
Abstract
Aims/hypothesis
The aim of this study was to understand the role of CXC chemokine receptor 3 (CXCR3), a T-helper 1(Th1) type chemokine receptor, in the pathogenesis of type 1 diabetes.
Methods
We observed the incidence of diabetes in Cxcr3 homozygous knockout mice. We compared the expression pattern of various cytokines and chemokines and the frequency of FOXP3+ cells in the pancreas and pancreatic lymph nodes from Cxcr3 −/− NOD mice and wild-type NOD mice. In addition, we observed the migration ability of CXCR3+CD4+ cells to pancreatic islets upon adoptive transfer. Finally, we examined whether Cxcr3 + regulatory T cells (Tregs) actually suppressed the onset of diabetes in vivo.
Results
Cxcr3 −/− NOD mice developed spontaneous diabetes earlier than did wild-type NOD mice. In Cxcr3 −/− NOD mice, Tregs were more frequent in pancreatic lymph nodes and less frequent in pancreatic islets than in wild-type NOD mice. While transferred CXCR3−CD4+ cells from wild-type NOD mice did not infiltrate pancreatic islets of NOD–severe combined immunodeficiency (SCID) mice, CXCR3+CD4+ cells from the same mice migrated into the recipient islets and contained Forkhead box P3 (FOXP3) upon adoptive transfer. Moreover, CD4+CD25+ cells from wild-type NOD mice suppressed and delayed the onset of diabetes compared with those from Cxcr3 −/− NOD mice in a cyclophosphamide-induced diabetes model system.
Conclusions/interpretation
The mechanism of accelerated diabetes onset in Cxcr3 −/− NOD mice was considered to be due to the lack of hybrid Tregs (CXCR3+FOXP3+CD4+ cells), which could effectively migrate into and regulate Th1 inflammation in local lesions under Cxcr3 knockout conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes is an autoimmune disease that results from T cell-mediated destruction of insulin-producing pancreatic beta cells [1]. It is assumed that T-helper 1 (Th1) cells play an important role in the onset of type 1 diabetes [2].
In patients with type 1 diabetes, the serum level of CXC ligand (CXCL)10/IFN-γ-inducible protein of 10 kDa (IP-10), a Th1 type chemokine, is significantly higher than in healthy controls and patients with type 2 diabetes [3]. In NOD mice, CXCL10 is not constitutively expressed on pancreatic beta cells but is upregulated in response to a typical Th1-associated cytokine, IFN-γ, as insulitis progresses [4]. Administration of CXCL10 monoclonal antibodies to a cyclophosphamide-induced NOD diabetes mouse model resulted in delayed diabetes onset [5]. Moreover, induction of anti-CXCL10 antibodies using a gene transfer system in young NOD mice also resulted in disease suppression [4]. Therefore, we speculated that the receptor for CXCL10/IP-10, CXC chemokine receptor 3 (CXCR3), must be a key protein in the pathogenesis of type 1 diabetes.
CXCR3 is a G protein coupled receptor with seven transmembrane domains and its gene is encoded on the X chromosome in both mice and humans [6, 7]. Although this receptor is also expressed on Cd8 + T cells, B cells, macrophages, dendritic cells, natural killer (NK) cells and regulatory T cells (Tregs), it is expressed mainly on Th1 cells and can lead them to sites of tissue inflammation in response to CXCR3-binding chemokines, such as CXCL9/monokine induced by IFN-γ (Mig), CXCL10/IP-10 and CXCL11/IFN-inducible T cell α chemoattractant (I-TAC) [6–11]. Therefore, it is reasonable to assume that there is an amplification loop for Th1 immune responses between Th1 cells, which produce IFN-γ, and pancreatic beta cells, which produce CXCL10, in response to this cytokine, thereby attracting more Cxcr3-expressing Th1 cells to the affected site and amplifying the ongoing Th1 response. In fact, blockade of the CXCL10–CXCR3 interaction inhibited infiltration of lymphocytes into pancreatic islets and suppressed the onset of diabetes in a virus-induced diabetes model [12, 13]. Moreover, overexpression of CXCL10 in islets resulted in accelerated disease [14]. Thus, we speculated that knockout of CXCR3 would suppress the onset of diabetes. Surprisingly, however, instead we observed earlier diabetes onset in CXCR3-deficient NOD mice, and there was clear acceleration of diabetes development, compared with wild-type NOD mice, when an adoptive transfer system was used. In this paper we discuss the importance of CXCR3+ Tregs, so-called ‘hybrid Tregs’ [15], in the regulation of type 1 diabetes, providing a possible explanation for the observations discussed above.
