OsHOX16, OsHOX20, OsHOX22 and OsHOX25 are induced during the maturation of panicles (Fig. S3). In cereals, some members of HD-Zip I are also involved in developmental processes. For example, the paralogous genes VRS1 (HvHox1) and HvHox2 of barley have conserved transcriptional activity but show differences in expression, i.e. VRS1 is expressed in the lateral spikelet primordia in immature spikes, whereas HvHox2 is broadly induced in different tissues (Komatsuda et al. 2007; Sakuma et al. 2010).
The regulation of plant growth and development is a complex process that requires the integration of multiple factors and signaling pathways and is often controlled by external stimuli. The role of HD-Zip I genes in these processes has been investigated using several approaches, including overexpression or RNAi suppression of target genes. The AtHB1 gene was the first member of this subfamily to be identified in A. thaliana, and it proved to be a mediator in determining leaf cell fate in AtHB1-overexpressing tobacco lines (Aoyama et al. 1995). The authors found that leaf development controlled by AtHB1 was dependent on light conditions. They also observed that tobacco plants overexpressing AtHB1 and growing in the dark exhibited de-etiolated phenotype. However, subsequent studies with A. thaliana 35S::AtHB1 plants and athb1 mutants failed to confirm these results (Capella et al. 2015). The Capella and coworkers suggested that the de-etiolated phenotype observed in tobacco plants may be an artefact due to heterologous expression, controlled by other members of the HD-Zip I subfamily or the result of an interaction of AtHB1 and other HD-Zip I genes. In contrast, they showed that AtHB1 is expressed in the hypocotyl and roots of young seedlings and is involved in hypocotyl elongation under short-day conditions (Capella et al. 2014, 2015; Fig. 3). Based on the above studies, they proposed a model for the regulation of hypocotyl elongation in which PHYTOCHROME-INTERACTING FACTOR 1 (PIF1) and AtHB1 play key roles and which is dependent on light conditions (Fig. 4). According to this model, PIF1 binds to the promoter of AtHB1, which controls downstream expression of other genes to promote hypocotyl elongation under a short-day photoperiod. AtHB1 induces the expression of CRU3 and represses the expression of PLP4 (a patatin-related phospholipase A responsible for cell elongation modifications), XTH26 (involved in cell wall loosening) and GAUT (belongs to the glycosyltransferase family 8 subgroup and is involved in secondary cell wall integrity). Other members of the HD-Zip I subfamily are also involved in the regulation of hypocotyl length in response to light conditions. For example, the AtHB16 and AtHB23 genes oppositely regulate hypocotyl elongation in response to blue and red light, respectively (Wang et al. 2003; Choi et al. 2014; Fig. 3). The HD-Zip I genes are also involved in other developmental processes such as leaf morphology and morphogenesis, root development and regulation of flowering time (Fig. 3). The A. thaliana AtHB1, AtHB6, AtHB13 and AtHB16 genes are involved in the regulation of leaf shape, including the promotion of leaf margins, but they control their different components (Wang et al. 2003; Lechner et al. 2011; Gao et al. 2014; Miguel et al. 2020). For example, AtHB1 regulates CUC2 expression by specifically repressing miR164 levels to promote leaf margins (Miguel et al. 2020; Fig. 4). The crucial role in regulating leaf development was also confirmed for AtHB3, AtHB13, AtHB20 and AtHB23 in transgenic A. thaliana overexpressing these genes (Henriksson et al. 2005). The paralogous genes AtHB13 and AtHB23 have partially similar functions in inhibiting inflorescence stem elongation but show no functional redundancy in other biological processes (Ribone et al. 2015). AtHB23 is expressed in the adaxial region of leaves and is involved in hypocotyl elongation and cotyledon expansion under red light (Kim et al. 2007; Choi et al. 2014). Whereas, AtHB13 is involved in stress responses, pollen hydration and seed germination (Cabello and Chan 2012; Gao et al. 2014; Ribone et al. 2015).

The model of regulation of various biological processes depending on HD-Zip I. Two-letter prefixes for sequence identifiers indicate species of origin: Le-Lycopersicon esculentum, At-Arabidopsis thaliana, Mt-Medicago truncatula
Many orthologs of A. thaliana AtHB genes identified in different plant species have been shown to be involved in the regulation of plant growth and development (Figs. 2 and S3). However, despite their evolutionary relationship, the orthologs exhibit only partial functional similarity at the transcriptional level. Many of them have evolved novel tissue preferences that have led to changes in morphological traits and facilitated adaptation to different environmental conditions. The Medicago truncatula MtHB1 gene, which is an ortholog of A. thaliana AtHB7 and AtHB12, is also induced in roots under exogenous ABA treatment and salt stress, resulting in inhibition of lateral root growth (Ariel et al. 2010). AtHB13 and its orthologous gene HaHB1 in sunflower are involved in leaf development. The transgenic plants overexpressing these genes show similar morphological characteristics such as serrated leaves and a differentiated cotyledon phenotype under sucrose 4% (Hanson et al. 2002). In contrast, orthologs of AtHB1 gene showed only partially conserved functions in organ development. The tomato LeHB-1 gene is involved in the flowering process by regulating floral organogenesis, carpel development and ripening (Lin et al. 2008). The grape VvHB58 gene controls fruit size, seed number and inhibits pericarp expansion in tomato by modulating various hormonal pathways (Li et al. 2019a, b). Whereas, the soybean GmHDZ20 gene is involved in regulating leaf morphology and the number of seeds per pod (Yang et al. 2020). Interestingly, the complex role of the Mict gene of Cucumis sativus, which belongs to the HD-Zip I subfamily, was found in multicellular trichome development (Zhao et al. 2015). At an early stage of trichome development, Mict negatively and positively controls trichome spacing and base cell morphogenesis, respectively. At later stages, Mict positively controls apical cell and stalk cell morphogenesis. A spectacular example of functional diversity becoming visible as phenotypic variation was observed in the paralogous genes VRS1 (HvHox1) and HvHox2 in barley (Sakuma et al. 2010). Phylogenetic analysis showed that VRS1 is a paralogue of HvHox2 that arose by duplication of an ancestral gene (Sakuma et al. 2010; Fig. 2). However, during the evolution of barley (after the separation of the taxa Brachypodium and Pooideae), VRS1 acquired a new function restricted to the immature inflorescence. By comparing the sequences of both genes, a mutation in the third exon of VRS1 was found to contain a conserved motif at the C-terminus of HvHox2, resulting in its new functions (Sakuma et al. 2010). More detailed studies showed that loss of function of the VRS1 gene led to a complete conversion of the rudimentary lateral spikelets from a two-row to a six-row phenotype (Sakuma et al. 2010, 2013).
