
Insight into Shiga toxin genes encoded by Escherichia coli O157 from whole genome sequencing
- Published
- Accepted
- Received
- Academic Editor
- Ramy Aziz
- Subject Areas
- Bioinformatics, Genetics, Genomics, Microbiology
- Keywords
- Stx, Genomics, Sequencing, O157, E. coli
- Copyright
- © 2015 Ashton et al.
- Licence
- This is an open access article distributed under the terms of the Open Government License.
- Cite this article
- 2015. Insight into Shiga toxin genes encoded by Escherichia coli O157 from whole genome sequencing. PeerJ 3:e739 https://doi.org/10.7717/peerj.739
Abstract
The ability of Shiga toxin-producing Escherichia coli (STEC) to cause severe illness in humans is determined by multiple host factors and bacterial characteristics, including Shiga toxin (Stx) subtype. Given the link between Stx2a subtype and disease severity, we sought to identify the stx subtypes present in whole genome sequences (WGS) of 444 isolates of STEC O157. Difficulties in assembling the stx genes in some strains were overcome by using two complementary bioinformatics methods: mapping and de novo assembly. We compared the WGS analysis with the results obtained using a PCR approach and investigated the diversity within and between the subtypes. All strains of STEC O157 in this study had stx1a, stx2a or stx2c or a combination of these three genes. There was over 99% (442/444) concordance between PCR and WGS. When common source strains were excluded, 236/349 strains of STEC O157 had multiple copies of different Stx subtypes and 54 had multiple copies of the same Stx subtype. Of those strains harbouring multiple copies of the same Stx subtype, 33 had variants between the alleles while 21 had identical copies. Strains harbouring Stx2a only were most commonly found to have multiple alleles of the same subtype (42%). Both the PCR and WGS approach to stx subtyping provided a good level of sensitivity and specificity. In addition, the WGS data also showed there were a significant proportion of strains harbouring multiple alleles of the same Stx subtype associated with clinical disease in England.
Introduction
Shiga toxin-producing Escherichia coli (STEC) are a rare but potentially fatal cause of gastroenteritis. They are associated with a wide spectrum of disease ranging from mild to bloody diarrhoea, through to haemorrhagic colitis and haemolytic uraemic syndrome (HUS) (Pennington, 2010). The main reservoir of STEC in England is cattle, although it is carried by other animals, mainly ruminants. Transmission to humans occurs through direct or indirect contact with animals or their environments; consumption of contaminated food or water, and through person-to-person contact. Each year, there are approximately 900 cases of STEC O157 in England confirmed by the Gastrointestinal Bacteria Reference Unit (GBRU) at Public Health England.
The primary STEC virulence factor responsible for the most serious outcomes of human infection is Shiga toxin (Stx), an AB5 toxin that targets cells expressing the glycolipid globotriaosylceramide (Gb3), disrupting host protein synthesis and causing apoptotic cell death (Ethelberg et al., 2004). Renal epithelial cell membranes are enriched for Gb3, resulting in the kidneys bearing the brunt of Stx toxicity; in 5%–10% of cases, this leads to the development of Hemolytic Uremic Syndrome (HUS) (Pennington, 2010). There are two types of Stx: Stx1 and Stx2, and both have multiple subtypes. These subtypes can be differentiated using a PCR targeted at the encoding genes described by Scheutz et al. (2012). In addition, a web-based tool, VirulenceFinder, has been developed which uses a de novo assembly followed by BLAST approach to identify subtypes of Stx (Joensen et al., 2014). This system was shown to have good, but not perfect, agreement with PCR, although how it handles strains that encode both stx2a and stx2c is uncertain as no strains that encoded both these subtypes were examined (Joensen et al., 2014). The ability of STEC to cause severe illness in humans is determined by multiple bacterial factors (in addition to host factors), including Shiga toxin subtype. There is evidence that the Stx2a subtype is significantly associated with progression to HUS (Persson et al., 2007; Luna-Gierke et al., 2014).
As part of a project investigating the utility of whole genome sequencing (WGS) for public health surveillance and outbreak investigation of foodborne pathogens, high throughput, short read Ilumina GAII sequence data for 444 strains of STEC O157 isolated in England between 2009 and 2013 was obtained. We determined the presence, or absence, of the Stx encoding genes stx1 and stx2 in all 444 isolates of STEC O157 from the genome sequence data. Given the link between Stx subtype and disease severity, we also sought to identify the stx subtypes present using bioinformatics methods and to compare the results with those obtained using the PCR scheme of Scheutz et al. (2012).
WGS high throughput short read technologies are rapid and low cost compared to Sanger sequencing, but it was recognised early on that assembling short reads would be problematic (Chaisson, Pevzner & Tang, 2004). A major difficulty in assembly is the presence of repeat sequences that are longer than the read length. Furthermore, the study by Scheutz et al. (2012) clearly demonstrated that as well as a high level of similarity between stx2a, stx2c and stx2d there is also considerable diversity within each of these subtypes. The assembly of stx into one contig in strains of STEC O157 containing both stx2a and stx2c is difficult because the regions of variation between these subtypes are concentrated at the 5′ and 3′ ends of the coding DNA sequence (CDS), with a largely homogenous region in the centre. Existing methods for subtyping stx from short read data have not been tested against strains encoding stx2a and stx2c (Joensen et al., 2014). This region of 100% identity is often longer than the typical read length of short read sequencing technologies, so contiguous assembly of both subtypes relies on information from the paired end reads, which has a limited ability to resolve repeats up to the average fragment size (550–700 bp for Illumina Nextera mate-pair). The STEC O157 Sakai reference genome encodes 18 pro-phage that show a large degree of modularity and similarity (Asadulghani et al., 2009); this further complicates assembly of these regions (Hayashi et al., 2001). These difficulties have led to a relative paucity of data on the presence of subtypes of Stx within the E. coli population despite large WGS projects.
In this study a dual bioinformatic approach was taken, using both mapping and de novo assembly to determine stx subtype. The results of the bioinformatic analysis were compared to the results from the PCR typing method (Scheutz et al., 2012). In addition, the diversity within and between the stx subtype genes were investigated and evidence that certain strains contained multiple copies of the same stx subtype was assessed.
Methods
Strain selection
A total of 444 isolates of STEC O157 submitted to GBRU for confirmation and typing were selected for sequencing, 365 from 2012 representing approximately one third of the culture positive isolates (1,002 total isolates) received by the reference laboratory that calendar year from laboratories in England, Wales and Northern Ireland, and 67 English historical isolates submitted to GBRU between 1990 and 2011 and 12 isolates from 2013. The collection contained strains from sporadic cases, known outbreaks, household clusters, and serial strains isolated from the same patient. However, only sporadic strains and a single strain from any related cases (e.g., household, outbreak) were included in the diversity and multiple allele analysis. A total of 18 phage types were represented.
Sequencing
Genomic DNA was fragmented and tagged for multiplexing with Nextera XT DNA Sample Preparation Kits (Illumina) and sequenced at the Animal Health Veterinary Laboratory Agency (Weybridge) using the Illumina GAII platform with 2 × 150 bp reads. Multiplexing allowed 96 samples to be sequenced per run. Sequencing data with a phred score below 30 or a read length below 50 were removed from the data set using Trimmomatic (Bolger, Lohse & Usadel, 2014). FASTQ data is available from the NCBI Short Read Archive, BioProject accession PRJNA248064.
Subtyping of stx by assembly
High quality reads were assembled using Velvet v1.2.03 (Zerbino & Birney, 2008) with k-mer chosen using VelvetK (http://bioinformatics.net.au/software.velvetk.shtml). The resulting contigs were then compared against a set of stx reference genes (stx1a, L04539.1; 1c, Z36901.1; 1d, AY170851.1; 2a, X07865.1; 2b, X65949.1; 2c, AB071845.1; 2d, AY095209.1; 2e, AJ249351.2; 2f, AB472687.1; 2g, AY286000.1) using BLASTn within the BioPython framework (Cock et al., 2009). Only matches with an E-value less than 1 × 10−20 were included in further analysis. For each strain, the length of the best-matched sequence (in terms of the BLAST score) between the contigs and each stx reference gene was calculated. For example where both stx2a and stx2c were present, there may be five query sequences each of 600 bp. If three of them matched stx2a with the highest BLAST score, and two of them matched stx2c with the best BLAST score, then stx2a would score 1,800 and stx2c would score 1,200.
Subtyping of stx by mapping
An alignment of stx1a, stx1c, 1d, 2a, 2b, 2c, 2d, 2e, 2f and 2g sequences (taken from Scheutz et al., 2012) was generated using ClustalW within the MEGA 5 software package (Tamura et al., 2011). Three bases for each reference subtype that, when combined, had 100% sensitivity and specificity for each subtype were identified. High quality sequencing reads were mapped to a set of reference stx genes (same genes as BLAST approach described above) using BWA-MEM (http://bio-bwa.sourceforge.net/). Reads that mapped to more than one place in the reference set (i.e., ambiguous reads) were removed from the resultant SAM file using Samtools (Li et al., 2009). If at least 10 reads and 90% of the total reads concordantly mapped to all three discriminatory positions for a specific subtype, then a positive match was returned for that subtype.
