
Genome analysis of the ubiquitous boxwood pathogen Pseudonectria foliicola
- Published
- Accepted
- Received
- Academic Editor
- Irene Newton
- Subject Areas
- Genomics, Mycology
- Keywords
- Volutella blight, Boxwood, Comparative genomics, Nectriaceae
- Copyright
- © 2018 Rivera et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2018. Genome analysis of the ubiquitous boxwood pathogen Pseudonectria foliicola. PeerJ 6:e5401 https://doi.org/10.7717/peerj.5401
Abstract
Boxwood (Buxus spp.) are broad-leaved, evergreen landscape plants valued for their longevity and ornamental qualities. Volutella leaf and stem blight, caused by the ascomycete fungi Pseudonectria foliicola and P. buxi, is one of the major diseases affecting the health and ornamental qualities of boxwood. Although this disease is less severe than boxwood blight caused by Calonectria pseudonaviculata and C. henricotiae, its widespread occurrence and disfiguring symptoms have caused substantial economic losses to the ornamental industry. In this study, we sequenced the genome of P. foliicola isolate ATCC13545 using Illumina technology and compared it to other publicly available fungal pathogen genomes to better understand the biology of this organism. A de novo assembly estimated the genome size of P. foliicola at 28.7 Mb (425 contigs; N50 = 184,987 bp; avg. coverage 188×), with just 9,272 protein-coding genes. To our knowledge, P. foliicola has the smallest known genome within the Nectriaceae. Consistent with the small size of the genome, the secretome, CAzyme and secondary metabolite profiles of this fungus are reduced relative to two other surveyed Nectriaceae fungal genomes: Dactylonectria macrodidyma JAC15-245 and Fusarium graminearum Ph-1. Interestingly, a large cohort of genes associated with reduced virulence and loss of pathogenicity was identified from the P. foliicola dataset. These data are consistent with the latest observations by plant pathologists that P. buxi and most likely P. foliicola, are opportunistic, latent pathogens that prey upon weak and stressed boxwood plants.
Introduction
Ascomycete fungi inhabit almost all known ecosystems, and play important roles as plant and insect pathogens, endophytes, mycoparasites, and saprobes (Arnold et al., 2009). Within the Ascomycota, the Nectriaceae family includes 55 genera with approximately 900 species (http://www.indexfungorum.org). Although best known as soil-borne saprobes or weak plant pathogens, several species in this family are responsible for extensive economic losses due to damage incurred to crops or in natural ecosystems (Halleen, Fourie & Crous, 2006; Malapi-Wight et al., 2016a; Windels, 2000). The systematics and taxonomy of the Nectriaceae family has been extensively studied (e.g., Salgado-Salazar et al., 2014; Lombard et al., 2015) however, outside of the genus Fusarium, only a small number of fungal species in this family have genome resources publicly available, including Calonectria pseudonaviculata, C. pseudoteaudii, Dactylonectria macrodidyma and Ilyonectria destructans (http://genome.jgi.doe.gov/Ilysp1/Ilysp1.home.html; Malapi-Wight et al., 2015; Malapi-Wight et al., 2016b; Ye et al., 2017). Whole genome resources are now commonly used to understand evolutionary characteristics of pathogenicity across fungi with different lifestyles (Lo Presti et al., 2015) and could become useful for the characterization and biosecurity analysis of undescribed pathogens (McTaggart et al., 2016).
Pseudonectria foliicola and P. buxi (the latter formerly known as Volutella buxi or P. rousseliana) are nectriaceous species causing a ubiquitous leaf and stem blight disease on boxwood (Buxus spp.), known as volutella blight (Fig. 1). To date, this disease has been reported worldwide, throughout the US, Armenia, Belgium, Bulgaria Canada, China, Greece, Portugal, Spain, UK and Ukraine, among others (Farr & Rossman, 2018), although its distribution may extend further along with the distribution of boxwood plants. Infected plants may lack any disease symptoms, or they may manifest visually discernable physiological changes such as leaf discoloration, stem dieback and extensive pink fungal sporulation on the surface of leaves and twigs (Shi & Hsiang, 2014). The causal agent of volutella blight has been described as the species P. buxi (and synonyms) since the early nineteenth century. However, on the basis of morphological and molecular distinctiveness, Lombard et al. (2015) recently described P. foliicola as a second species of Pseudonectria that infects boxwood in New Zealand and the US. It is currently unclear to what extent previous sightings of volutella blight prior to the discovery of P. foliicola were actually caused by P. buxi, P. foliicola, or both of these pathogens.