Hormone Signal Transduction and Stress Responses
As sessile organisms, plants had to develop new mechanisms to adapt to the rapidly changing environment. Plant stress responses controlled by transcription factors such as HD-Zip I are often associated with developmental plasticity resulting from the interplay of morphology and growth rate and hormone signaling pathways. The HD-Zip I genes show expression changes mainly in response to treatment with abscisic acid (ABA), known stress hormone in plants (Henriksson et al. 2005). In A. thaliana, most members of this subfamily are induced or repressed in response to exogenous ABA and various stresses such as drought, heat, salinity and cold (Fig. S4). However, due to their functional diversity, they show different responses to these stimuli. Some reports have highlighted the complex interactions between the HD-Zip I transcription factors and stress, which can be attributed to evolutionary events (Lee et al. 2001; Himmelbach et al. 2002; Olsson et al. 2004). There is a correlation between the function and evolutionary relatedness of the members of this subfamily. The paralogous gene pair AtHB40 and AtHB53 is induced by high temperature, osmotic stress and salinity (Perotti et al. 2021; Fig. 3). The paralogous gene pairs AtHB7 and AtHB12 and AtHB5 and AtHB6 are regulated by water deficit conditions in an ABA-dependent pathway (Söderman et al. 1999; Lee et al. 2001; Himmelbach et al. 2002; Olsson et al. 2004; Fig. 3). The AtHB7 and AtHB12 genes are induced by exogenous treatment with ABA, but their expression patterns differ depending on the developmental stage of the plant (Olsson et al. 2004). Only AtHB12 shows sensitivity of ABA during seed germination and reduces inflorescence stem growth by inhibiting hormone synthesis of gibberellic acid (GA) (Son et al. 2010). Further studies showed that they act as negative regulators in the ABA-dependent response to drought (Johannesson et al. 2003; Deng et al. 2006; Valdes et al. 2012). Thus, AtHB7 and AtHB12 are regulators of plant growth and development controlled by ABA and drought stress. The Arabidopsis HD-Zip I genes are also expressed under other stress conditions and show opposite expression patterns. The paralogous genes AtHB7 and AtHB12 are up-regulated under cold and salinity, whereas the paralogous genes AtHB13 and AtHB23 are repressed under osmotic stress and salinity (Perotti et al. 2021; Fig. 3). The authors also concluded that AtHB23 is essential for plant survival and adaptation to salt stress conditions by controlling the gravitropic response mediated by starch granule turnover. These results confirm that duplication followed by structural rearrangements of the HD-Zip I genes is responsible for their partial functional diversity toward opposing activities in repressing the ABA signaling network. Other members of HD-Zip I are also up- or down-regulated depending on stress conditions. AtHB1, for example, is induced in response to osmotic stress, UV-B, and high temperature, but is suppressed upon NaCl treatment (Perotti et al. 2021).
The biological functions of HD-Zip I genes associated with the stress response have also been studied in other monocotyledonous and dicotyledonous plants. Expression profiling of these genes revealed that many of them are induced by ABA, drought, cold and salt stresses. Despite their evolutionary relationship with the Arabidopsis HD-Zip I genes, they show only partially similar expression patterns under these stress conditions (Fig. S5). The orthologous genes of AtHB7 and AtHB12 have been identified and functionally characterized under abiotic stress conditions in other species such as sunflower, rice, tobacco and chickpea (Gago et al. 2002; Agalou et al. 2008; Ré et al. 2011; Sen et al. 2017). The studies based on the overexpression and repression of these genes show that only some of them retained their ancestral functions in regulating stress responses. The HaHB4 gene of H. annuus is involved in hormone-dependent responses (Gago et al. 2002). Similar to the AtHB7 and AtHB12 genes of A. thaliana, it is up-regulated in response to ABA and drought (Lee and Chun 1998). However, later studies show that only the HAHB4 gene confers drought tolerance to transgenic Arabidopsis plants under the control of constitutive or drought-inducible promoters (Dezar et al. 2005). This is related to its ability to protect plants from photooxidative stress by down-regulating the biogenesis of photosynthetic machinery (Manavella et al. 2006). Its expression also changes in response to jasmonic acid, mechanical damage, mannitol and ethylene during Spodoptera sp. attack (Dezar et al. 2005; Manavella et al. 2006, 2008). Detailed analyzes revealed that HAHB4 is an integral component of the phytohormone signaling pathways JA, ET and SA that regulate the response to biotic stress. Overexpression of HaHB4 also resulted in the decreased expression of ethylene signaling and biosynthesis genes confirming its involvement in the regulation of flower and fruit development (Manavella et al. 2006; Fig. 3). The Nicotiana attenuata NaHD12 gene is highly expressed in roots and leaves under exogenous ABA treatment and drought stress (Ré et al. 2011). The nahd20 mutant showed decreased expression of the NaNCED1 (ABA biosynthesis gene) and NaOSM1 (osmotin 1) genes under water deficit. In rice, the orthologous OsHOX6, OsHOX22 and OsHOX24 genes of the AtHB7 and AtHB12 genes of A. thaliana are also induced by drought stress, but their expression differs from that of their counterparts in Arabidopsis and also from each other (Agalou et al. 2008). The OxHOX6 gene shows high expression in all tissues under well-watered conditions and is upregulated under drought in the drought-sensitive rice cultivars. Whereas, the more closely related OxHOX22 and OsHOX24 genes are strongly expressed in leaves and panicles and only weakly expressed in other tissues under well-watered conditions (Fig. S3). Overexpression of these genes in the A. thaliana background resulted in the susceptibility of these plants to ABA, drought and salt at different developmental stages, confirming their negative regulation of response to these stressors (Agalou et al. 2008; Bhattacharjee et al. 2016). The negative role of HD-Zip I genes in stress responses has also been confirmed in other species. Overexpression of Jatropha curcas JcHDZ07 in transgenic A. thaliana plants increases their sensitivity to salt stress (Tang et al. 2019). RNAi suppression of the tomato SlHB2 gene also confirmed its role as a negative regulator of salt and drought stress responses (Hu et al. 2017). On the other hand, the positive role of HD-Zip I genes in regulation of salt stress response was also confirmed, but they are controlled by other mechanisms. Overexpression of the maize ZmHDZ10 gene in both A. thaliana and rice leads to increased tolerance to drought and salinity in these plants by ABA-dependent signal transduction (Zhao et al. 2014). Similarly, overexpression of the MtHB1 gene in M. truncatula plants had a positive effect on increasing their salinity tolerance through the root system control (Ariel et al. 2010). The main regulatory mechanism of this process is associated with the repression of the LOB BINDING DOMAIN (LBD1) gene by MtHB1 under salt stress and ABA treatment during root formation (Fig. 4). In apple, overexpression and RNAi suppression of the Malus domestica MdHB7-like gene showed that it is involved in the regulation of salt tolerance by controlling sugar accumulation (Zhao et al. 2021). In contrast, overexpression of chickpea CaHDZ12 in tobacco increased tolerance to osmotic stress by reducing the accumulation of ROS and sensitivity to ABA (Sen et al. 2017). The complex functions of the HD-Zip I transcription factors are also observed in the “resurrection plant” Craterostigma plantagineum. The CpHB4 and CpHB5 genes grouped in the phylogenetic tree with AtHB5, AtHB6, and AtHB16 and AtHB1, respectively, show down-regulation in leaves and roots during drought stress (Deng et al. 2002; Harris et al. 2011). Interestingly, the CpHB6 and CpHB7 genes grouped with AtHB52 and AtHB54 show different expression patterns (Deng et al. 2002; Harris et al. 2011). They are upregulated under drought stress, and the expression of CpHB7 is similar to that of AtHB6 under normal growth conditions (Deng et al. 2006). These results indicate that the function of some members of HD-Zip I is partially conserved in Arabidopsis and Craterostigma. In contrast, no correlation between phylogenetic relationship and functional conservation of HD-Zip I genes was found in Marchantia polymorpha, an early-divergent bryophyte, and A. thaliana. The MpC1HDZ gene is the single-copy ortholog with all the features of the HD-Zip I genes found in A. thaliana. This gene plays a specific role in biotic stress response not found in its orthologs in flowering plants (Romani et al. 2020). Romani and coworkers found that the functions of this gene are related to oil body formation, terpenoid biosynthesis and response to herbivory, but not to the regulation of response to abiotic stress. These studies clearly show that phylogenetic relationship of genes based on sequence similarity does not correlate with their functional conservation in evolutionarily distant species such as early-diverging land plants and angiosperms. One of the reasons for this functional diversity could be the WGD events that were followed by diploidization and genome rearrangements in flowering plant lineage, where newly formed genes acquired new functions.