Determination of the presence of multiple alleles of the same stx subtype by mapping depth
Multiple copies of the same stx allele could be identified using two complementary approaches. In the first approach, reads were mapped to the stx reference genes, with ambiguous mapping allowed. Then the coverage of each stx allele, which had been identified by the mapping and assembly methods described above, was calculated using the Samtools ‘depth’ option. A distribution of mapping depth in all the strains that were positive for one particular Stx subtype was plotted revealing a bimodal distribution with the higher mode approximately twice the lower mode. The lower mode represented strains with only one copy of stx and the higher mode represented strains with multiple alleles of stx. There was no bimodal distribution of mapping depth for strains that encoded both stx2a and stx2c, due to the redundant mapping between these two subtypes. For example, if a strain encoded stx2a only and mapped to an stx2c reference gene, it showed approximately one third of the average coverage compared to if it were mapped to an stx2a gene. This cross-mapping meant that multiple alleles of the same stx subtype could not be detected in strains that encoded both stx2a and stx2c.
In the second approach, the bam file resulting from the mapping of the reads to the stx reference set was parsed for mixed positions, with the minority variant present in at least 25% of reads i.e., one position in the reference gene was mapped by two different bases. Only strains that were known to encode only one of stx2a or stx2c from the subtyping results were analysed, as the high similarity between stx2a and stx2c can result in pseudo-mixed bases when compared with stx reference genes. If there were mixed bases present in an alignment (where the depth was greater than 20x and minority variant present in greater than 15% of reads), from a strain encoding only one of stx2a or stx2c, the presence of multiple alleles of a specific stx subtype that vary by at least one base was assumed to be present (Fig. S1).
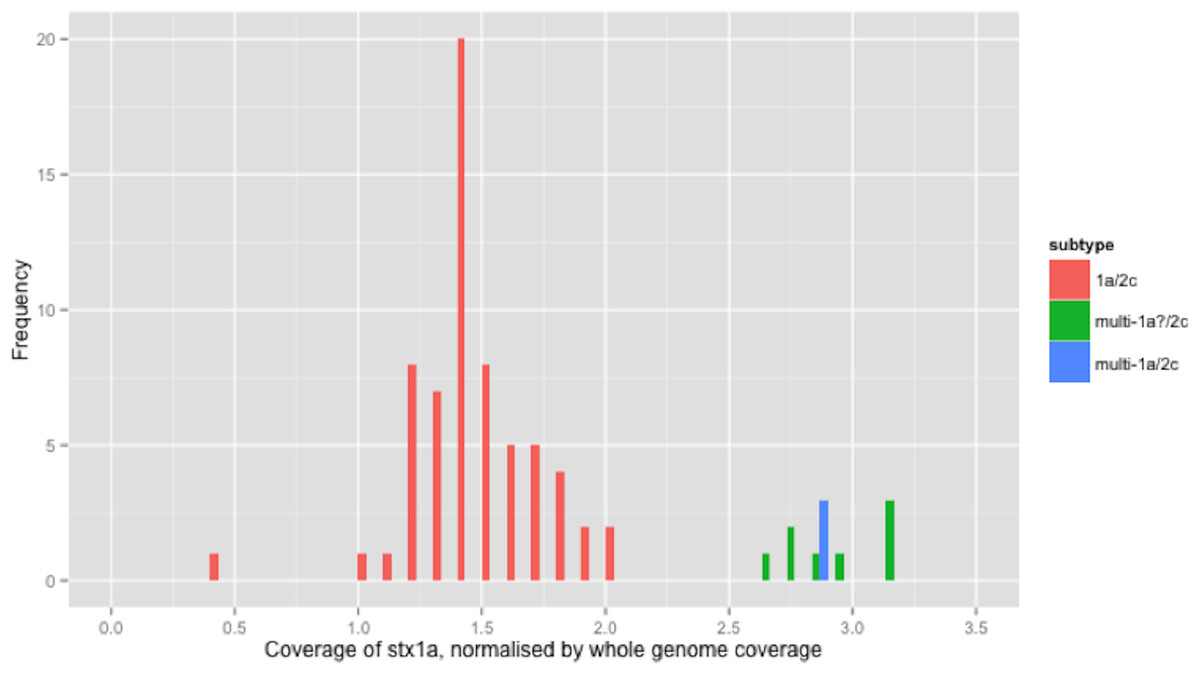
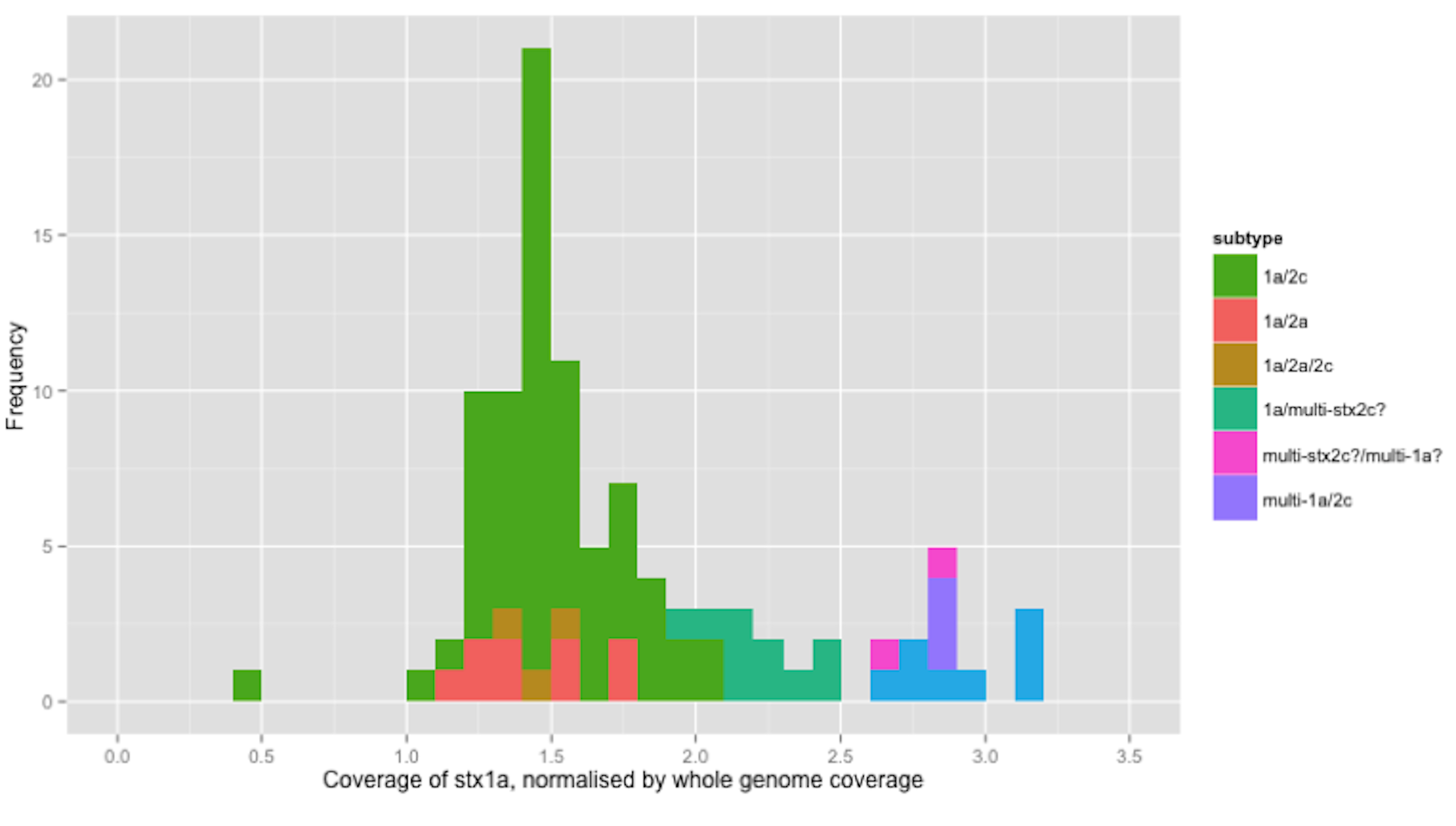
Figure 1: Coverage of stx1a, normalised by whole genome coverage.