Methods
Animals
We purchased NOD and NOD–SCID mice from CLEA Japan (Tokyo, Japan). A female C57Bl/6 background CXCR3-deficient mouse was generated as previously described [16]. The mice were backcrossed to male NOD mice, and F1 female mice were again backcrossed to male NOD mice. From the F2 generation, heterozygous female mice were chosen and then backcrossed to male NOD mice. This manipulation was repeated until the sixth generation. At the fifth generation (F5), mice were screened for polymorphic microsatellite markers to determine whether they had the NOD gene background and we confirmed that they had NOD type on all markers shown in the electronic supplementary material (ESM) Fig. 1 [17]. Cxcr3 homozygous knockout female NOD mice were generated by crossing F6 knockout male and heterozygous female mice (speed congenic method). We housed all mice under specific pathogen-free conditions. All studies were approved by the Animal Care and Use Committee of Keio University.
Genotyping
DNA was extracted from a small portion of the tail of experimental mice. A QIAGEN DNeasy kit (Qiagen, Courtaboeuf, France) was used for DNA isolation, followed by PCR using TaKaRa Ex Taq (TAKARA BIO, Shiga, Japan). Genotyping was performed using primers recognising Cxcr3 genomic DNA sequences or inserted genomic DNA sequences named Neu. The primers shown in ESM Table 1 were used. The wild-type allele yielded a 589 bp DNA fragment and the knockout allele a 320 bp fragment, which were analysed using agarose gels and detected by ethidium bromide (EtBr) staining. Flow cytometric analysis confirmed that CXCR3 on splenocytes was decreased in heterozygous mice and was undetectable in knockout mice (ESM Fig. 2).
Definition of diabetes
All animals were assessed for glycosuria at least once a week in the evening, and plasma glucose was measured when glycosuria was observed. Being positive for glycosuria and having a plasma glucose level higher than 13.9 mmol/l confirmed diabetes.
Histological examination
The pancreas was removed from mice, fixed in 10% formaldehyde and embedded in paraffin. Thin sections at four levels, 200 μm apart, were cut for hematoxylin–eosin (H&E) staining to evaluate the degree of cell infiltration in the islets by light microscopy. The pancreatic islets were scored using the following rankings: grade 0, normal; grade 1, peri-insulitis (mononuclear cells surrounding islets and ducts, but no infiltration of the islet architecture); grade 2, moderate insulitis (mononuclear cells infiltrating <50% of the islet architecture); and grade 3, severe insulitis (mononuclear cells infiltrating >50% of the islet architecture).
Quantitative
RT-PCR Total RNA was extracted from the pancreas, pancreatic lymph nodes (pLN) and spleen using an RNeasy Mini kit (Qiagen). During the procedure, DNase treatment was performed according to the manufacturer’s protocol. The extracted RNA was reverse transcribed using Not I-d(T)18 primer and a First-Strand cDNA synthesis kit (GE Healthcare, Little Chalfont, UK), according to the manufacturer’s instructions. Quantitative RT-PCR was conducted for Il4, Il10, Tgf-β (also known as Tgfb1), Foxp3, Ifn-γ (also known as Ifng), Il2, Gzmb, Ccr5 and Gapdh (internal control) in an ABI Prism 7700 sequence detector (Applied Biosystems, Foster City, CA, USA). The primers were purchased from Applied Biosystems. All reactions were performed using TaqMan Universal MasterMix (Applied Biosystems). The expression of each gene was normalised to that of Gapdh.
Immunohistochemical study
The pancreases removed from NOD mice were inflated with optimal cutting temperature (OCT) compound and snap-frozen in liquid nitrogen. Following this, 4% paraformaldehyde (PFA)-fixed 6 μm thick fresh frozen tissue sections were blocked with 5% goat serum diluted in PBS. The sections were incubated with polyclonal guinea pig anti-swine insulin antibody (DakoCytomation, Kyoto, Japan), fluorescein isothiocyanate (FITC)-labelled rat anti-mouse CD4 monoclonal antibody (mAb) (H129.19; BD Pharmingen, San Diego, CA, USA), rat anti-mouse FOXP3 antibody (MF333F; Alexis Biochemicals, Plymouth Meeting, PA, USA), rabbit anti-mouse FOXP3 antibody (PA1-46126; Thermo Fisher Scientific, Rockford, IL, USA), and phycoerythrin (PE)-labelled rat anti-mouse CXCR3 mAb (R&D Systems, Minneapolis, MN, USA), followed by Alexa Fluor-594-labelled goat anti-guinea pig IgG, Alexa Fluor-488-labelled goat anti-rat IgG and Alexa Fluor-568-labelled goat anti-rabbit IgG (Molecular Probes, Eugene, OR, USA). The sections were then examined by fluorescence microscopy. We ensured staining specificity by using control slides in which incubation with the primary antibody was skipped. Thin sections at three levels, 200 μm apart, were cut to evaluate the islets.