Photomorphogenesis
Changes in plant growth and development occur frequently under light stimuli and are controlled by both the amount and wavelength of light. They are caused by information received from photoreceptors and the cascade of light-induced (or light-independent) responses leads to changes in gene transcription. Some members of HD-Zip I are involved in the modulation of light-dependent signaling networks (Henriksson et al. 2005; Fig. 3). The authors found that the expression of the AtHB52 gene is regulated by light, therefore it may be involved in photomorphogenesis (Fig. 3). Its expression was increased 30-fold in darkness compared to white light and four-fold in blue light. In contrast, the expression of AtHB53 is upregulated in darkness but not under blue light conditions. Transcript levels of AtHB5, AtHB6, AtHB7, AtHB12, AtHB13 and AtHB20 are higher in seedlings grown under white light than under blue light or in darkness. In other work, AtHB16 was found to be a component of light sensitization mechanisms in plants (Wang et al. 2003). This study confirmed the role of AtHB16 as a suppressor of flowering time sensitivity to photoperiod. The transgenic Arabidopsis plants with reduced levels of AtHB16 showed enhanced response of flowering time to photoperiod (Fig. 3). Moreover, this transcription factor may control plant development as a mediator of a blue light response by positively regulating blue light-dependent inhibition of hypocotyl growth. Other HD-Zip I, AtHB23 interacts with phytochrome B (phyB) and is involved in phyB-mediated light signal transduction in Arabidopsis (Choi et al. 2014). Molecular analysis of the Arabidopsis athb23 mutant revealed altered hypocotyl growth under R light and defects in phyB-dependent seed germination and cotyledon expansion. Choi and coworkers (2014) hypothesized that ATHB23 is a novel component of the phyB-mediated R-light signaling pathway.
Recent reports have shown that interactions between different HD-Zip families link both morphogenesis and environmental responses through phytohormone signaling pathways. Brandt and coworkers (2014) found that members of the HD-Zip I subfamily AtHB7 and AtHB12 and REV, which belong to HD-Zip III, oppositely regulate ABA-receptor proteins in a cell type specific manner.
Molecular Mechanisms of Target Gene Regulation by HD-Zip I Transcription Factors
The main role of TFs is to regulate cell- and tissue-specific gene expression by binding to the specific sequence in the promoters of target genes or by physically interacting with other proteins. The major functional elements of the HD-Zip I proteins are the well-characterized HD and LZ, which are responsible for DNA binding and dimerization, respectively. The HD-Zip I transcription factor specifically recognizes and binds the pseudopalindromic sequence CAAT(A/T)ATTG in promoters of target genes (Palena et al. 2001; Table S6). Recently, using the DAP-seq technique for in vitro TF-DNA binding assays, the pseudopalindromic sequence AAT(N)ATT was identified as a target for HD-Zip I (O’Malley et al. 2016). The DNA-binding process requires dimerization by Zip. Many of the HD-Zip I proteins are capable of forming homodimers, but some of them such as AtHB5, AtHB6, AtHB7, AtHB12 and AtHB16 also have the ability to form heterodimers with different affinities, depending on structural constraints (Johannesson et al. 2001). DNA-binding site preference for HD-Zip I is determined by residues in the helix III and the N-terminal arm of HD (Palena et al. 2001). Interestingly, the different binding preferences of the closely related AtHB7 and AtHB12 for this consensus motif have been demonstrated in planta and in vitro (Johannesson et al. 2001; Henriksson et al. 2005; Valdes et al. 2012). AtHB7 and AtHB12 do not interact in vitro with the CAATNATTG sequence recognized by AtHB1, AtHB3, AtHB5, AtHB6, AtHB13 and AtHB16, despite high sequence similarity in the DNA binding assay (Johannesson et al. 2001). On the other hand, they bind to this sequence in planta and activate transcription (Henriksson et al. 2005; Valdes et al. 2012). Johannesson and coworkers suggest that the differences in phosphorylation sites in HD between AtHB7 and AtHB12 and other HD-ZIP I proteins may involve posttranslational modifications and affect DNA binding preference. Although most HD-Zip I proteins bind to the same DNA sequence, they exhibit different expression patterns and regulatory properties. Therefore, other functional elements are also considered that may be involved in the interaction with target proteins and affect the different functions of these proteins. These include additional motifs in the CTR and NTR regions of the HD-Zip I proteins that could play an activating or regulatory role by physical interaction with different specific partner proteins (Fig. S1B).
Recently, a general model for the organization of functional regions in the HD-Zip I proteins was proposed (Arce et al. 2011). According to this model, they characterize the presence of HD, Zip, AHA motif in the C-terminal region and regulatory motifs in the N- and C-terminal regions. Although HD and LZ are highly conserved, the N- and C-terminal regions show differences and not all motifs were found in each protein (Fig. S1B). The variability of the N- and C-terminal regions could possibly lead to the differences in the activity of the members of HD-Zip I. The number and distribution of regulatory motifs correlate with phylogenetically distinct groups within the HD-Zip I subfamily (Fig. 2). The importance of HD and LZ for protein-protein interaction has been experimentally confirmed for AtHB6 and AtABI1 (Abscisic acid Insensitive 1) and AtHB6 and BPM3, which belongs to the MATH-BTB family, among others (Himmelbach et al. 2002; Lechner et al. 2011). The functional domain of AtABI1 and a serine residue within the HD of AtHB6 were found to be responsible for their physical interaction. The LZ of AtHB6 is recognized by BPM3 and this complex is identified by Cullin-RING E3 ubiquitin ligases 3 (CRL3), which facilitates the transfer of ubiquitin moieties to AtHB6 and initiates its degradation by the 26S proteasome. A similar direct physical interaction was confirmed in vivo between AtHB12 and AtABI2 (Valdes et al. 2012). These results suggest that post-translational modifications and protein–protein interactions influence the functions of HD-Zip I. Regulatory motifs located in the N- and C-terminal regions also play important role in these processes. For example, the different versions of the AHA motif were also found in the C-terminal region of the Arabidopsis proteins AtHB1, AtHB7, AtHB12 and AtHB13, which act as transcriptional activators (Lee et al. 2001; Fig. S1B). Moreover, this motif may be involved in the transactivation process, as exemplified by AtHB1 by using a yeast one-hybrid approach (Arce et al. 2011). Previous studies have shown that the AHA motif is involved in interactions with proteins representing the basal transcription machinery (Döring et al. 2000; Kotak et al. 2004). The later experimental studies showed that AtHB1 interacts with AtTBP2, AtHB12 with AtTFIIB and AtHB7 with AtTBP2 and AtTFIIB (Capella et al. 2014). In barley, amino acids characteristic of the AHA motif were detected in the C-terminal region of the HvHox2 protein (Sakuma et al. 2010). Interestingly, this sequence has been lost in the closely related VRS1 protein, contributing to a tl mutation mimicking effect (Hofer et al. 2009). Therefore, motifs such as AHA could be a potential source of functional divergence between members of the HD-Zip I TFs. Many acidic serine-rich motifs have also been found in the C- and-N-terminal regions of HD-Zip I proteins, which may be involved in activation and phosphorylation or sumoylation processes (Arce et al. 2011). The proximal region of the C-terminal region (adjacent to the LZ) is rich in serine residues that are putative phosphorylation sites (Arce et al. 2011). In addition, putative sumoylation sites were found (Arce et al. 2011). Some potential residues for these posttranslational modifications were also found in the N-terminal region of these proteins. The functions of the C-terminal region in activation and phosphorylation processes for HD-Zip I proteins have been studied experimentally. For example, the phosphorylation of AtHB6 by PKA kinase inhibits its DNA-binding activity in vitro (Himmelbach et al. 2002). These studies suggest that posttranslational modifications such as protein phosphorylation and sumoylation may be important in controlling the activities of HD-Zip I proteins.