Histogram of coverage of stx1a normalised by whole genome coverage.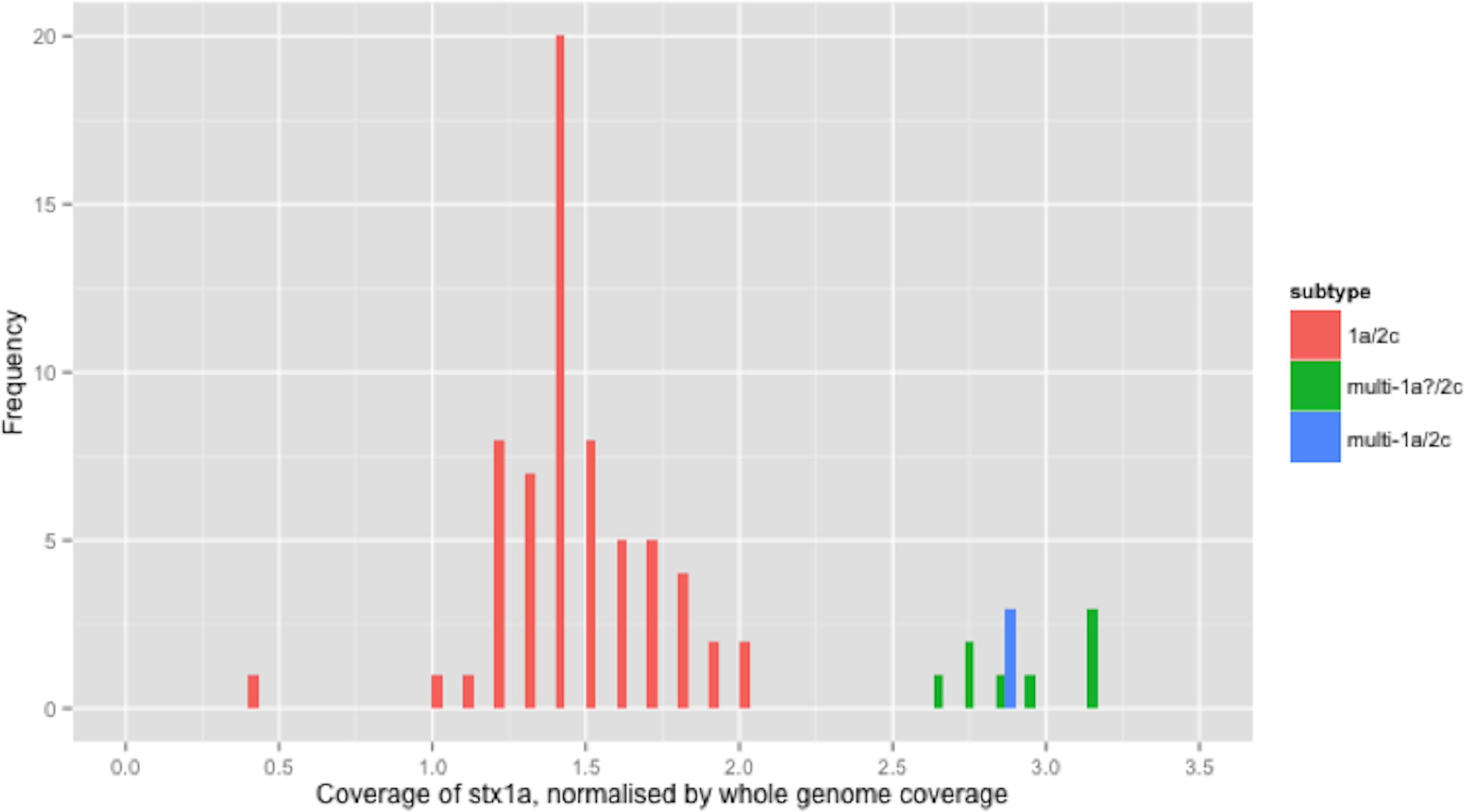{kind=link}
Diversity of stx associated with STEC O157 in the England, Wales and Northern Ireland
The stx genes that were successfully assembled into a single contig were extracted from the de novo genome assemblies using BLAST and aligned. Only strains that subtyping had shown to encode one of stx2a or stx2c were included in this part of the study. At the location where the complete sequence of both stxA and stxB, including the intergenic region, was assembled into a single contig, the CDSs were aligned and represented in minimum spanning trees generated using Bionumerics v6 (http://www.applied-maths.com/bionumerics). Strains where the stxA and stxB subunits could not be assembled into a single contig (e.g., due to the presence of multiple copies of the same stx subtype with sequence variation between them), were aligned against a reference gene and the resulting Sam file was parsed using custom python scripts to identify variant positions. The sequences of stx1a, stx2a and stx2c present in the strains investigated here were compared with a representative sample of stx subtype sequences in the National Centre for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) nucleotide database to assess diversity and identify novel alleles.
Stx real-time qPCR and block-based subtyping PCR
DNA was prepared by inoculating single colonies into 490ul distilled water, which was then boiled in a water bath for 10 min. The real-time qPCR described by the European Union Reference Laboratory (EURL) for stx1 and stx2 was performed as previously described (Jenkins et al., 2012). For the block-based subtyping PCR, DNA was amplified on a block-based DNA Engine platform using the stx subtyping primers and amplification parameters described by Scheutz et al. (2012). Amplified DNA was electrophoresed on a 2% gel, stained with ethidium bromide and visualised with UV light.
Results
Stx subtyping of 444 STEC O157 in the UK—comparison of NGS and PCR
Subtyping results from PCR and WGS were identical in 422/444 strains (Table 1); there was agreement for 85 stx2a encoding strains, 153 stx2a/stx2c strains and 187 stx2c strains. When the subtyping PCR was repeated for the 22 discordant strains, results for 442/444 strains were identical. Of the two strains where PCR and sequencing were discordant, one strain was positive for stx2c by PCR but no stx2c was identified in the sequencing data by the bioinformatics algorithms described here, and one strain was positive for stx2a by sequencing that was not detected by PCR. The strain that had a positive PCR result for stx2c but no corresponding result in the WGS data had a very low level of mapping (54 reads, <7x average coverage) to stx2c. This was not enough to definitively identify stx2c by either the mapping or assembly algorithms, although is indicative of its presence. The stx2a gene sequence of the strain that was PCR negative but that had stx2a reads identified in the WGS data was analysed for mutations in the primer binding sites, but none were identified.
| Subtype | Sequencing results |
Subtyping PCR results—1st pass |
Subtyping PCR results—2nd pass |
|---|---|---|---|
| 2a | 82 | 89 | 82 |
| 2c | 194 | 196 | 196 |
| 2a/2c | 167 | 155 | 166 |
| No result | 1 | 4 | 0 |
| Total | 444 | 444 | 444 |
Detection of multiple alleles of stx
A subset of 349 sporadic strains of STEC O157 (i.e., not from same person, household or outbreak) was investigated for the presence of multiple alleles of the same subtype of stx. The detection of multiple copies of the same stx subtype was performed using two complimentary methods (i) mapping and determining the short read coverage of a particular stx subtype relative to the coverage of the whole genome and (ii) the detection of mixed bases (coverage >20x, minority variant >15%, see Fig. S1) in an alignment to a single reference gene.
The stx1a gene was detected in 6 different combinations with other subtypes/alleles, in 100 strains from independent sources (Table 2). For clarity, the relative coverage of stx1a in three of the observed combinations (totalling 77 strains) is presented (Fig. 1). The relative coverage of stx1a in all 6 combinations observed in the 100 stx1a strains can be seen in Fig. S2. In Fig. 1, a bimodal distribution was clear, with the higher mode being approximately twice as high as the lower mode. There were 11 strains in the higher mode (Fig. 1). When the stx1a alignments were examined for the presence of mixed bases, there were 97 strains with no mixed bases and three strains that had at least one mixed base position. The relative coverage was examined in the context of the presence of mixed bases; strains with no mixed bases had a median relative coverage of 1.7x, whereas strains with at least one mixed base had a mean coverage of 2.8x (Fig. 1). There were nine strains without mixed bases that had relative stx1a coverage closer to the average of mixed base position strains than the average of no mixed bases suggesting that two identical copies of the stx1a gene were present.