Flow cytometry
Cell surfaces were stained with the following directly conjugated antibodies specific for mouse proteins: FITC or PE-labelled anti-mouse CD4 mAb (H129.19; BD Pharmingen) or PE-labelled anti-mouse CXCR3 mAb (R&D Systems). Mouse FOXP3 staining was performed with a FITC anti-mouse FOXP3 staining set (eBioscience, San Diego, CA, USA).
To perform flow cytometric analyses of pancreases, approximately 2.5 ml of cold HBSS (Sigma-Aldrich, St Louis, MO, USA) containing collagenase XI (1 mg/ml; Sigma-Aldrich) was injected into the common bile duct. The distended pancreas was removed and incubated at 37°C for 17 min with shaking. Cold HBSS was added to stop the digestion. The pancreas was then dissected using a cell strainer and stained with the same monoclonal antibodies as the spleen cell preparations. The stained cells were analysed with an Epics XL-MCL (Beckman Coulter, Brea, CA, USA).
Suppression assay
We evaluated the suppressive capacity of Tregs (CD4+CD25+ cells) against proliferation of effector cells (CD4+CD25− cells) using a carboxyfluorescein diacetate succinimidyl ester (CFSE) labelling system in vitro [18]. We conducted this experiment using splenocytes and lymphocytes from pancreatic lymph nodes each separately. CD4+CD25+ and CD4+CD25− cells were purified using a MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany). We stimulated CD4+CD25− cells (spleen: 3.5 × 105 cells, pancreatic lymph nodes: 1.3 × 104 cells) from 12-week-old wild-type female NOD mice with a Dynabeads mouse CD3/CD28 T cell Expander (Invitrogen, Carlsbad, CA, USA) and simultaneously added Tregs (CD4+CD25+ cells; spleen: >3.5 × 104 cells, pancreatic lymph nodes: >6.5 × 103 cells) from wild-type female NOD or Cxcr3 −/− female NOD mice. These cells were cultured for 48 h in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and penicillin/streptomycin. These cells were then analysed with an Epics XL-MCL (Beckman Coulter).
Cell transfer
For the adoptive transfer experiments, the spleens of non-diabetic and diabetic Cxcr3 −/− and wild-type 14–18-week-old NOD mice were removed. After lysis of erythrocytes, the splenocytes were washed with PBS. The splenocytes were then resuspended at an appropriate concentration for transfer (8.0 × 106 cells/ml), and 250 μl of the cell suspension (including 2.0 × 106 cells) was injected intraperitoneally into each NOD–SCID recipient.
For the migration study, the spleens of 27-week-old diabetic wild-type NOD mice were removed. After lysis of erythrocytes, the splenocytes were washed with PBS and incubated with FITC-labelled anti-mouse CD4 mAb (H129.19; BD Pharmingen) and PE-labelled anti-mouse CXCR3 mAb (R&D Systems). CXCR3−CD4+ and CXCR3+CD4+ cells were isolated by sorting using an Altra HyperSort System (Beckman Coulter). The splenocytes were then resuspended at an appropriate concentration for transfer (8.0 × 105 cells/ml), and 250 μl of the cell suspension (including 2.0 × 105 cells) was injected intraperitoneally into each NOD–SCID recipient. Eleven weeks later, the recipients were killed and the pancreas was examined histologically.
For regulatory T cell transfer study, we used MACS (Miltenyi Biotec) sorted CD4+CD25+ cells as regulatory T cells. Nine to 12-week-old female non-diabetic NOD mice were injected intraperitoneally with cyclophosphamide (Shionogi, Osaka, Japan, 225 mg/kg body weight) on day 0 and injected intraperitoneally with CD4+CD25+ cells (0.6–1.0 × 106 cells/mouse) of Cxcr3 −/− or wild-type 12–14-week-old female NOD mice on day 3. All animals were assessed for glycosuria daily in the evening, and plasma glucose was examined when glycosuria was observed. Diabetes was confirmed when glycosuria was positive and plasma glucose level was higher than 13.9 mmol/l.