The binding of HD-Zip I to the promoters of target genes has been studied experimentally, but little is known about the downstream target genes regulated by these transcription factors. However, protein-DNA binding is dependent on external conditions. For example, the binding of HD-Zip I proteins to the upstream regulatory sequences of key components of the ABA signaling pathway, such as PP2CA, ABA receptors and SNF1-related Ser/Thr kinases (SnRKs) is ABA-dependent (Valdes et al. 2012). Valdes and coworkers showed that the binding of AtHB7 and AtHB12 proteins to the promoters of AtABI1 and AtABI2 is restricted to plants treated with ABA (Fig. 4). Moreover, the AtHB12 protein binds to the upstream regulatory sequences of AtHAB1, AtHAB2, AtAHG3 AtPYL5, AtPYL8, AtSnRK2.3 and SnRK2.8 only under the same stimulus conditions. In contrast, binding of the AtHB7 protein to the promoters of these genes is ABA-independent. In other studies, the binding of tomato LeHB-1 to the promoter of LeACO1 (ACC oxidase), a component of the ethylene biosynthesis pathway, was detected by a gel retardation assay (Lin et al. 2008). Further experimental analyses showed that LeHB-1 positively controls LeACO1 and that this interaction is important for the regulation of the ripening process (Fig. 3). Bioinformatic analyzes indicated putative LeHB-1 binding sites in the promoters of other ripening related genes such as LePG1, LeMADS-RIN, and LeNAC-NOR. This suggests that LeHB-1 may be involved in the control of flower development by other regulatory factors such as MADS-box genes (Fig. 3). In Medicago, MtHB1 represses LOB-binding domain 1 (LBD1), which forms the lateral root emergence phenotype (Ariel et al. 2010, Fig. 4). LBD1 is involved in auxin-regulated initiation of lateral roots and root formation in Arabidopsis and rice, respectively (Liu et al. 2005; Okushima et al. 2007). Recently, AtHB23 was also found to be involved in the gene regulatory network controlling root branching (Perotti et al. 2019). Perotti and coworkers (2019) observed by chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) that AtHB23 directly controls the functions of Lateral Organ Boundary (LBD16) and the auxin transporter gene LAX3 (Fig. 4). It suppresses the expression of LBD16, which is a critical element of lateral root development. Perotti and coworkers (2019) observed that AtHB23 is expressed in the early stages of secondary primordia and later represses LBD16 in the tertiary primordium lateral root development, further inhibiting root formation. AtHB23 also induces the auxin transporter gene LAX3. The researchers hypothesized that AtHB23 mediates the regulation of LAX3 by ARF7/19 (auxin response factors) (Fig. 4). Previously, AtHB5 was known to be a negative regulator of BDL (BODENLOS) expression and to control the BDL-dependent auxin response (De Smet et al. 2013; Fig. 4). These data clearly demonstrate that at least some HD-Zip I transcription factors are involved in regulating developmental processes via the auxin signaling pathway.
The HD-Zip I transcription factors can act as positive or negative regulators, which was confirmed in an in vivo promoter-reporter gene assay (Henriksson et al. 2005; Harris et al. 2011; Valdes et al. 2012). For example, AtHB5 induces transcription, and its close relative AtHB6 and the other paralogous gene pair AtHB7 and AtHB12 are both transcriptional activators and repressors (Himmelbach et al. 2002; Valdes et al. 2012). The authors reported the transcriptional relationships between group A protein phosphatase 2C (PP2CA) and HD-Zip I genes in drought- and ABA-mediated responses. These studies showed that AtHB6, AtHB7, and AtHB12 regulate the ABA signaling pathway in the primary drought response. They positively regulate the expression of PP2CA genes, including ABI1, and are required for their full transcriptional activity. In addition, AtHB7 and AtHB12 repress transcription of the ABA receptor genes PYL5 and PYL8, becoming a negative regulator of the ABA signaling pathway (Fig. 3). Taken together, these data provided evidence for the role of HD-Zip I genes in negatively regulating the ABA signaling pathway by upregulating PP2CA genes and downregulating ABA-receptor genes. Of note, other members of the HD-Zip I subfamily are also involved in the regulation of ABA signaling. However, this process might be more complex because their expression patterns differ depending on the stage of plant development and external conditions. For example, AtHB5 and AtHB20 are also negative regulators of ABA signal transduction during germination (Johannesson et al. 2003; Barrero et al. 2010). These overlapping functions of these genes in regulating ABA signal perception should be further investigated.
Conclusions and Future Perspectives
The HD proteins act as transcription factors and activate or repress the expression of targets in all living organisms. Their characteristic structural feature is the highly conservative HD domain of 60 amino acids. Together with access to numerous genome sequences, we have explored their evolutionary fate over hundreds of millions of years. Of the 11 families in the HD superfamily, only the HD-Zip family is unique in plants. The major regulatory elements of the HD-Zip proteins are the HD domain and the adjacent LZ motif, which are involved in regulating gene expression by binding to targets in gene promoters and interacting with other proteins, respectively. Recent reports suggest the presence of additional domains, motifs, or cofactors that may be required to increase the specificity of their functions. This suggests that there is not only one mechanism and one region in the HD-Zip proteins that regulate the functions of target genes. In plants, whole genome duplications (WGDs) and additional mechanisms also play important roles in the functional renewal of these transcription factors, which in turn may be responsible for drastic changes in an entire cascade of downstream target genes. Most HD-Zip proteins interact with multiple partners that are the same or different members of this subfamily (homo- or hetero-interactions) or with other regulatory factors such as other transcription factors, signaling molecules, or regulatory RNAs. In addition, the functional divergence of paralogs forms a complex network of interactions, for example, during stress. Therefore, the regulation of gene function is still poorly understood. Further studies should provide new insights into the complexity of gene regulation resulting from the functional differentiation of transcription factors such as homeobox genes.