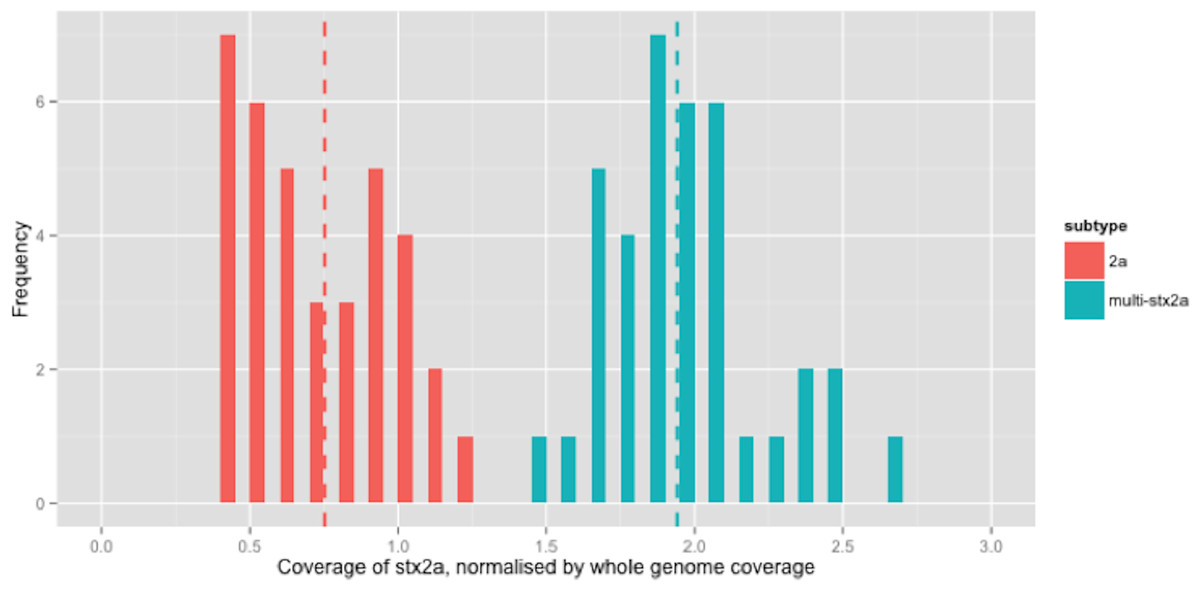
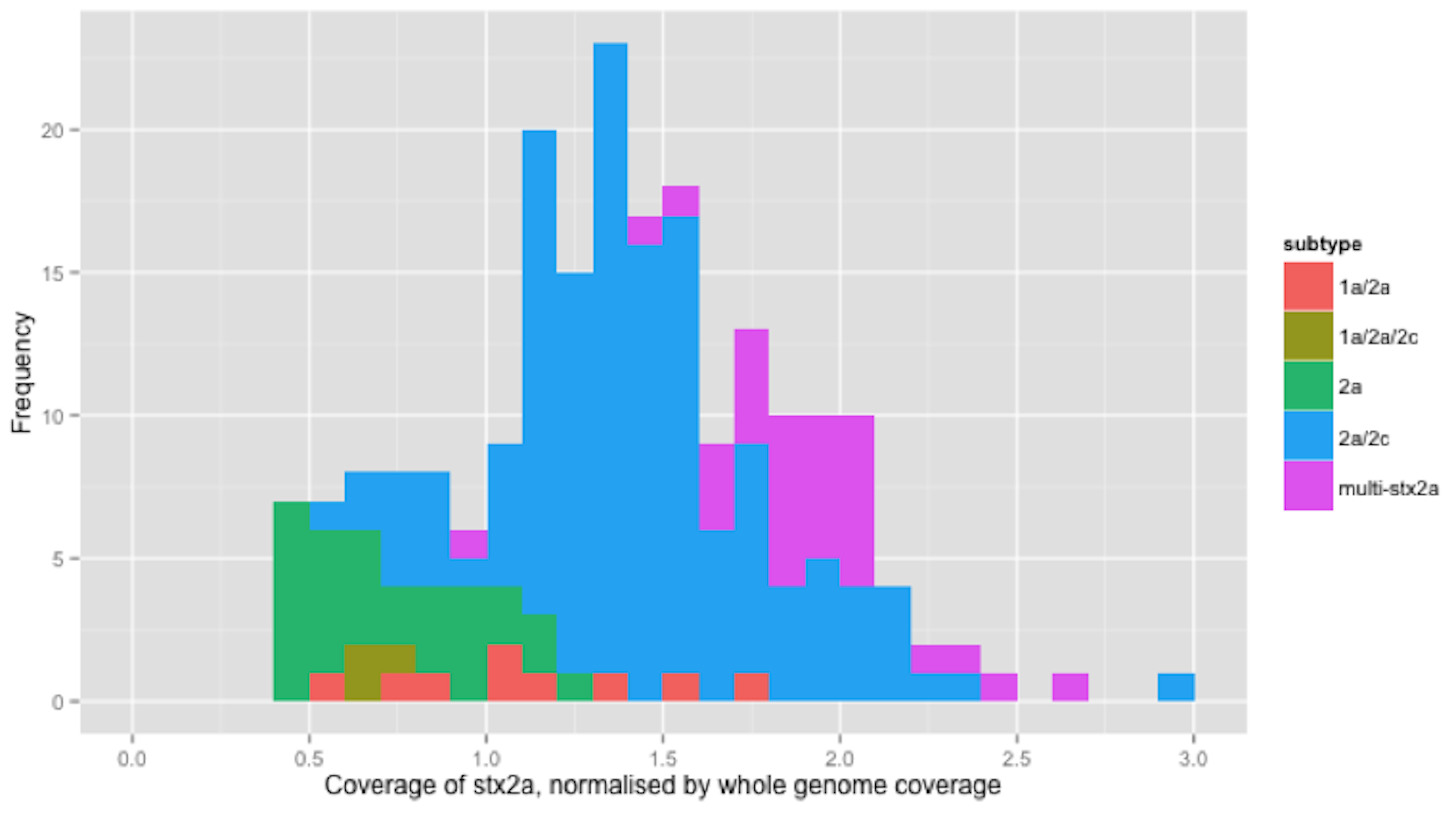
Figure 2: Coverage of stx2a, normalised by whole genome coverage.
Histogram of coverage of stx2a normalised by whole genome coverage.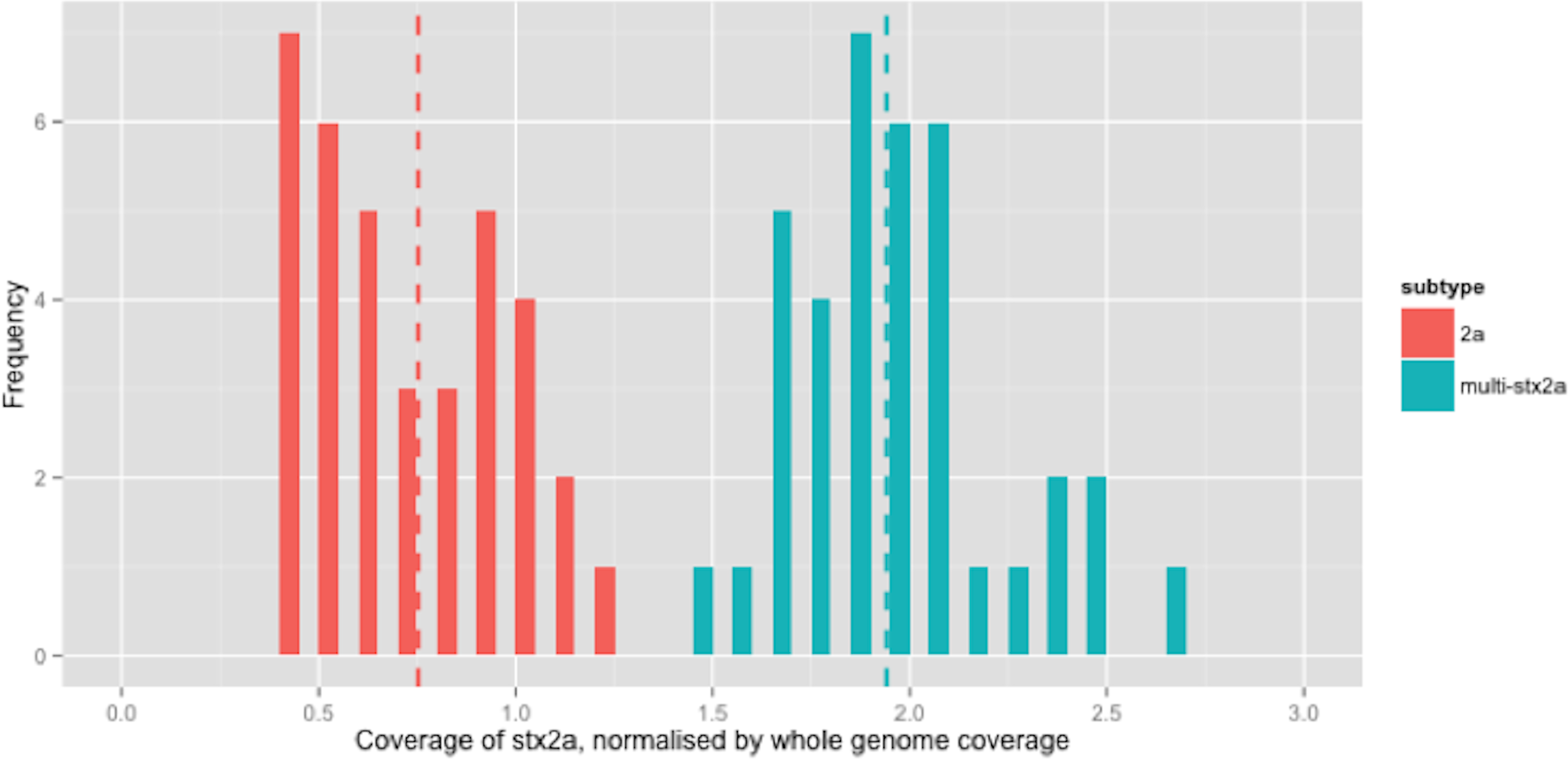{kind=link}
| stx profile | Frequency |
|---|---|
| 1a/2a | 9 |
| 1a/2a/2c | 3 |
| 1a/2c | 64 |
| 1a/multi-stx2c? | 10 |
| 2a | 30 |
| 2a/2c | 136 |
| 2c | 51 |
| multi-1a?/2c | 9 |
| multi-1a/2c | 3 |
| multi-stx2a | 31 |
| multi-stx2c?/multi-1a? | 2 |
| No stx detected | 1 |
There were 210 isolates that encoded stx2a, either alone or in combination with other subtypes (Table 2). For clarity, only the relative coverage of stx2a from the 73 strains that encoded only stx2a were presented in Fig. 2 (the relative coverage of stx2a in all strains which encoded this subtype can be seen in Fig. S3). Inspection of the distribution of coverage of the short reads in stx2a revealed at least two modes within the relative coverage of stx2a, with the upper mode (1.8x) being twice that of the lower (0.9x) (Fig. 2). Of the 70 strains that encoded stx2a but not stx2c, 31 (42%) had short read coverage in the upper mode (1.8x), of which all 31 had mixed base positions in their alignments, indicating the presence of two alleles of stx2a. When the mixed position data was compared with the relative coverage distribution, the mean relative coverage of the strains with mixed positions of stx2a was 1.9x, while the coverage in strains with no mixed positions was 0.75x (Fig. 2). There were 1 (n = 29), 2 (n = 1) or 3 (n = 1) positions with mixed bases between the alleles in the 31 strains with multiple copies.