Statistical analysis
Results are presented as mean ± SEM. The χ2 test or Fisher’s exact probability test were used to compare the incidence of diabetes. The χ2 test was used to compare the frequency of FOXP3+ cells among CD4+ cells in and around the islets. Other mean values in the groups of Cxcr3 −/− NOD mice and wild-type NOD mice were compared using the Mann–Whitney U test. A p value less than 0.05 was considered to indicate a statistically significant difference.
Results
Earlier onset of diabetes in Cxcr3 −/− NOD mice
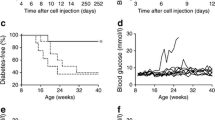
First, we compared the incidence of diabetes between Cxcr3 −/− NOD and wild-type NOD mice. As shown in Fig. 1, the onset of diabetes was significantly earlier in Cxcr3 −/− NOD than in wild-type NOD mice (55% vs 24% at 28 weeks of age, p = 0.04). Moreover, histological examination of the pancreas revealed a higher degree of insulitis in 12-week-old Cxcr3 −/− NOD mice than in age-matched wild-type NOD mice (pancreatic islet number with score grade 3/total pancreatic islet number: Cxcr3 −/− 27.3% (38/139) vs wild-type 8.9% (16/180), p < 0.0001, ESM Fig. 3). Furthermore, when we transferred Cxcr3 −/− NOD splenocytes into NOD–SCID mice, the incidence of diabetes was higher (Cxcr3 −/− non-diabetic 100% vs wild-type non-diabetic 10% at 9 weeks after transfer, p < 0.01, Cxcr3 −/− diabetic 100% vs wild-type diabetic 42.1% at 9 weeks after transfer, p = 0.01, Fig. 2) and the onset was earlier than in NOD–SCID recipients of wild-type NOD splenocytes, confirming that Cxcr3 −/− NOD spleen cells were clearly more immunologically active than wild-type NOD spleen cells.

Cxcr3 −/− NOD mice developed diabetes earlier than Cxcr +/− or wild-type NOD mice. Kaplan–Meier plot of cumulative diabetes incidence in female wild-type (white circles, n = 21), Cxcr3 +/− (black triangles, n = 31) and Cxcr3 −/− NOD mice (black circles, n = 20). * p < 0.05 vs wild-type NOD group at 28 weeks of age, by χ2 test

Splenocytes from Cxcr3 −/− NOD mice are more diabetogenic than those from wild-type NOD mice. Kaplan–Meier plot of cumulative diabetes incidence in female NOD–SCID mice after adoptive transfer. White circles: Cxcr3 −/− non-diabetic donors (n = 7); black circles: Cxcr3 −/− diabetic donors (n = 7); white triangles: wild-type non-diabetic donors (n = 10); black triangles: wild-type diabetic donors (n = 19) * p < 0.01 vs wild-type non-diabetic group and p = 0.01 vs wild-type diabetic group at 9 weeks after adoptive transfer, by Fisher’s exact probability test
Mirror image expressions of Treg element in pancreatic lymph nodes and pancreas
To gain insight into the earlier onset of diabetes in Cxcr3 −/− NOD mice, we examined cytokine expression levels in the pancreatic lymph nodes and pancreas from prediabetic 12-week-old Cxcr3 −/− NOD mice, which are considered to represent the stage immediately before the onset of diabetes, and compared the findings with those in age-matched wild-type NOD mice. As shown in Fig. 3, expression levels of Tgf-β, Foxp3, Il10 and Il4 were higher in pancreatic lymph nodes from Cxcr3 −/− NOD mice, whereas expression levels were lower in the pancreas, with these levels being a ‘mirror image’ of each other. Moreover, immunohistochemical examination of pancreatic islets revealed that the frequency of FOXP3+ cells among CD4+ cells, which surrounded the pancreatic islets, was higher in wild-type than in Cxcr3 −/− islets (FOXP3+CD4+ cells/ CD4+ cells: Cxcr3 −/− 13.7 ± 1.7% vs wild-type 23.5 ± 2.8%, p < 0.05, Figs 4 and 5). In addition, the frequency of FOXP3+ cells among CD4+ cells was higher in pancreatic lymph nodes from Cxcr3 −/− NOD mice, as demonstrated by flow cytometry (Cxcr3 −/− 10.01 ± 0.60% vs wild-type 8.51 ± 0.43%, p < 0.05, n = 9/group, ESM Fig. 4). These results suggest that CXCR3 is required for effective migration of FOXP3+ cells into the pancreas itself.