References
Agalou A, Purwantomo S, Overnaes E et al (2008) A genome-wide survey of HD-Zip genes in rice and analysis of drought-responsive family members. Plant Mol Biol 66:87–103. https://doi.org/10.1007/s11103-007-9255-7
Aoyama T, Dong CH, Wu Y et al (1995) Ectopic expression of the Arabidopsis transcriptional activator Athb-1 alters leaf cell fate in tobacco. Plant Cell 7:1773–1785. https://doi.org/10.1105/tpc.7.11.1773
Arce AL, Raineri J, Capella M et al (2011) Uncharacterized conserved motifs outside the HD-Zip domain in HD-Zip subfamily I transcription factors; a potential source of functional diversity. BMC Plant Biol 11:42. https://doi.org/10.1186/1471-2229-11-42
Ariel FD, Manavella PA, Dezar CA, Chan RL (2007) The true story of the HD-Zip family. Trends Plant Sci 12:419–426. https://doi.org/10.1016/j.tplants.2007.08.003
Ariel F, Diet A, Verdenaud M et al (2010) Environmental regulation of lateral root emergence in Medicago truncatula requires the HD-Zip I transcription factor HB1. Plant Cell 22:2171–2183. https://doi.org/10.1105/tpc.110.074823
Arsovski AA, Pradinuk J, Guo XQ et al (2015) Evolution of cis-regulatory elements and regulatory networks in duplicated genes of Arabidopsis. Plant Physiol 169:2982–2991. https://doi.org/10.1104/pp.15.00717
Aso K, Kato M, Banks JA, Hasebe M (1999) Characterization of homeodomain-leucine zipper genes in the fern, Ceratopteris richardii and the evolution of the homeodomain-leucine zipper gene family in vascular plants. Mol Biol Evol 16:544–551. https://doi.org/10.1093/oxfordjournals.molbev.a026135
Barrero JM, Millar AA, Griffiths J et al (2010) Gene expression profiling identifies two regulatory genes controlling dormancy and ABA sensitivity in Arabidopsis seeds. Plant J 61:611–622. https://doi.org/10.1111/j.1365-313X.2009.04088.x
Bharathan G, Janssen BJ, Kellogg EA, Sinha N (1997) Did homeodomain proteins duplicate before the origin of angiosperms, fungi, and metazoa? Proc Natl Acad Sci USA 94:13749–13753. https://doi.org/10.1073/pnas.94.25.13749
Bhattacharjee A, Khurana JP, Jain M (2016) Characterization of rice homeobox genes, OsHOX22 and OsHOX24, and over-expression of OsHOX24 in transgenic Arabidopsis suggest their role in abiotic stress response. Front Plant Sci 7:627. https://doi.org/10.3389/fpls.2016.00627
Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13:137–144. https://doi.org/10.1101/gr.751803
Bowman JL, Kohchi T, Yamato KT et al (2017) Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 171:287-304.e15. https://doi.org/10.1016/j.cell.2017.09.030
Brandt R, Cabedo M, Xie Y, Wenkel S (2014) Homeodomain leucine-zipper proteins and their role in synchronizing growth and development with the environment. J Integr Plant Biol 56:518–526. https://doi.org/10.1111/jipb.12185
Cabello J, Chan RL (2012) The homologous homeodomain-leucine zipper transcription factors HaHB1 and AtHB13 confer tolerance to drought and salinity stresses via the induction of proteins that stabilize membranes. Plant Biotechnol J 10:815–825. https://doi.org/10.1111/j.1467-7652.2012.00701.x
Capella M, Re DA, Arce AL, Chan RL (2014) Plant homeodomain-leucine zipper I transcription factors exhibit different functional AHA motifs that selectively interact with TBP or/and TFIIB. Plant Cell Rep 33:955–967. https://doi.org/10.1007/s00299-014-1576-9
Capella M, Ribone PA, Arce AL, Chan RL (2015) Arabidopsis thaliana HomeoBox 1 (AtHB1), a Homedomain-Leucine Zipper I (HD-Zip I) transcription factor, is regulated by PHYTOCHROME-INTERACTING FACTOR 1 to promote hypocotyl elongation. New Phytol 207:669–682. https://doi.org/10.1111/nph.13401
Catarino B, Hetherington AJ, Emms DM et al (2016) The stepwise increase in the number of transcription factor families in the Precambrian predated the diversification of plants on land. Mol Biol Evol 33:2815–2819. https://doi.org/10.1093/molbev/msw155
Chan RL, Gago GM, Palena CM, Gonzalez DH (1998) Homeoboxes in plant development. Biochim Biophys Acta 1442:1–19. https://doi.org/10.1016/S0167-4781(98)00119-5
Chen X, Chen Z, Zhao H, Zhao Y, Cheng B, Xiang Y (2014) Genome-wide analysis of soybean HD-Zip gene family and expression profiling under salinity and drought treatments. PLoS One 9:e87156. https://doi.org/10.1371/journal.pone.0087156
Choi H, Jeong S, Kim DS et al (2014) The homeodomain-leucine zipper ATHB23, a phytochrome B-interacting protein, is important for phytochrome B-mediated red light signaling. Physiol Plant 150:308–320. https://doi.org/10.1111/ppl.12087
Deng X, Phillips J, Meijer AH et al (2002) Characterization of five novel dehydration-responsive homeodomain leucine zipper genes from the resurrection plant Craterostigma plantagineum. Plant Mol Biol 49:601–610. https://doi.org/10.1023/A:1015501205303
Deng X, Phillips J, Bräutigam A et al (2006) A homeodomain leucine zipper gene from Craterostigma plantagineum regulates abscisic acid responsive gene expression and physiological responses. Plant Mol Biol 61:469–489. https://doi.org/10.1007/s11103-006-0023-x
De Smet I, Lau S, Ehrismann JS et al (2013) Transcriptional repression of BODENLOS by HD-ZIP transcription factor HB5 in Arabidopsis thaliana. J Exp Bot 64:3009–3019. https://doi.org/10.1093/jxb/ert137
Desplan C, Theis J, O’Farrell PH (1988) The sequence specificity o homeodomain-DNA interaction. Cell 54:1081–1090. https://doi.org/10.1016/0092-8674(88)90123-7
Dezar CA, Gago GM, González DH et al (2005) HAHB-4, a sunflower homeobox-leucine zipper gene, confers drought tolerance to Arabidopsis thaliana plants. Transgenic Res 14:429–440. https://doi.org/10.1007/s11248-005-5076-0
Döring P, Treuter E, Kistner C et al (2000) The role of AHA motifs in the activator function of tomato heat stress transcription factors HsfA1 and HsfA2. Plant Cell 12:265–278
Gago GM, Almoguera C, Jordano J, Gonzalez DH, Chan RL (2002) Hahb-4, a homeobox-leucine zipper gene potentially involved in abscisic acid-dependent responses to water stress in sunflower. Plant Cell Environ 25:633–640. https://doi.org/10.1046/j.1365-3040.2002.00853.x
Gao D, Appiano M, Huibers RP et al (2014) Activation tagging of ATHB13 in Arabidopsis thaliana confers broad-spectrum disease resistance. Plant Mol Biol 86:641–653. https://doi.org/10.1007/s11103-014-0253-2
Gong S, Ding Y, Hu S et al (2019) The role of HD-Zip class I transcription factors in plant response to abiotic stresses. Physiol Plant 167:516–525. https://doi.org/10.1111/ppl.12965
Hanson J, Regan S, Engström P (2002) The expression pattern of the homeobox gene ATHB13 reveals a conservation of transcriptional regulatory mechanisms between Arabidopsis and hybrid aspen. Plant Cell Rep 21:81–89. https://doi.org/10.1007/s00299-002-0476-6
Harris JC, Hrmova M, Lopato S, Langridge P (2011) Modulation of plant growth by HD-Zip class I and II transcription factors in response to environmental stimuli. New Phytol 190:823–837. https://doi.org/10.1111/j.1469-8137.2011.03733.x
Henriksson E, Olsson ASB, Johannesson H et al (2005) Homeodomain leucine zipper class I genes in Arabidopsis. Expression patterns and phylogenetic relationships. Plant Physiol 139:509–518. https://doi.org/10.1104/pp.105.063461
Himmelbach A, Hoffmann T, Leube M et al (2002) Homeodomain protein ATHB6 is a target of the protein phosphatase ABI1 and regulates hormone responses in Arabidopsis. EMBO J 21:3029–3038. https://doi.org/10.1093/emboj/cdf316
Hofer J, Turner L, Moreau C et al (2009) Tendril-less regulates tendril formation in pea leaves. Plant Cell 21:420–428. https://doi.org/10.1105/tpc.108.064071
Hu R, Chi X, Chai G et al (2012) Genome-wide identification, evolutionary expansion, and expression profile of homeodomain-leucine zipper gene family in poplar (Populus trichocarpa). PLoS One 7:e31149. https://doi.org/10.1371/journal.pone.0031149
Hu J, Chen G, Yin W et al (2017) Silencing of SlHB2 improves drought, salt stress tolerance, and induces stress-related gene expression in tomato. J Plant Growth Reg 36:578–589. https://doi.org/10.1007/s00344-017-9664-z