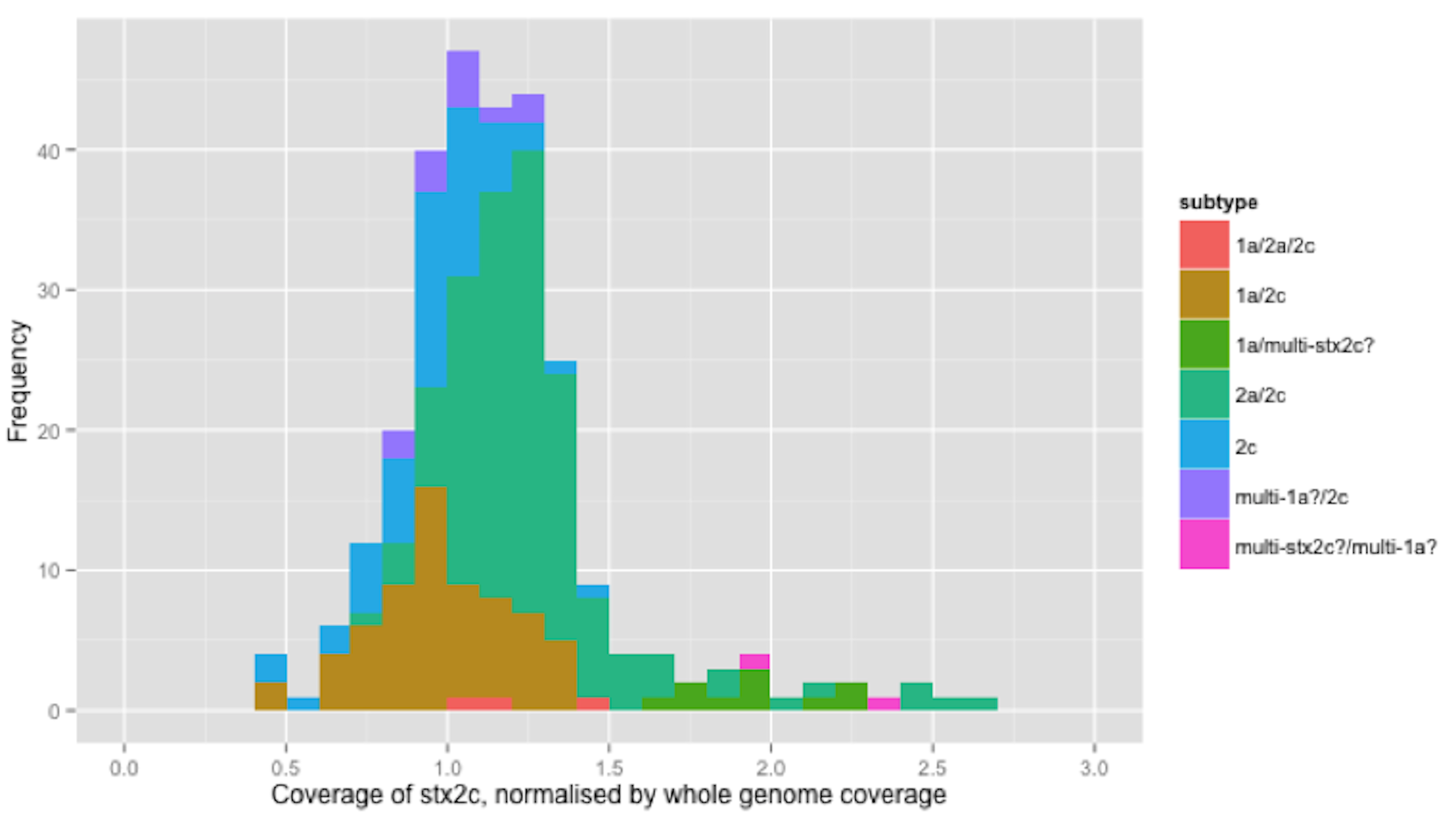
Figure 3: Coverage of stx2c, normalised by whole genome coverage.
Histogram of coverage of stx2c normalised by whole genome coverage.{kind=link}
The relative coverage of the 279 isolates that encoded stx2c was calculated. For clarity, only the relative coverage of the 139 strains that encoded stx2c but not stx2a are presented in Fig. 3. The relative coverage of stx2c in all 279 strains can be seen in the Supplemental Information and analysis of the distribution of relative stx2c coverage showed that the majority of these strains fell into an approximately normal distribution around 1x relative coverage (Fig. 3). Twelve (8.6%) of the 139 strains had a relative coverage >1.5x but no mixed base positions were found.
Diversity of stx associated with STEC O157 in the UK
The diversity of stx found in a subset of 349 sporadic strains of STEC O157 (i.e., not from same person, household or outbreak) was investigated. Ninety-seven complete stx1a genes from this study were compared with nine stx1a alleles from NCBI, and a total of 16 variant positions were identified along the 1392 bp length of the gene. Of the five different alleles present in the strains investigated here, three were not present in the NCBI database (as of 06/23/14, Fig. 4). The most frequently observed allele accounted for 76 (78.3%) of the 97 assembled stx1a genes from this study, while the second most frequently observed allele accounted for 16 (16.5%) stx1a genes. Both the most frequently observed alleles had been previously identified in E. coli O103:H2 (BAI33872.1) and E. coli O157:H7 (EF079675.1), respectively. The five remaining stx1a genes comprised three different alleles, none of which had been previously submitted to the NCBI database (as of 06/23/14), although they were all within a single variant of previously observed alleles (Fig. 4).
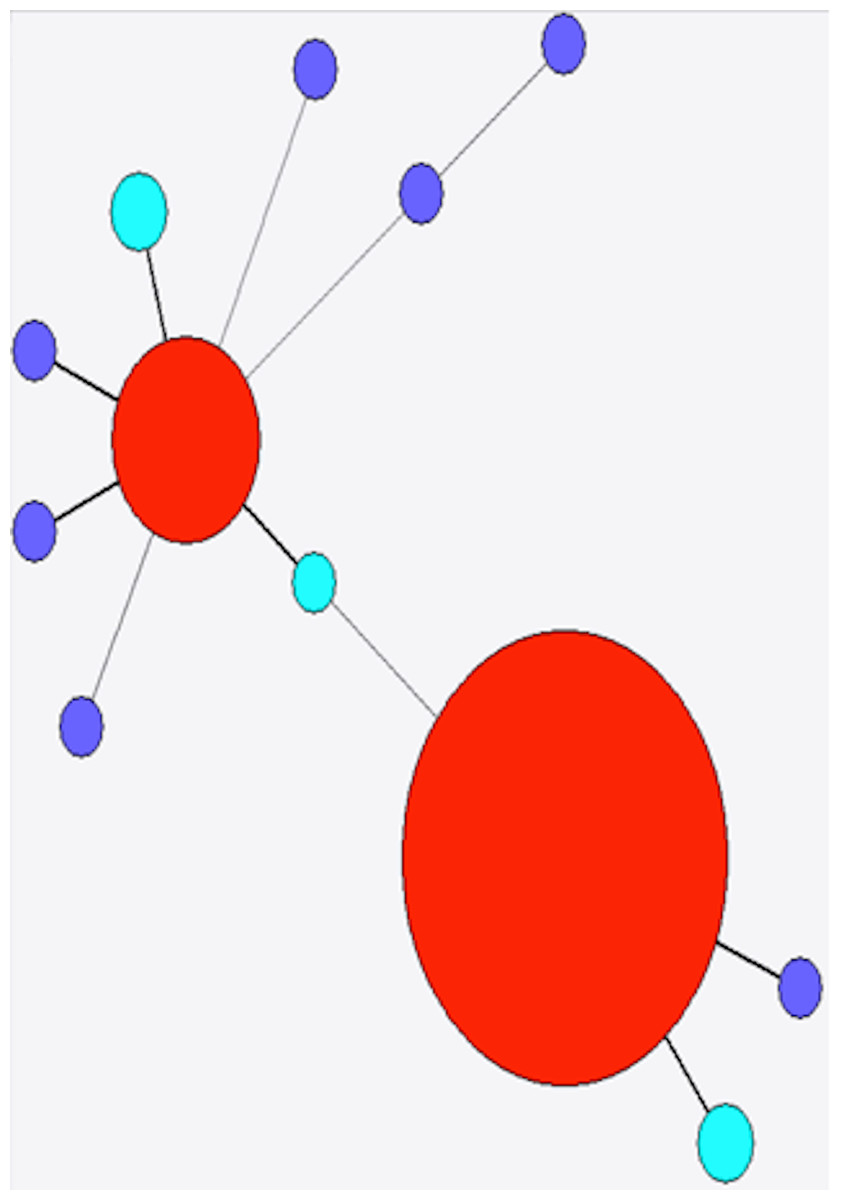
Figure 4: Minimum spanning tree of stx1a.