In Cxcr3 −/− NOD mice (KO), regulatory T cells accumulate in pancreatic lymph nodes (pLN) but not in pancreas (Panc), compared with wild-type (WT) NOD mice. Expression of Treg-associated cytokines and chemokines in pLN and pancreas in 12-week-old Cxcr3 −/− mice (white bars) and WT NOD mice (black bars) (n = 4/group). a Tgf-β, (b) Foxp3, (c) Il10, (d) Il4. Data are shown as mean ± SEM. * p < 0.05 by Mann–Whitney U test

FOXP3+ cells are rare in pancreatic islets from Cxcr3 −/− NOD mice (a) but are present in those from wild-type NOD mice (b). FOXP3-positive cells (green, shown by arrows) infiltrating around pancreatic islets (red) were evaluated at 12 weeks of age (magnification, ×200) in both groups (n = 5/group). Representative islets are shown

Comparison of FOXP3+CD4+ cell staining of Cxcr3 −/− NOD pancreas (a) and wild-type NOD pancreas (b). Representative flow cytometric data are shown. X-axis: CD4-PE, Y-axis: FOXP3-FITC. Numbers in quadrants indicate the percentage of cells in each
CXCR3+CD4+ cells migrate into recipient islets with adoptive transfer
First of all, we confirmed the presence of CXCR3+FOXP3+ cells in prediabetic (20 weeks old) wild-type NOD islets (ESM Fig. 5). We transferred CXCR3+CD4+ cells from 27-week-old diabetic wild-type NOD mice into NOD–SCID recipients, and found that CXCR3+CD4+ cells migrated into the recipient islets (Fig. 6a). These infiltrating cells were confirmed to be CD4+ cells (Fig. 6c). Furthermore, a portion of these cells contained FOXP3 (Fig. 6d). Thus, CXCR3+FOXP3+CD4+ cells were confirmed to migrate into recipient islets. On the other hand, when we transferred CXCR3−CD4+ cells from the same donor into NOD–SCID recipients, there was no cell infiltration in the recipient islets by 11 weeks post-transfer (Fig. 6b), indicating that CXCR3 is required for CD4+ cell migration into islets (pancreatic islet number in which insulitis occurred/total pancreatic islet number: CXCR3+ cells: 22/100 vs CXCR3− cells: 0/94, p < 0.0001).

CXCR3+FOXP3+CD4+ cells migrate into recipient islets with adoptive transfer. CD4+ CXCR3+ cells (positive group) or CD4+CXCR3− cells (negative group) from 27-week-old diabetic NOD mice were injected intraperitoneally into NOD–SCID recipients. a,b H&E staining of pancreatic islets of positive group (a) and negative group (b) (magnification, ×400). Arrows indicate infiltrating lymphocytes. c Immunohistochemical staining in positive group (magnification, ×200). CD4+ cells (green, arrows) have infiltrated pancreatic islets (red). d Immunohistochemical staining in positive group (magnification, ×200). Infiltration of FOXP3+ cells (green, arrows) are observed around pancreatic islets (red)
Suppressive function of Tregs is similar in Cxcr3 −/− NOD mice and Cxcr3 +/+ NOD mice
In order to evaluate the difference in Treg function between Cxcr3 −/− NOD mice and wild-type NOD mice, we used CD4+CD25+ cells from spleen or pancreatic lymph nodes as Tregs. These cells were cultured with purified CD4+CD25− cells (effector cells) from a wild-type NOD mouse to evaluate their suppressive capacity against the proliferation of the effector cells. We found no significant difference in the suppressive capacity of Tregs from either spleen or pancreatic lymph nodes between Cxcr3 −/− and wild-type NOD mice; the suppression rate for proliferation of effector cells by Tregs, which were added at a ratio of one tenth or more of effector cells, was as follows: spleen; Cxcr3 −/− 23.4 ± 3.2% (n = 4), wild-type 27.9 ± 2.4% (n = 4), pancreatic lymph nodes; Cxcr3 −/− 63.0%, wild-type 51.9%.