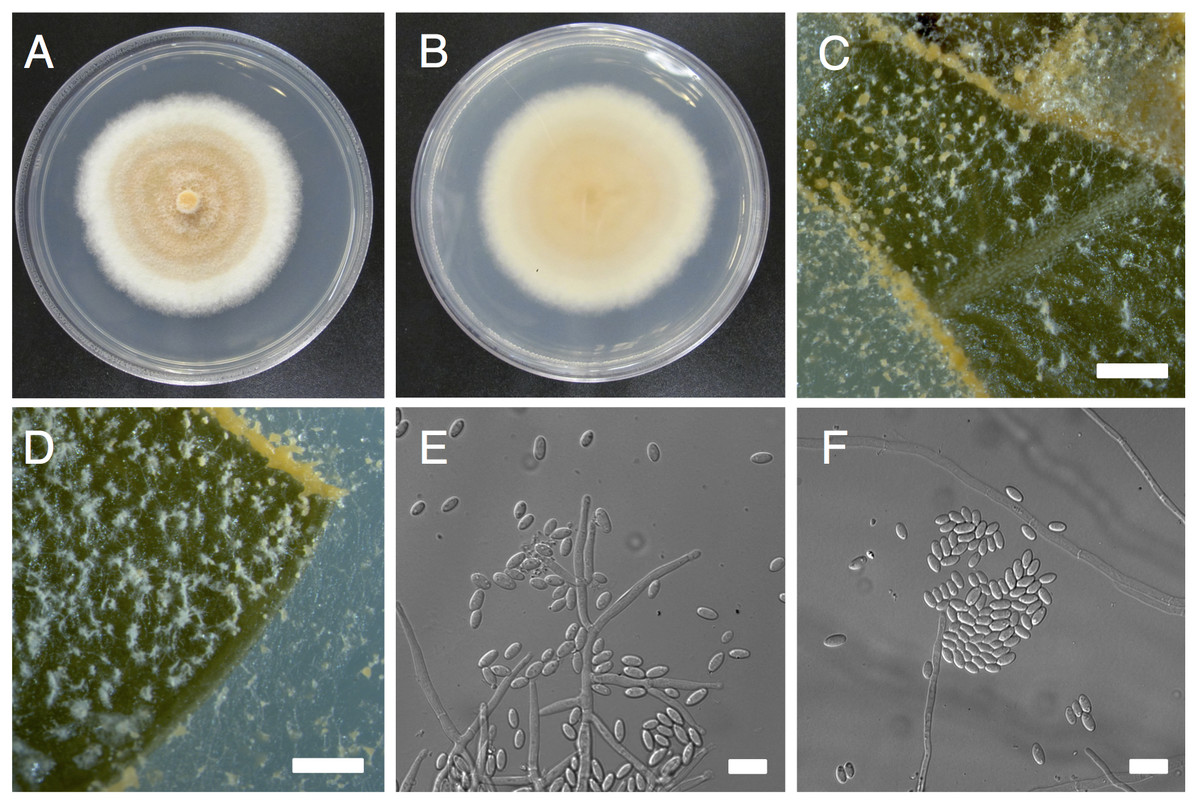
Figure 1: Morphological characters of Pseudonectria foliicola (ATCC 1354).