Red, previously identified and observed in this study; purple, previously identified but not observed in this study; light blue, novel allele.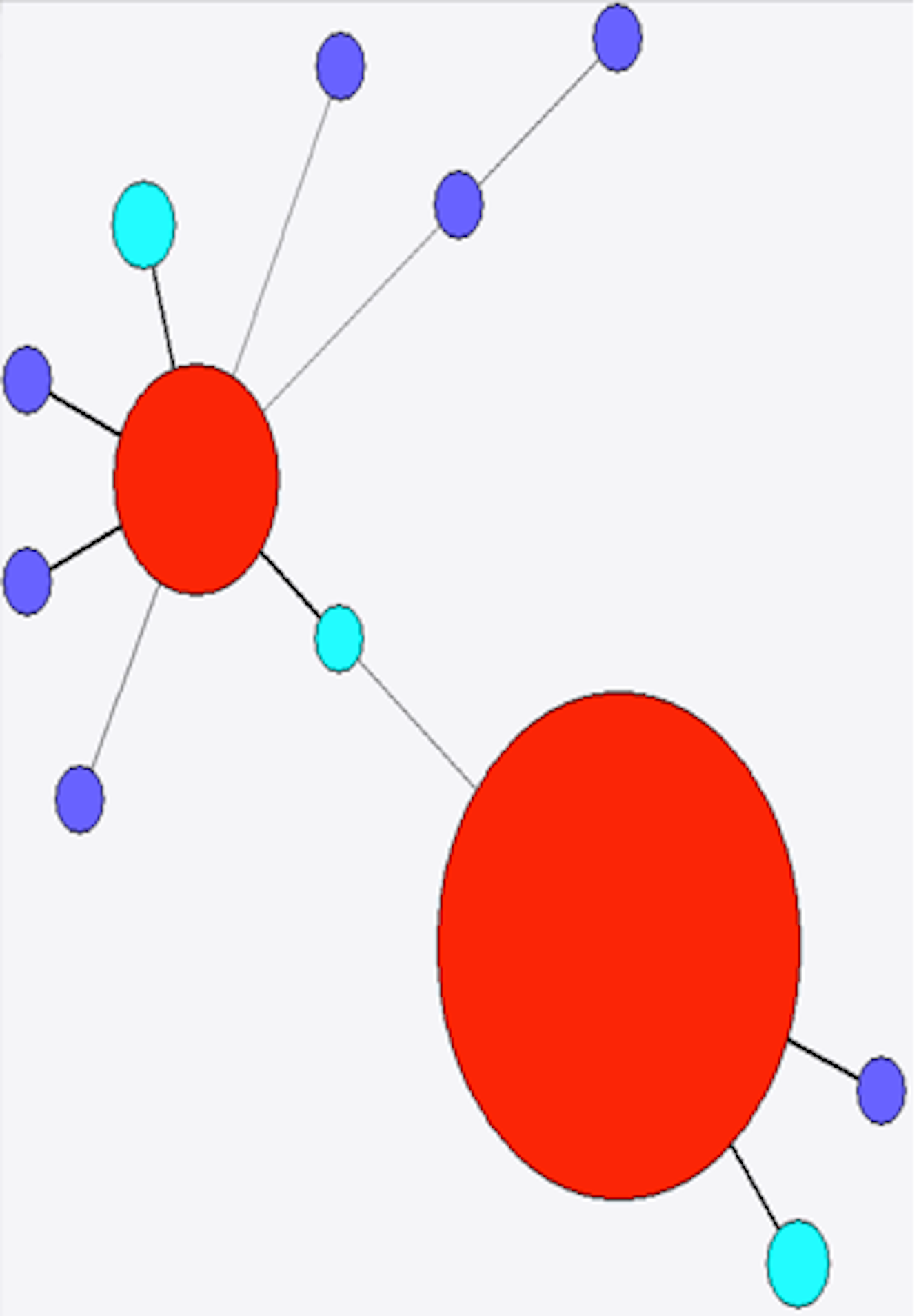{kind=link}
The 38 fully assembled stx2a genes from this study were compared with 22 stx2a alleles from the NCBI nucleotide database. There were a total of 48 variant positions in a 1,442 bp alignment of the 59 stx2a genes that included 25 different alleles (Fig. 5). Of these 25 alleles, six were present in the strains investigated here. The most frequently observed allele was a single variant from a stx2a allele observed in E. coli O157 (AF524944.1), E. coli O111 and E. coli O145 and was present in 18 (47.3%) of the strains in this study. The second most frequently observed allele was present in 11 (28.5%) strains and was widely distributed, including in Bacteriophage 933W (X07865). The other nine strains represented four alleles, two of which had been identified before. The remaining allele (from strain H124840173) was highly divergent from the other stx2a alleles, with six SNPs compared to any previously identified stx2a gene and 11 variants compared to the closest stx2a observed in this study. Interestingly, this strain was a sorbitol-fermenting (SF) STEC O157, the only SF strains to be included in this study.
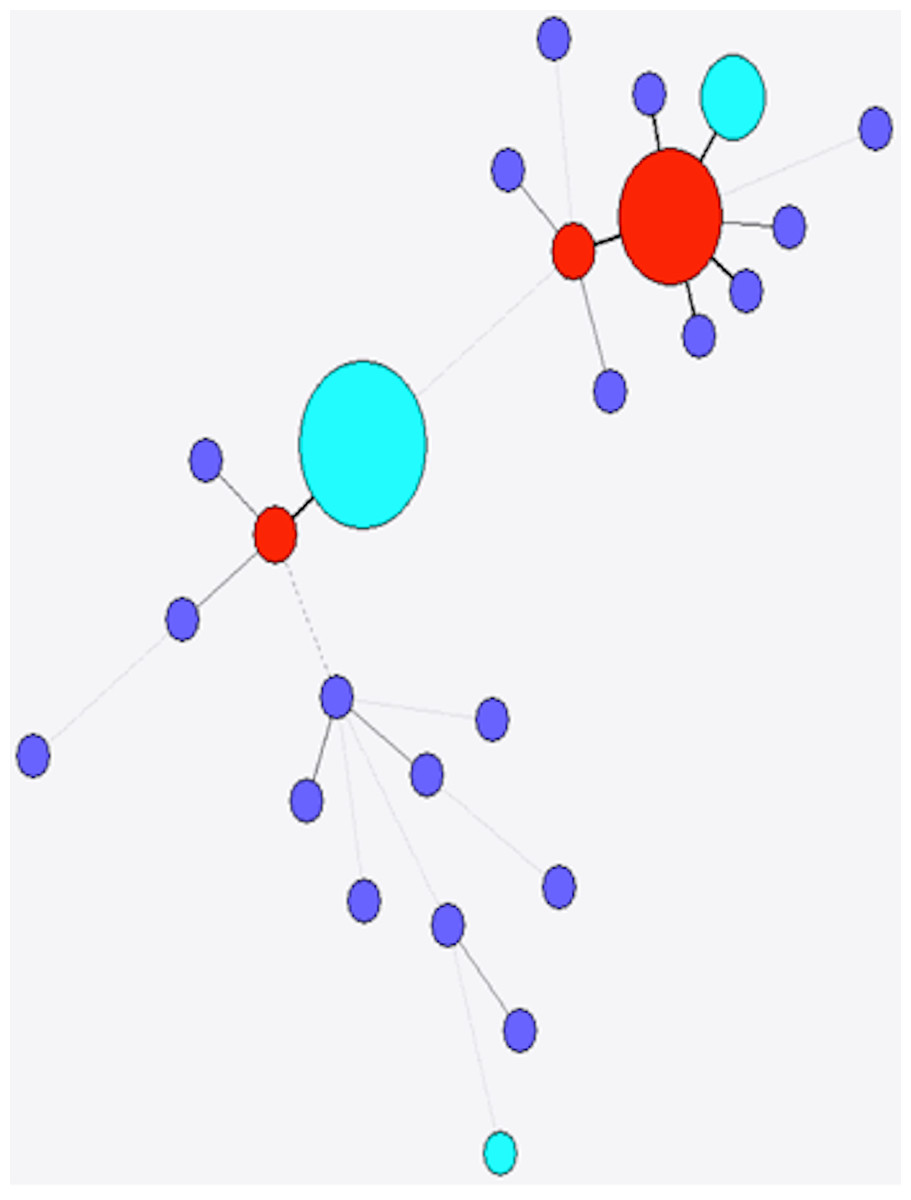
Figure 5: Minimum spanning tree of stx2a.