Regulatory T cells from Cxcr3 +/+ NOD mice suppress and delay the onset of diabetes compared with those from Cxcr3 −/− NOD mice
To evaluate whether CXCR3+ Tregs actually suppress the onset of diabetes in vivo, we transferred CD4+CD25+ cells (0.6–1.0 × 106 cells) from wild-type mice into cyclophosphamide (225 mg/kg/mouse) injected 9–12-week-old female NOD mice 3 days post cyclophosphamide injection and compared the response to that with transfer of a matched number of Tregs from Cxcr3 −/− NOD mice. As a result, we observed significantly delayed onset of diabetes in wild-type Treg-transferred mice (13.0 ± 0.6 days) compared with Cxcr3 −/− Treg-transferred mice (11.1 ± 0.5 days, p < 0.05, ESM Fig. 6). Moreover, 6 of 20 Cxcr3 −/− Treg-transferred mice developed diabetes by 11 days post cyclophosphamide injection, whereas none of the wild-type Treg-transferred mice developed diabetes at the same time point (p = 0.02). The incidence of diabetes at 18 days post cyclophosphamide treatment in NOD mice without transfer was 42% (8/19) in this system. These results indicate that the presence of CXCR3 in Tregs effectively suppresses diabetes in vivo.
Discussion
Numerous reports [2, 19–22] have clarified that Th1 cells are intimately involved in the onset of type 1 diabetes. In this study, we generated NOD mice lacking CXCR3, which has been considered to make a major contribution to the cytotoxic functions of Th1 cells, and we anticipated that diabetes onset in these mice would be delayed or suppressed. The results, however, were entirely the opposite of our expectations.
Other researchers have investigated the role of CXCR3 by using Cxcr3 gene deficient mice. Gao et al. [23] reported that, in a bleomycin-induced lung injury model, Cxcr3 knockout mice were protected from lung injury as evidenced by lower accumulation of inflammatory cells in the airways and lung interstitium than in their wild-type littermates. As with our initial expectation, they also assumed that CXCR3 signalling would promote inflammatory cell recruitment and initiate an inflammatory cytokine cascade following endotracheal bleomycin administration. In this study we examined the mRNA expression level of a Th1-type cytokine, Ifn-γ, in the pancreases from Cxcr3 −/− mice, and found the level to be similar to that in wild-type mice (data not shown). This result suggested that CXCR3-deficient Th1 cells migrated in the same manner as wild-type Th1 cells, using other chemokine receptors independent of CXCR3, such as CCR5, which we confirmed to be present in pancreases from Cxcr3 −/− NOD mice (data not shown).
On the other hand, Müller et al. [11] reported that, in the central nervous system (CNS) in experimental autoimmune encephalomyelitis (EAE), the times to onset and peak disease severity were similar in Cxcr3 knockout and wild-type animals. However, Cxcr3 knockout mice had more severe chronic disease, with increased demyelination and axonal damage. Although the numbers of CD4 + and CD8 + T cells infiltrating the CNS were similar in Cxcr3 knockout and wild-type mice, FOXP3+ regulatory T cells were significantly reduced in number and were more dispersed in Cxcr3 knockout mice. The authors concluded that, in EAE, CXCR3 signalling kept T cells within the perivascular space in the CNS and augmented regulatory T cell recruitment and interaction between regulatory T cells and effector T cells, thereby limiting autoimmune-mediated tissue damage. In our experimental system, mRNA expression patterns clarified the marked differences in location of regulatory T cells between Cxcr3 −/− and wild-type NOD mice. In Cxcr3 −/− mice, regulatory T cells accumulated in pancreatic lymph nodes, with fewer being detected in pancreatic islets. These findings suggest that CXCR3 is a chemokine receptor for regulatory T cell migration into pancreatic islets, and that the earlier onset of diabetes in Cxcr3 −/−mice may be due to lack of regulatory T cells in pancreatic islets.
Very recently, CXCR3+ Tregs, so-called ‘hybrid Tregs’ [15], were reported to efficiently downregulate the Th1 inflammatory state. This concept is entirely consistent with our observations. According to the report on hybrid Tregs, CXCR3+FOXP3+CD4+ cells were generated from FOXP3+CD4+ cells in response to strong Th1 inflammation, particularly IFN-γ. These new-born CXCR3+FOXP3+CD4+ cells (hybrid Tregs) are unique cells, having both the migratory capacity of Th1 cells and the regulatory function of regulatory T cells, and these cells migrate into Th1-inflammatory sites depending on the Th1–chemokine gradient, as do Th1 cells, and effectively suppress the function of Th1 cells as regulatory T cells. It is efficient that naive Tregs differentiate in response to an inflammatory environment and express the chemokine receptor of their target cells. In our system, CXCR3-deficient regulatory T cells did not have the capacity to migrate into Th1-inflammatory sites (pancreatic islets), and accumulated in pancreatic lymph nodes and could not effectively suppress Th1 activity.