(A) and (B) Mycelial growth on potato dextrose agar (A, front; B, back). (C) and (D) Sporulation on infected boxwood leaf tissue. E, Conidiophores. (E) and (F) conidia. Scale bars: B–D = 50 µm, E–F = 10 µm.{kind=link}
The pathogens responsible for volutella blight disease have long been considered saprophytes or secondary invaders, however, recent studies by Shi & Hsiang (2014) identified P. buxi causing primary infection on wounded tissue contributing to boxwood decline. Reports from China and Italy confirm the impact of P. buxi as a primary pathogen of Buxus spp. (Shi & Hsiang, 2014; Garibaldi et al., 2016). Unlike boxwood blight disease caused by C. pseudonaviculata and C. henricotiae (Gehesquière et al., 2016), volutella blight primarily affects the ornamental value of boxwood, as plants are typically not killed by the fungal infection. Nonetheless, financial losses due to volutella blight may be considerable. For example, in 2008, economic losses in a single nursery in southern Ontario due to volutella blight of boxwood exceeded $60,000 (Shi & Hsiang, 2014), and similar economic burdens could be expected across the ornamental industry in other regions.
Despite being one of the most commonly observed diseases affecting boxwood, little is known about the genetics, biology and etiology of the causal agents of volutella blight. In this study, we report the first draft genome sequence assembly and annotation of P. foliicola and compare the genome characteristics against two other plant pathogenic fungi in the Nectriaceae, Fusarium graminearum and Dactylonectria macrodidyma. Fusarium graminearum is the hemibiotrophic pathogen that causes head blight on wheat and barley, responsible for substantial economic losses in these industries (Goswami & Kistler, 2004). Dactylonectria macrodidyma is a destructive necrotrophic pathogen that causes the black foot rot of grapevine and root rots of avocado and olive trees (Urbez-Torres, Peduto & Gubler, 2012; Vitale et al., 2012). Our goal in this study was to compare the genome sequence of P. foliicola to these organisms to reveal genome-wide characteristics that may help us better understand the lifestyle of this important fungal pathogen.
Materials and Methods
Fungal isolate and nucleic acid isolation
An axenic culture of P. foliicola isolate ATCC13545® (also known as isolate A.R. 2711) was used for genome sequencing. This fungal isolate was originally cultured from B. sempervirens in Maryland, US. The isolate was grown on potato dextrose agar (BD Difco™, Sparks, MD, USA) for 5-days under 12-h white light photoperiod and then transferred to yeast extract potato dextrose liquid media at 25 °C for 2-days under continuous light. Genomic DNA was extracted from hyphal tissue harvested from liquid media using the OmniPrep DNA kit (G-Biosciences, St. Louis, MO, USA) according to manufacturer’s instructions, and subsequently purified using the Zymo Genomic DNA Clean and Concentrator kit (Zymo Research, Irvine, CA, USA).
Whole genome sequencing and de novo assembly
A genomic DNA library was constructed using the TruSeq Nano DNA Library Prep kit (Illumina, Inc., San Diego, CA, USA) and quantified using the Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY) and the LabChipXT DNA 750 (Caliper Life Sciences, Hopkinton, MA, USA). The library was sequenced on an Illumina MiSeq in two independent runs using paired-end 600-cycle reagent cartridge v.3 (Illumina, Inc.). Reads were processed and assembled using CLC Genomics Workbench v.7.5.1 (CLC Bio, Boston, MA, USA) with a k-mer of 24 and a minimum contig length of 500 bp. Illumina adapters were trimmed and low quality reads (Phred score < 0.05) were removed. Summary statistics for the draft genome were generated using CLC Genomics Workbench and QUAST (Gurevich et al., 2013).
Completeness of the P. foliicola draft genome assembly was evaluated using BUSCO v.1.1b1 (Simão et al., 2015). The genome assembly of P. foliicola used in this study was deposited in NCBI GenBank under accession LMTV00000000, and datasets are also available at the US National Agricultural Library on AgData Commons (http://dx.doi.org/10.15482/USDA.ADC/1408094).
Nuclear genome annotation
Ab initio gene predictions for the draft genome assembly of P. foliicola were performed using the MAKER2 v.2.31.6 annotation pipeline (Holt & Yandell, 2011). Gene training was performed after running three rounds of the program according to the program documentation. Gene boundaries were assigned using protein homology evidence from Fusarium graminearum strain PH-1 (NCBI BioProject Acc: PRJNA13839; Cuomo et al., 2007). Additional ab initio gene predictions were made using the program SNAP (http://korflab.ucdavis.edu/software.html) and AUGUSTUS v.3.2.1 (Stanke et al., 2004) with F. graminearum set as the prediction species model organism. New gene predictions using MAKER2 were also performed for the previously published genome assembly of D. macrodidyma (Malapi-Wight et al., 2015) using the same parameters as above.