Hur YS, Um JH, Kim S et al (2015) Arabidopsis thaliana homeobox 12 (ATHB12), a homeodomain-leucine zipper protein, regulates leaf growth by promoting cell expansion and endoreduplication. New Phytol 205:316–328. https://doi.org/10.1111/nph.12998
Johannesson H, Wang Y, Engström P (2001) DNA-binding and dimerization preferences of Arabidopsis homeodomain-leucine zipper transcription factors in vitro. Plant Mol Biol 45:63–73. https://doi.org/10.1023/A:1006423324025
Johannesson H, Wang Y, Hanson J et al (2003) The Arabidopsis thaliana homeobox gene ATHB5 is a potential regulator of abscisic acid responsiveness in developing seedlings. Plant Mol Biol 51:719–729. https://doi.org/10.1023/a:1022567625228
Johnson AD (2017) The rewiring of transcription circuits in evolution. Curr Opin Genet Devel 47:121–127. https://doi.org/10.1126/science.1152398
Khan N, Hu CM, Khan WA et al (2018) Genome-wide identification, classification and expression pattern of homeobox gene family in Brassica rapa under various stresses. Sci Rep 8:16265. https://doi.org/10.1038/s41598-018-34448-x
Kim YK, Son O, Kim MR et al (2007) ATHB23, an Arabidopsis class I homeodomain-leucine zipper gene, is expressed in the adaxial region of young leaves. Plant Cell Rep 26:1179–1185. https://doi.org/10.1007/s00299-007-0340-9
Komatsuda T, Pourkheirandish M, He C et al (2007) Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc Natl Acad Sci USA 104:1424–1429. https://doi.org/10.1073/pnas.0608580104
Kotak S, Port M, Ganguli A, Bicker F, von Koskull-Döring P (2004) Characterization of C-terminal domains of Arabidopsis heat stress transcription factors (Hsfs) and identification of a new signature combination of plant class A Hsfs with AHA and NES motifs essential for activator function and intracellular localization. Plant J 39:98–112. https://doi.org/10.1111/j.1365-313X.2004.02111.x
Lee YH, Oh HS, Cheon CI et al (2001) Structure and expression of the Arabidopsis thaliana homeobox gene ATHB-12. Biochem Biophys Res Commun 284:133–141. https://doi.org/10.1006/bbrc.2001.4904
Lee YH, Chun JY (1998) A new homeodomain-leucine zipper gene from Arabidopsis thaliana induced by water stress and abscisic acid treatment. Plant Mol Biol 37:377–384. https://doi.org/10.1023/A:1006084305012
Lechner E, Leonhardt N, Eisler H et al (2011) MATH/BTB CRL3 receptors target the Homeodomain-Leucine Zipper ATHB6 to modulate abscisic acid signaling. Dev Cell 21:1116–1128. https://doi.org/10.1016/j.devcel.2011.10.018
Landschulz WH, Johnson PF, McKnight SL (1988) The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240:1759–1764. https://doi.org/10.1126/science.3289117
Li Y, Bai B, Wen F et al (2019a) Genome-wide identification and expression analysis of HD-ZIP I gene subfamily in Nicotiana tabacum. Genes 10:575. https://doi.org/10.3390/genes10080575
Li Y, Zhang S, Dong R et al (2019b) The grapevine homeobox gene VvHB58 influences seed and fruit development through multiple hormonal signaling pathways. BMC Plant Biol 19:523. https://doi.org/10.1186/s12870-019-2144-9
Li X, Hou Y, Zhang F et al (2022) Identification and characterization of stress responsive homeodomain leucine zipper transcription factors in Medicago truncatula. Mol Biol Rep 49:3569–3581. https://doi.org/10.1007/s11033-022-07197-4
Lin Z, Hong Y, Yin M et al (2008) A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J 55:301–310. https://doi.org/10.1111/j.1365-313X.2008.03505.x
Liu HJ, Wang SF, Yu XB et al (2005) ARL1, a LOB-domain protein required for adventitious root formation in rice. Plant J 43:47–56. https://doi.org/10.1111/j.1365-313X.2005.02434.x
Liu W, Fu R, Li Q, Li J, Wang L, Ren Z (2013) Genome-wide identification and expression profile of homeodomain-leucine zipper Class I gene family in Cucumis sativus. Gene 531:279–287. https://doi.org/10.1016/j.gene.2013.08.089
Luscombe NM, Austin SE, Berman HM, Thornton JM (2000) An overview of the structures of protein-DNA complexes. Genome Biol 1:001. https://doi.org/10.1186/gb-2000-1-1-reviews001
Maere S, De Bodt S, Raes J et al (2005) Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA 102:5454–5459. https://doi.org/10.1073/pnas.0501102102
Manavella PA, Arce AL, Dezar CA et al (2006) Cross-talk between ethylene and drought signalling pathways is mediated by the sunflower Hahb-4 transcription factor. Plant J 48:125–137. https://doi.org/10.1111/j.1365-313X.2006.02865.x
Manavella PA, Dezar CA, Ariel FD et al (2008) The sunflower HD-Zip transcription factor HAHB4 is up-regulated in darkness, reducing the transcription of photosynthesis-related genes. J Exp Bot 59:3143–3155. https://doi.org/10.1093/jxb/ern170
Mao H, Yu L, Li Z, Liu H, Han R (2016) Molecular evolution and gene expression differences within the HD-Zip transcription factor family of Zea mays L. Genetica 144:243–257. https://doi.org/10.1007/s10709-016-9896-z
Miguel V, Manavella PA, Chan RL, Capella M (2020) The AtHB1 transcription factor controls the miR164-CUC2 regulatory node to modulate leaf development. Plant Cell Physiol 61:659–670. https://doi.org/10.1093/pcp/pcz233
Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) AP2/ERF family transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819:86–96. https://doi.org/10.1016/j.bbagrm.2011.08.004
Mukherjee K, Brocchieri L, Burglin TR (2009) A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol 26:2775–2794. https://doi.org/10.1093/molbev/msp201
Okushima Y, Fukaki H, Onoda M, Theologis A, Tasaka M (2007) ARF7 and ARF19 regulate lateral root formation via direct activation of LBD/ASL genes in Arabidopsis. Plant Cell 9:118–130. https://doi.org/10.1105/tpc.106.047761
Olsson AS, Engström P, Söderman E (2004) The homeobox genes ATHB12 and ATHB7 encode potential regulators of growth in response to water deficit in Arabidopsis. Plant Mol Biol 55:663–677. https://doi.org/10.1007/s11103-004-1581-4
O’Malley RC, Huang SC, Song L et al (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 165:1280–1292. https://doi.org/10.1016/j.cell.2016.04.038
Palena CM, Gonzalez DH, Chan RL (1999) A monomer-dimer equilibrium modulates the interaction of the sunflower homeodomain leucine-zipper protein Hahb-4 with DNA. Biochem J 341:81–87
Palena CM, Tron AE, Bertoncini CW et al (2001) Positively charged residues at the N-terminal arm of the homeodomain are required for efficient DNA binding by homeodomain-leucine zipper proteins. J Mol Biol 308:39–47. https://doi.org/10.1006/jmbi.2001.4563
Paris M, Kaplan T, Li XY et al (2013) Extensive divergence of transcription factor binding in Drosophila embryos with highly conserved gene expression. PLoS Genet 9:e1003748. https://doi.org/10.1371/journal.pgen.1003748
Perotti MF, Ribone PA, Cabello JV, Ariel FD, Chan RL (2019) AtHB23 participates in the gene regulatory network controlling root branching, and reveals differences between secondary and tertiary roots. Plant J 100:1224–1236. https://doi.org/10.1111/tpj.14511
Perotti MF, Arce AL, Chan RL (2021) The underground life of homeodomain-leucine zipper transcription factors. J Exp Bot 72:4005–4021. https://doi.org/10.1093/jxb/erab112
Prigge MJ, Clark SE (2006) Evolution of the class III HD-Zip gene family in land plants. Evol Dev 8:350–361. https://doi.org/10.1111/j.1525-142X.2006.00107.x
Prud’homme B, Gompel N, Carroll SB (2007) Emerging principles of regulatory evolution. Proc Natl Acad Sci USA 104:8605–8612. https://doi.org/10.1073/pnas.0700488104
Ré DA, Dezar CA, Chan RL, Baldwin IT, Bonaventure G (2011) Nicotiana attenuata NaHD20 plays a role in leaf ABA accumulation during water stress, benzylacetone emission from flowers, and the timing of bolting and flower transitions. J Exp Bot 62:155–166. https://doi.org/10.1093/jxb/erq252
Ré DA, Capella M, Bonaventure G, Chan RL (2014) Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol 14:1471–2229. https://doi.org/10.1186/1471-2229-14-150