As described above, when we transferred CXCR3+CD4+ cells from 27-week-old diabetic NOD mice, considered to be in the so-called ‘malignant phase’ [24] (in other words an active diabetic phase) into NOD–SCID recipients, FOXP3+CD4+ cells were observed in the recipient islets. On the other hand, when we transferred the same phenotypic population, CXCR3+CD4+ cells from 10-week-old NOD mice, considered to be in a ‘benign phase’ [24] (in other words a less active non-diabetic phase) no FOXP3+CD4+ cells were observed in islets, although CD4+ cells infiltrated pancreatic islets (data not shown). Moreover, the frequency of CXCR3+FOXP3+ cells was significantly higher in both pancreatic lymph nodes and among splenocytes from 29-week-old (malignant phase) NOD mice than in those from 10-week-old (benign phase) NOD mice (ESM Fig. 7). Taken together, these results suggest that CXCR3+FOXP3+CD4+ cells are generated mainly in the malignant phase, in response to IFN-γ. As we reported previously, the IFN-γ response increased immediately prior to (or after) the onset of diabetes [2]. In general, the IFN-γ response in NOD mice during the non-diabetic phase is lower than that in the normal strain, whereas the IFN-γ response recovers to a ‘normal’ level at approximately the time of diabetes onset [2]. Our previous observations support our current hypothesis that CXCR3+FOXP3+CD4+ cell generation coincides with or occurs shortly after the onset of diabetes.
As mentioned in the Introduction, blockade of the CXCL10–CXCR3 interaction inhibited infiltration of lymphocytes into pancreatic islets and suppressed the onset of diabetes [14, 25]. These data are from a virus-induced diabetes model in which CXCL10 is acutely produced after infection, and therefore the migration of possibly both effector as well as regulatory T cells has been blocked. Both effector and regulatory T cells might be attracted in excess, leading to disease acceleration, despite the possible presence of a higher absolute number of ‘hybrid Tregs’. Hybrid Tregs producing CXCR3 might be more important in a spontaneous model with slow disease progression.
One might argue that the function of Tregs might be different between Cxcr3 −/− and wild-type NOD mice. However, Tgf-β, Il10, Il4, Ifn-γ, Il-2 and Gzmb expression levels of sorted FOXP3+ cells (data not shown), as well as the suppressive function of CD4+CD25+ cells in vitro, were essentially the same in the two strains. Therefore, we consider that the function of Tregs must be the same in the two strains.
Considering the clinical situation, after sorting this CXCR3+ Treg population at the onset of diabetes, and the expansion of this population, these cells might be used to regulate the active disease state through transfer back into the donor. We examined whether Tregs from wild-type mice, which were considered to include CXCR3+ Tregs, could suppress diabetes development compared with Tregs from Cxcr3 −/− mice (all CXCR3− Tregs), using a cyclophosphamide-induced diabetes model to create the same condition in the recipients, and found that Tregs from wild-type mice suppressed the onset of diabetes in comparison with Tregs from Cxcr3 −/− mice. Therefore, CXCR3+ Tregs (hybrid Tregs) seem to work in vivo to control diabetes development. We believe hybrid Tregs to be a very promising candidate cell population for the control of type 1 diabetes.