Colour as in Fig. 4.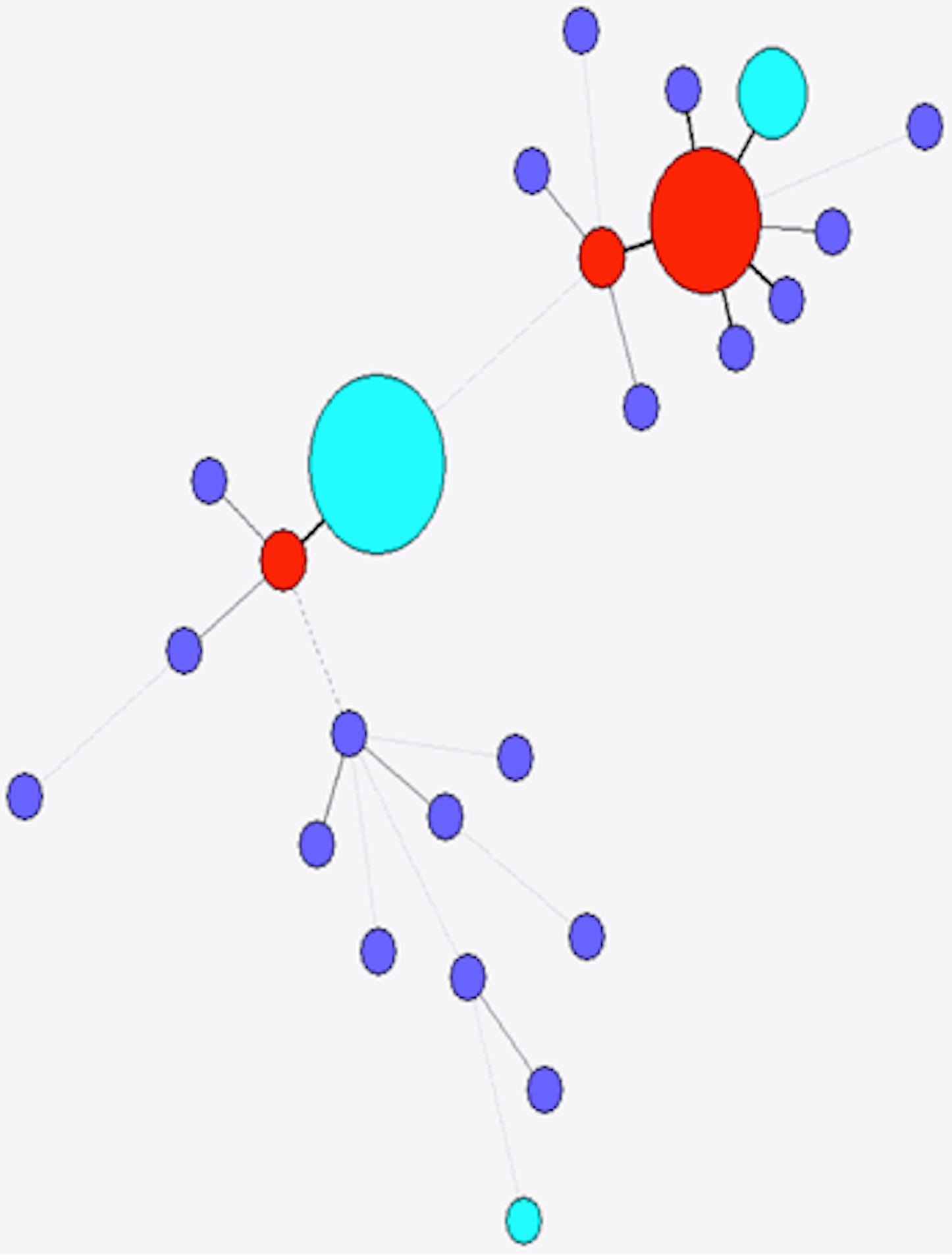{kind=link}
There were 132 fully assembled stx2c genes from this study that were compared with 18 previously identified stx2c alleles from NCBI. There was a total of 59 variant positions along the 1441 bp gene alignment of the 150 stx2c sequences, comprising 22 unique alleles, of which seven were identified in the strains analysed here (Fig. 6). The most frequently observed allele accounted for 115 (87.1%) of the 132 fully assembled stx2c genes. This allele had been previously observed in a single E. coli O157:H- strain (AB015057.1). The 17 other stx2c genes represented six distinct alleles that, with one exception, were within two variants of the most frequently observed allele (Fig. 6). There were two strains encoding the most divergent observed stx2c allele, with six variant positions compared with the most frequently observed allele. This divergent allele had been previously identified in E. coli RM10648 (KF932369.1).
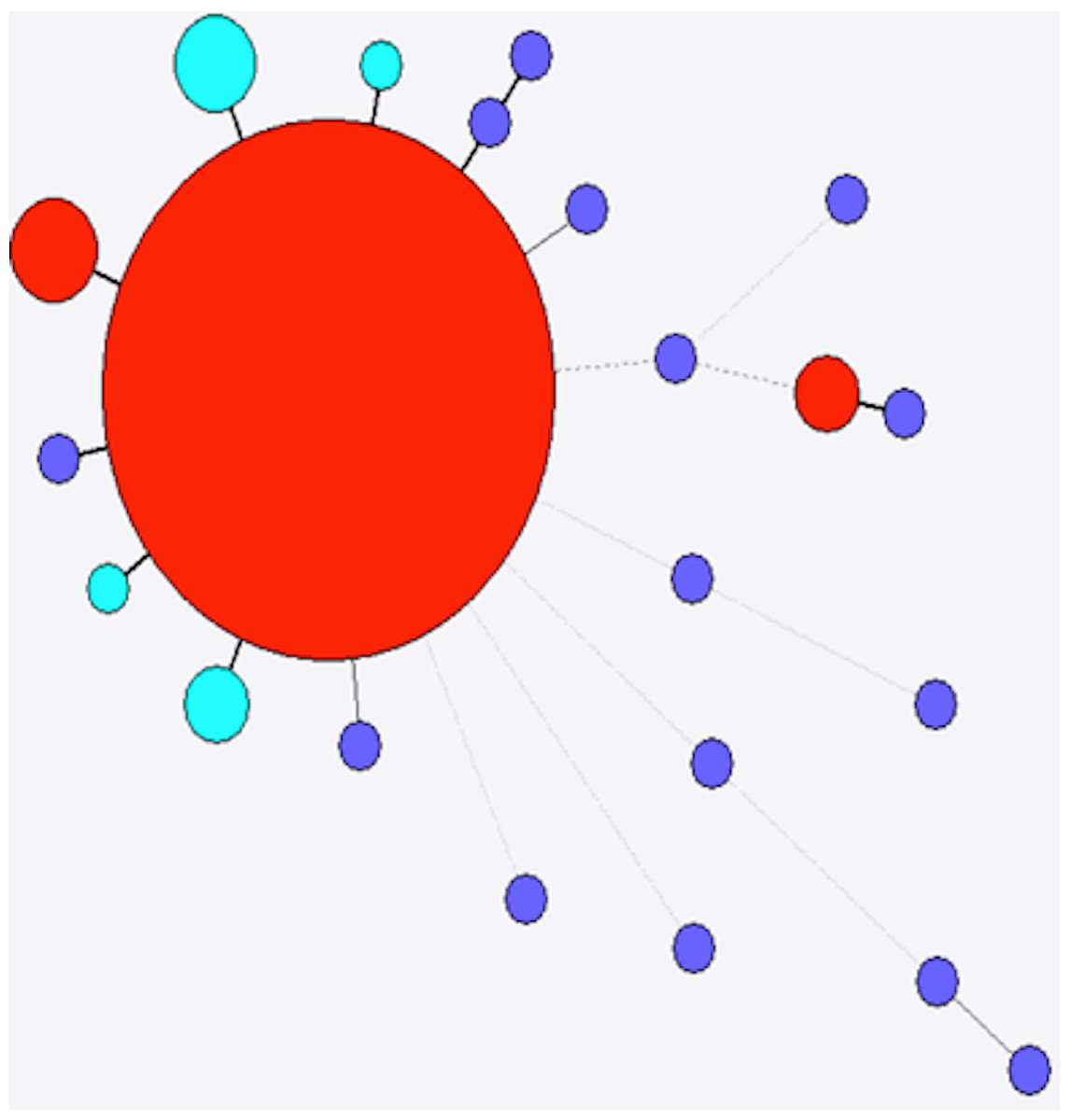
Figure 6: Minimum spanning tree of stx2c.