Rensing SA (2014) Gene duplication as a driver of plant morphogenetic evolution. Curr Opin Plant Biol 17:43–48. https://doi.org/10.1016/j.pbi.2013.11.002
Ribone PA, Capella M, Chan RL (2015) Functional characterization of the homeodomain leucine zipper I transcription factor AtHB13 reveals a crucial role in Arabidopsis development. J Exp Bot 66:5929–5943. https://doi.org/10.1093/jxb/erv302
Romani F, Reinheimer R, Florent SN et al (2018) Evolutionary history of HOMEODOMAIN LEUCINE ZIPPER transcription factors during plant transition to land. New Phytol 219:408–421. https://doi.org/10.1111/nph.15133
Romani F, Banić E, Florent SN et al (2020) Oil body formation in Marchantia polymorpha is controlled by MpC1HDZ and serves as a defense against arthropod herbivores. Curr Biol 30:2815–2828. https://doi.org/10.1016/j.cub.2020.05.081
Romani F, Moreno JE (2021) Molecular mechanisms involved in functional macroevolution of plant transcription factors. New Phytol 230:1345–1353. https://doi.org/10.1111/nph.17161
Rosas U, Mei Y, Xie Q et al (2014) Variation in Arabidopsis flowering time associated with cis-regulatory variation in CONSTANS. Nat Commun 5:3651. https://doi.org/10.1038/ncomms4651
Sakakibara K, Nishiyama T, Kato M, Hasebe M (2001) Isolation of homeodomain-leucine zipper genes from the moss Physcomitrella patens and the evolution of homeodomain-leucine zipper genes in land plants. Mol Biol Evol 18:491–502. https://doi.org/10.1093/oxfordjournals.molbev.a003828
Sakakibara K, Nishiyama T, Sumikawa N et al (2003) Involvement of auxin and a homeodomain-leucine zipper I gene in rhizoid development of the moss Physcomitrella patens. Develop 130:4835–4846. https://doi.org/10.1242/dev.00644
Sakuma S, Pourkheirandish M, Matsumoto T et al (2010) Duplication of a well-conserved homeodomain-leucine zipper transcription factor gene in barley generates a copy with more specific functions. Funct Integr Genomics 10:123–133. https://doi.org/10.1007/s10142-009-0134-y
Sakuma S, Pourkheirandish M, Hensel G et al (2013) Divergence of expression pattern contributed to neofunctionalization of duplicated HD-Zip I transcription factor in barley. New Phytol 197:939–948. https://doi.org/10.1111/nph.12068
Schena M, Davis RW (1992) HD-Zip proteins: members of an Arabidopsis homeodomain protein superfamily. Proc Nat Acad Sci USA 89:3894–3898. https://doi.org/10.1073/pnas.89.9.3894
Sen S, Chakraborty J et al (2017) Chickpea WRKY70 regulates the expression of a Homeodomain-Leucine Zipper (HD-Zip) I transcription factor CaHDZ12, which confers abiotic stress tolerance in transgenic tobacco and chickpea. Plant Cell Physiol 11:1934–1952. https://doi.org/10.1093/pcp/pcx126
Sharif R, Raza A, Chen P et al (2021) HD-ZIP gene family: potential roles in improving plant growth and regulating stress-responsive mechanisms in plants. Genes 12:1256. https://doi.org/10.3390/genes12081256
Shiu SH et al (2005) Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol 139:18–26. https://doi.org/10.1104/pp.105.065110
Sessa G, Morelli G, Ruberti I (1993) The Athb-1 and Athb-2 HD-ZIP domains homodimerize forming complexes of different DNA-binding specificities. EMBO J 12:3507–3517. https://doi.org/10.1002/j.1460-2075.1993.tb06025.x
Sessa G, Steindler C, Morelli G, Rubert I (1998) The Arabidopsis Athb-8, -9 and -14 genes are members of a small gene family coding for highly related HD-Zip proteins. Plant Mol Biol 38:609–622. https://doi.org/10.1023/a:1006016319613
Son O, Hur YS, Kim YK et al (2010) ATHB12, an ABA-inducible homeodomain-leucine zipper (HD-Zip) protein of Arabidopsis, negatively regulates the growth of the inflorescence stem by decreasing the expression of a gibberellin 20-oxidase gene. Plant Cell Physiol 51:1537–1547. https://doi.org/10.1093/pcp/pcq108
Söderman E, Hjellström M, Fahleson J, Engström P (1999) The HD-Zip gene ATHB6 in Arabidopsis is expressed in developing leaves, roots and carpels and up-regulated by water deficit conditions. Plant Mol Biol 40:1073–1083. https://doi.org/10.1023/A:1006267013170
Tang Y, Bao X, Wang S et al (2019) A physic nut stress-responsive HD-Zip transcription factor, JcHDZ07, confers enhanced sensitivity to salinity stress in transgenic Arabidopsis. Front Plant Sci 10:942. https://doi.org/10.3389/fpls.2019.00942
Tron AE, Bertoncini CW, Palena CM, Chan RL, Gonzalez DH (2001) Combinatorial interactions of two amino acids with a single base pair define target site specificity in plant dimeric homeodomain proteins. Nucl Acids Res 29:4866–4872. https://doi.org/10.1093/nar/29.23.4866
Tron AE, Welchen E, Gonzalez DH (2004) Engineering the loop region of a homeodomain-leucine zipper protein promotes efficient binding to a monomeric DNA binding site. Biochem 43:15845–15851. https://doi.org/10.1021/bi048254a
Valdes AE, Overnas E, Johansson H, Rada-Iglesias A, Engstrom P (2012) The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol Biol 80:405–418. https://doi.org/10.1007/s11103-012-9956-4
Vlad D, Kierzkowski D, Rast MI et al (2014) Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene. Science 343:780–783. https://doi.org/10.1126/science.1248384
Vollbrecht E, Veit B, Sinha N et al (1991) The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 350:241–243. https://doi.org/10.1038/350241a0
Wang Y, Henriksson E, Söderman E et al (2003) The Arabidopsis homeobox gene, ATHB16, regulates leaf development and the sensitivity to photoperiod in Arabidopsis. Dev Biol 264:228–239. https://doi.org/10.1016/j.ydbio.2003.07.017
Wilhelmsson PKI, Mühlich C, Ullrich K, Rensing SA (2017) Comprehensive genome-wide classification reveals that many plant-specific transcription factors evolved in streptophyte algae. Genome Biol Evol 9:3384–3397. https://doi.org/10.1093/gbe/evx258
Wray G (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8:206–216. https://doi.org/10.1038/nrg2063
Yang C, Liu J, Dong X, Cai Z, Tian W, Wang X (2014) Short-term and continuing stresses differentially interplay with multiple hormones to regulate plant survival and growth. Mol Plant 7:841–855. https://doi.org/10.1093/mp/ssu013
Yang Y, Al-Baidhani HHJ, Harris J et al (2020) DREB/CBF expression in wheat and barley using the stress-inducible promoters of HD-Zip I genes: Impact on plant development, stress tolerance and yield. Plant Biotechnol J 18:829–844. https://doi.org/10.1111/pbi.13252
Yue H, Shu D, Wang M et al (2018) Genome-wide identification and expression analysis of the HD-Zip gene family in wheat (Triticum aestivum L). Genes 9:70. https://doi.org/10.3390/genes9020070
Zhang J, Zhu Q, Moncuquet P, Llewellyn D, Wilson I (2016) Genome-wide identification and characterization of the homeodomain-leucine zipper I family of genes in cotton (Gossypium spp.). Plant Gene 7:51–60. https://doi.org/10.1016/j.plgene.2016.05.002
Zhang Z, Chen X, Guan X et al (2014) A genome-wide survey of homeodomain-leucine zipper genes and analysis of cold-responsive HD-Zip I members expression in tomato. Biosci Biotechnol Biochem 78:1337–1349. https://doi.org/10.1080/09168451.2014.923292
Zhang Z, Zhu R, Ji X et al (2021) Genome-wide characterization and expression analysis of the HD-ZIP gene family in response to salt stress in pepper. Int J Genomics 2021:8105124. https://doi.org/10.1155/2021/8105124
Zhao JL, Pan JS, Guan Y et al (2015) Micro-trichome as a class I homeodomain-leucine zipper gene regulates multicellular trichome development in Cucumis sativus. J Integr Plant Biol 57:925–935. https://doi.org/10.1111/jipb.12345
Zhao S, Wang H, Jia X et al (2021) The HD-Zip I transcription factor MdHB7-like confers tolerance to salinity in transgenic apple (Malus domestica). Physiol Plant 172:1452–1464. https://doi.org/10.1111/ppl.13330
Zhao Y, Xing L, Wang X et al (2014) The ABA receptor PYL8 promotes lateral root growth by enhancing MYB77-dependent transcription of auxin-responsive genes. Sci Signal 7:ra53. https://doi.org/10.1126/scisignal.2005051
Acknowledgements
This work was funded by grant from the National Science Centre 2016/23/B/N29/02175 to DBS.