Abbreviations
- CNS:
-
Central nervous system
- CXCL9/Mig:
-
CXC ligand 9/monokine induced by IFN-γ
- CXCL10/IP-10:
-
CXC ligand 10/IFN-γ-inducible protein of 10 kDa
- CXCL11/I-TAC:
-
CXC ligand 11/IFN-inducible T cell α chemoattractant
- CXCR3:
-
CXC chemokine receptor 3
- EAE:
-
Experimental autoimmune encephalomyelitis
- FITC:
-
Fluorescein isothiocyanate
- FOXP3:
-
Forkhead box P3
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- H&E:
-
Hematoxylin–eosin staining
- mAb:
-
Monoclonal antibody
- PE:
-
Phycoerythrin
- SCID:
-
Severe combined immunodeficiency
- Th1:
-
T-helper 1
- Treg:
-
Regulatory T cell
References
Atkinson MA, Maclaren NK (1994) The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med 331:1428–1436
Shimada A, Rohane P, Fathman CG, Charlton B (1996) Pathogenic and protective roles of CD45RB(low) CD4+ cells correlate with cytokine profiles in the spontaneously autoimmune diabetic mouse. Diabetes 45:71–78
Shimada A, Morimoto J, Kodama K et al (2001) Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care 24:510–515
Shigihara T, Shimada A, Oikawa Y et al (2005) CXCL10 DNA vaccination prevents spontaneous diabetes through enhanced beta cell proliferation in NOD mice. J Immunol 175:8401–8408
Morimoto J, Yoneyama H, Shimada A et al (2004) CXC chemokine ligand 10 neutralization suppresses the occurrence of diabetes in non obese diabetic mice through enhanced beta cell proliferation without affecting insulitis. J Immunol 173:7017–7024
Lacotte S, Brun S, Muller S, Dumortier H (2009) CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 1173:310–317
Rotondi M, Chiovato L, Romagnani S, Serio M, Romagnani R (2007) Role of chemokines in endocrine autoimmune diseases. Endocrine Rev 28:492–520
Christensen JE, de Lemos C, Moos T, Christensen JP, Thomsen AR (2006) CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol 176:4235–4243
Nicholas MW, Dooley MA, Hogan SL et al (2008) A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE. Clin Immunol 126:189–201
Wilk E, Kalippke K, Buyny S, Schmidt RE, Jacobs R (2008) New aspects of NK cell subset identification and inference of NK cells’ regulatory capacity by assessing functional and genomic profiles. Immunobiology 213:271–283
Müller M, Carter SL, Hofer MJ et al (2007) CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol 179:2774–2786
Frigerio S, Junt T, Lu B et al (2002) Beta cells are responsible for CXCR3-mediated T cell infiltration in insulitis. Nat Med 8:1414–1420
Christen U, McGavern DB, Luster AD, von Herrath NG, Oldstone MB (2003) Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol 171:6838–6845
Rhode A, Pauza ME, Barral AM et al (2005) Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol 175:3516–3524
Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ (2009) The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10:595–602
Hancock WW, Lu B, Gao W et al (2000) Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 192:1515–1520
Yamamoto T, Yamato E, Tashiro F et al (2004) Development of autoimmune diabetes in glutamic acid decarboxylase 65 (GAD65) knockout NOD mice. Diabetologia 47:221–224
Turner MS, Kane LP, Morel PA (2009) Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol 183:4895–4903
Rabinovitch A (1994) Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Therapeutic intervention by immunostimulation? Diabetes 43:613–621
Katz JD, Benoist C, Mathis D (1995) T helper cell subsets in insulin-dependent diabetes. Science 268:1185–1188
Toyoda H, Formby B (1998) Contribution of T cells to the development of autoimmune diabetes in the NOD mouse model. Bioessays 20:750–757
Delovitch TL, Singh B (1997) The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 7:727–738
Gao JM, Lu B, Guo ZJ (2006) CXC chemokine receptor 3 modulates bleomycin-induced pulmonary injury via involving inflammatory process. Chin Med Sci J 21:152–156
Yamada S, Irie J, Shimada A et al (2003) Assessment of beta cell mass and oxidative peritoneal exudate cells in murine type 1 diabetes using adoptive transfer system. Autoimmunity 36:63–70
Christen U, Benke D, Wolfe T et al (2004) Cure of prediabetic mice by viral infections involves lymphocyte recruitment along an IP-10 gradient. J Clin Invest 113:74–84
Funding
This study was partly funded by a Grant-in-Aid for Scientific Research.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
All authors contributed to the conception and design, or analysis and interpretation of data, drafting the article or revised it critically, and approved the final version of the paper for publication. YY and YO (Y. Okubo) contributed to the experimental work. YY, YO (Y. Okubo) and AS drafted the article and revised it most critically.
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Yamada and Y. Okubo contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 95 kb)
ESM Fig. 2
(PDF 255 kb)
ESM Fig. 3
(PDF 158 kb)
ESM Fig. 4
(PDF 125 kb)
ESM Fig. 5
(PDF 178 kb)
ESM Fig. 6
(PDF 252 kb)
ESM Fig. 7
(PDF 100 kb)
ESM Table 1
(PDF 63 kb)
Rights and permissions
About this article
Cite this article
Yamada, Y., Okubo, Y., Shimada, A. et al. Acceleration of diabetes development in CXC chemokine receptor 3 (CXCR3)-deficient NOD mice. Diabetologia 55, 2238–2245 (2012). https://doi.org/10.1007/s00125-012-2547-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2547-8