Colour as in Fig. 4.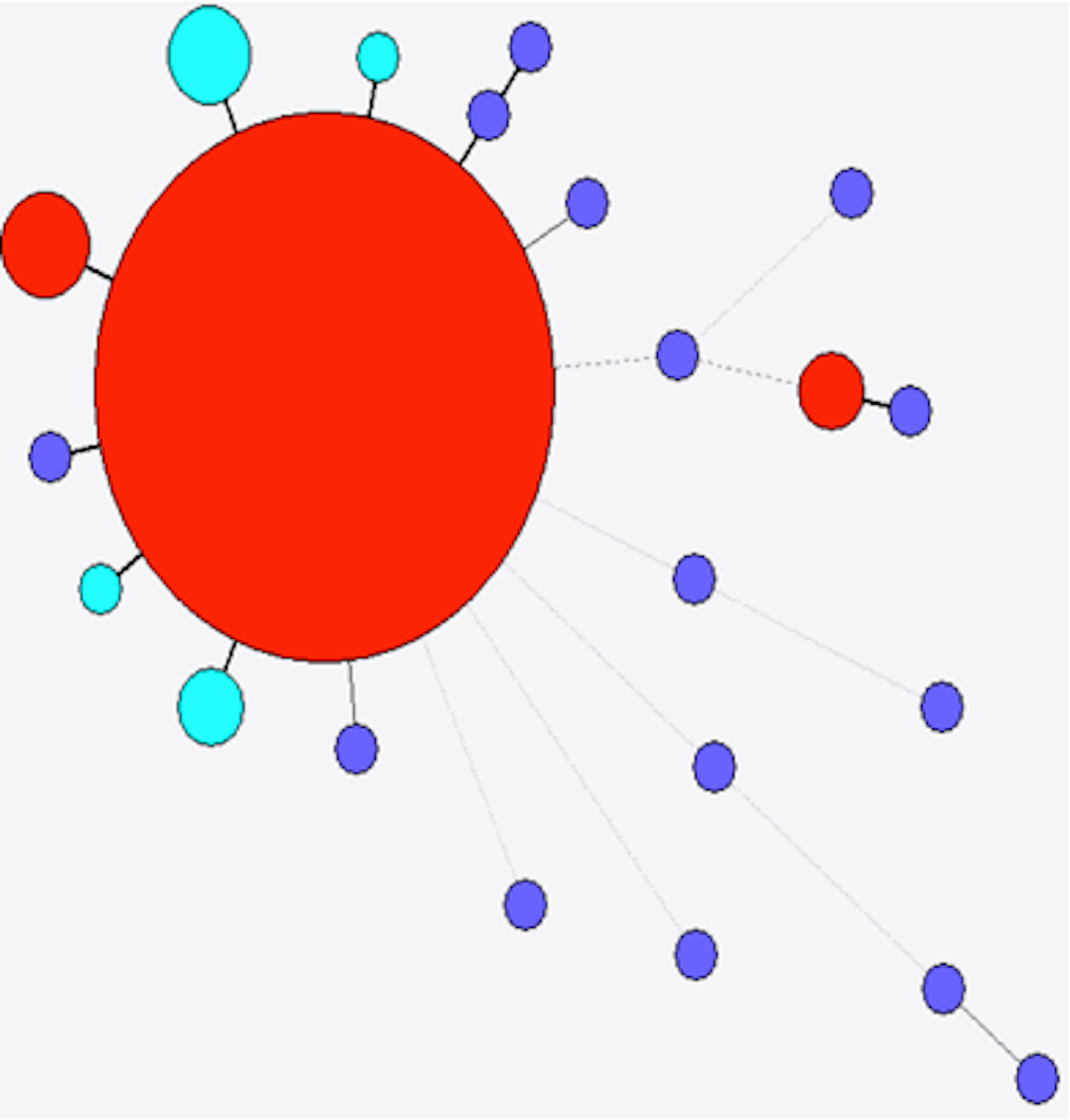{kind=link}
Although the complete gene sequence could not be determined for strains that had more than one copy of a stx subtype, an alignment of the reads against a reference was analysed to identify variant positions. Of the three strains with multiple alleles of stx1a, all three had the same four variant positions. There was one SNP in all three multiple-stx1a strains that was not previously identified in the stx1a sequences described above or in the NCBI reference sequences. Of the 30 strains with multiple copies of stx2a, 28 had only a single variant position that was the same in all 28 strains and that had been previously identified. Of the other two strains, one had the same SNP as the 28 other mixed position strains and an additional SNP that had not been previously observed in the strains described in this study above or in the NCBI reference strains. The final strain had three unique mixed positions, all of which had been previously observed in this study.
Discussion
In this study we have developed novel, robust and highly accurate methods for subtyping of stx from short read sequence data, validating this method against PCR for 444 STEC O157 isolates. Furthermore, we have mined the WGS data to show that a significant proportion of strains encode multiple copies of the same subtype of Shiga-toxin gene. The diversity of stx genes from STEC O157 in England was also elucidated.
There was over 95% initial agreement (422 of 444 strains) between WGS subtyping and PCR subtyping in determining subtypes of stx2, which shows that WGS is an acceptable method for subtyping stx in O157. The strains where there were discrepancies between WGS and PCR were subjected to a repeat subtyping PCR, after which all but two of the discrepancies became concordant. One possible reason for the discrepancy between the initial and repeat PCR results is the high stringency of the subtyping PCR. During a multi-centre evaluation of the subtyping PCR, there were differences observed in the subtyping results obtained between different laboratories, and these were ascribed to the use of different reagents and thermocyclers, with the main source of variability thought to be the use of different polymerase. While the taq polymerase recommended by Scheutz et al. (2012) was used here, variations in other laboratory reagents and equipment may have resulted in the discrepancies. The excellent concordance between the PCR and WGS results, even despite the problems associated with analysis of homologous genes using short read data, provides evidence of the accuracy of the bioinformatics algorithm showing that WGS could replace PCR for subtyping.
Using mapping coverage to detect multiple copies of the Stx phage has been described previously using more challenging metagenome data (Loman et al., 2013). The novelty of this work is to use the mapping coverage of stx relative to the average coverage of the whole genome to identify strains encoding multiple alleles of the same stx subtype. There are stx sequences in the NCBI database that indicate that multiple alleles of the same subtype encoded by the same strain have been previously observed i.e., these sequences contain ambiguous bases. However, the studies associated with these sequences make no mention of the possibility of multiple alleles (De Baets et al., 2004; Lee et al., 2007; Asakura et al., 2001; Hegde, Ballal & Shenoy, 2012). The presence of multiple alleles of the same stx type has been previously identified by WGS (Eppinger et al., 2011); however, this is the first study to present a large sale comparison of this method with PCR subtyping. Some of the ambiguous positions in sequences in the NCBI database were the same positions in stx as the mixed positions observed in this study, supporting the evidence that multiple alleles exist and are present in the same strain. While mapping of short reads has been successful at detecting multiple copies of the same subtype, it has not been possible in strains that encode stx2a and stx2c due to ambiguous mapping between these types (see Figs. S2–S4). For characterisation of these stx2a/stx2c strains, and full characterisation of the insertion sites and genomic context of the stx alleles in strains encoding multiple copies of the same subtype, longer sequencing reads (from e.g. PacBio) are needed. There was also evidence that some strains of STEC O157 encoded multiple alleles of both stx1a and stx2c; further characterisation of these strains to determine whether they had a genetic determinant that made Stx phage acquisition more likely would be interesting.
The functional implication of encoding multiple alleles of the same stx subtype remains unclear. Three hypotheses to explain the 9% prevalence of strains encoding multiple alleles of the same subtype are (i) these strains are more likely to cause symptomatic human disease (ii) these strains have a fitness advantage which increases their chance of being present in the environment (iii) carrying multiple alleles of the same subtype is ‘merely’ a side effect of the recombinogenic capacity of Stx phage, which confers no phenotype. It is interesting that while the multiple alleles of stx2a and stx2c always seem to have nucleotide differences, this is a minority in the strains that encode multiple copies of stx1a. The close sequence relationship between the multiple copies of the same subtypes raises the question whether they are derived from multiple insertions by different Stx phage, or a phage or stx gene duplication.
This study reports on the diversity of stx observed in STEC O157 in the UK (except Scotland) between 1990 and 2013, with a focus on 2012. Although there are 10 described subtypes of stx1 and stx2 combined (Scheutz et al., 2012), in an examination of 444 strains covering a wide temporal spread and range of phage types only three subtypes (stx2a, stx2c and stx1a) were observed. Previous studies examining strains from cattle and humans similarly found only stx2a, stx2c and stx1a (Mellor et al., 2013). The most diverse stx identified here was stx2a, followed by stx1a and then stx2c (Figs. 4–6). The majority of stx2c were of a single genotype, and all the novel alleles identified were within a single SNP of the majority genotype. This difference in diversity observed between stx2a and stx2c is interesting, considering that the background diversity of these two subtypes is largely similar (Scheutz et al., 2012). Further studies in this laboratory aim to determine the phylogenetic context of isolates encoding these two subtypes. This study also described 10 novel alleles of stx, with the most diverse being an stx2a sequence 6 SNPs from any previously described stx2a. The fact that this diverse stx was observed in a sorbitol fermenting strain indicates that there may be a significant reservoir of stx diversity in other serotypes of STEC. The majority of novel alleles had sequences that were single nucleotide variants to previously described sequences.
This study is the first to describe stx subtyping by both PCR and WGS methods in a large number of strains of STEC O157. Both the PCR and WGS approaches to stx subtyping provided a good level of sensitivity and specificity. The WGS data also showed that a significant proportion of strains of STEC O157 harbour multiple alleles of the same Stx subtype. The functional significance of multiple alleles of the same subtype remains unclear, although this is the subject of ongoing work. Furthermore, the WGS analysis highlighted 10 novel alleles of stx identified in this study and enabled us to study the diversity of stx sequences in a population of STEC O157 associated with clinical disease in England.
Supplemental Information
Mixed position in an stx2a gene
Variant from reference highlighted.
{kind=link}
{kind=link}
{kind=link}
{kind=link}