Author information
Authors and Affiliations
Contributions
NŻ and DBS: designed research, prepared the documentations and wrote the manuscript. Both authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling editor: Bojian Zhong.
Supplementary Information
Below is the link to the electronic supplementary material.
239_2023_10121_MOESM1_ESM.docx
Supplementary Fig. S1. The phylogenetic and structural characterization of the HD-Zip I subfamily in A. thaliana. A. The phylogenetic tree of Arabidopsis HD-Zip I subfamily based on alignment of full-length protein sequences by using MAGA 11, B. Structural domain organizations of Arabidopsis HD-Zip I proteins including the conserved domain (HD and Zip) and additional motifs. Uncharacterized motifs outside the HD and Zip as a potential source of functional diversity are numbered according to Arce et al. (2011). C. Schematic diagram representing the structure of Arabidopsis HD-Zip I genes. Yellow boxes represent exons and spaces between boxes correspond to introns. The 5’ and 3’ UTRs are marked by blue boxes. The exon/intron structure of each HD-Zip I gene was defined by comparison of their genomic and cDNA sequences. The sizes of exons, introns and UTRs are drawn to scale as indicated below. The gene structures were illustrated using the Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/ (DOCX 295 KB)
239_2023_10121_MOESM2_ESM.jpg
Supplementary Fig. S2. Expression profiles of 17 HD-Zip I genes in different tissues and stages of development of A. thaliana. The data are based on Arabidopsis eFP Browser (The BAR and other Data Analysis Tools for Plant Biology (utoronto.ca). The heatmap shows the log2-transformed TPM values of each gene. The expression level of AtHB genes is represented using color scale ranging from green (low expression) to red (high expression (JPG 109 kb)
239_2023_10121_MOESM3_ESM.pptx
Supplementary file3 Fig. S3. Expression profiles of HD-Zip I genes in five tissues of the selected plant species. The tissues include leaves, stem, roots, flowers and silique/seeds. The Reads/Kb/Million (RPKM) normalized values of expressed genes were log2-transformed and visualized as heatmap. The order of genes in the heatmap is consistency with the phylogeny in Figure 2. The data were from the different databases or the published studies. Data were obtained from: H. vulgare - Barley RNA-seq Database morexGenes - Barley Assembly and RNA-seq (hutton.ac.uk); G. max - soybean expression atlas (Soybean expression atlas | Home (uenf.br)); S. lycopersicum - Tomato BARePlant (ePlant (utoronto.ca)); L. tuberosum - the PGSC database (Spud DB (uga.edu)); Z. mays - Maize eFP Browser (Maize eFP Browser (utoronto.ca)); M. truncatula - Medicago eFP Browser (Medicago eFP Browser (utoronto.ca)); B. napus - BrassicaEDB - A Gene Expression Database for Brassica Crops (BrassicaEDB - A Gene Expression Database for Brassica Crops (biodb.org)); B. rapa - the Brassicaceae Database (BRAD)(BRAD (brassicadb.cn)); O. sativa - Rice eFP Browser (https://bar.utoronto.ca/efprice/cgi-bin/efpWeb.cgi) (PPTX 968 KB)
239_2023_10121_MOESM4_ESM.jpg
Supplementary file4 Fig. S4. Expression profiles of HD-Zip I genes under abiotic stresses and ABA treatment of A. thaliana. The data are based on Arabidopsis eFP Browser (The BAR and other Data Analysis Tools for Plant Biology (utoronto.ca). The expression level are based on the log2 values and color scale represent the expression levels of AtHB genes from green (downregulated) to red (upregulated) (JPG 99 kb)
239_2023_10121_MOESM5_ESM.pptx
Supplementary file5 Fig S5. Expression profiles of HD-Zip I genes in response to cold, heat, drought and salt stresses in different species. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmap. The order of genes in the heatmap is consistency with the phylogeny in Figure 2. Data were obtained from: B. napus - BrassicaEDB - A Gene Expression Database for Brassica Crops (BrassicaEDB - A Gene Expression Database for Brassica Crops (biodb.org)); B. rapa - based on data from Khan et al. (2018); G. max obtained from the NCBI GEO database under accession numbers GSE40627 (drought), GSE41125 (salinity) and GSE213479 (heat); H. vulgare: obtained from the NCBI GEO database under accession numbers PRJEB18276 (cold), PRJNA324116 (heat), PRJNA439267 (drought) and PRJEB13621 (salt); S. lycopersicum - Tomato BARePlant (ePlant (utoronto.ca)) and obtained from the NCBI GEO database under accession number PRJNA730730 (heat); L. tuberosum - the PGSC database (Spud DB (uga.edu)); Z. mays - Maize eFP Browser (Maize eFP Browser (utoronto.ca)); M. truncatula - based on data from Li et al. (2022); P. trichocarpa - Poplar eFP Browser https://bar.utoronto.ca/eplant_poplar/; C. sativa - Cucurbit Genomics Database: Cucurbit Genomics Database (CuGenDB); O. sativa - Rice eFP Browser (https://bar.utoronto.ca/efprice/cgi-bin/efpWeb.cgi) (PPTX 186 KB)
239_2023_10121_MOESM6_ESM.docx
Supplementary file6 Table S1. Number of transcription factors (TFs) in plant genomes based on data derived from PlantTFDB v5.0 (http://planttfdb.gao-lab.org/) and PLAZA project (https://bioinformatics.psb.ugent.be/plaza/) (DOCX 35 KB)
Supplementary Table S4. List of HD-Zip I proteins included in the phylogenetic analysis (XLSX 25 KB)
239_2023_10121_MOESM10_ESM.docx
Supplementary Table S5. Characterization of 17 A. thaliana HD-Zip I genes including gene and protein lengths, gene and protein organizations and biological functions (DOCX 16 KB)
239_2023_10121_MOESM11_ESM.docx
Supplementary Table S6. Characterization of 17 members of A. thaliana HD-Zip I subfamily including the consensus sequence of cis-regulatory element and their interacting partners. The sequence logo of cis-regulatory elements were generated by JASPAR (https://jaspar.genereg.net/) (DOCX 76 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Żyła, N., Babula-Skowrońska, D. Evolutionary Consequences of Functional and Regulatory Divergence of HD-Zip I Transcription Factors as a Source of Diversity in Protein Interaction Networks in Plants. J Mol Evol 91, 581–597 (2023). https://doi.org/10.1007/s00239-023-10121-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-023-10121-4