Identification of transposable elements and repeat-induced mutations
The presence of transposable elements (TEs) was evaluated from the P. foliicola and D. macrodidyma genomes using the REPET v2.5 (Flutre et al., 2011) pipeline, along with supporting databases Repbase v.20.05 (Kapitonov & Jurka, 2008; Bao, Kojima & Kohany, 2015) and Pfam v.27.0, and run according to the accompanying program documentation (https://urgi.versailles.inra.fr/Tools/REPET). The TEdenovo stage was used first to produce a database of four de novo identified TEs: an incomplete helitron, a MITE (miniature inverted-repeat TE), a TRIM (terminal-repeat retrotransposon in miniature), and one uncategorized TE sequence. These sequences were then used as initial input for the first of two runs of the TEannot pipeline stage, which produced a final genome-wide GFF annotation file using the TE classification scheme described by Wicker et al. (2007). A custom script was used to tabulate counts of TE classes, orders and superfamilies and filtered out fragments less than 80-bp in length according to Wicker et al. (2007) recommendations to avoid misclassification. Results from TEannot based on tblastx and blastx (BLAST+ vr. 2.2.31; Camacho et al., 2009) searches against Repbase were also filtered to remove hits sharing <70% sequence identity.
Individual TE families with ten or more sequences identified, including at least one sequence ≥ 300-bp in length, were checked for signatures of repeat-induced point (RIP) mutation activity using the RIPCAL vr. 2 program (Hane & Oliver, 2008; Hane, 2015). The 300-bp length cutoff was selected based on RIPCAL’s default setting for scanning subsequences and the program was run using both the alignment mode consensus model with ClustalW (Larkin et al., 2007) and di-nucleotide frequency-based methods. Evidence of the signature of RIP mutations was present if di-nucleotide frequencies matched the indexes: (TpA / ApT) ≥ 0.89 and (CpA + TpG) / (ApC + GpT) ≤ 1.03 (Margolin et al., 1998) and visual inspection of RIPCAL alignments showed one or more peaks for (CA ←→TA) + (TG ← →TA) mutations (Hane & Oliver, 2008).
Mitochondrial genome
The P. foliicola and D. macrodidyma mitogenomes were identified by performing tBLASTx searches of the complete genome assemblies using the 95.7 kb F. graminearum mitochondrial genome as a query (Al-Reedy et al., 2012), with the genetic code set to four (mold mitochondrial). Mitogenomes were annotated using MITOS (Bernt, Donath & Jühling, 2013). Comparative analysis of genes, rRNA and tRNA was performed in the program SimpleSynteny (Veltri, Malapi-Wight & Crouch, 2016) using the P. foliicola CDS file to annotate all genomes (e-value cutoff 1e-5, minimum query cutoff 10%), with circular genome mode implementation and genomes organized to minimize Euclidean line distance.
Identification of mating type idiomorphs
We assessed the presence of the MAT1-1 and MAT1-2 idiomorphs in the P. foliicola genome by performing a local BLASTn search (e-value cutoff 1e-5) against a MAT gene database. The database contained nucleotide sequences for the highly conserved alpha domain DNA binding motif (MAT1-1-1) and the high mobility group (HMG) box DNA binding motif (MAT1-2-1) for 13 different filamentous fungal species, retrieved from the NCBI GenBank database (Files S1 and S2).
Phylogenetic reconstruction
A phylogenetic analysis was used to illustrate the relationship between P. foliicola and 14 other fungal species. For this analysis, the predicted proteomes of Aspergillus nidulans FGSC A4 (ASM114v1; Galagan et al., 2005), Botrytis cinerea BcDW1 (Assembly GCA000349525; Blanco-Ulate et al., 2013), F. graminearum PH-1 (GCA000240135; Cuomo et al., 2007), Macrophomina phaseolina MS6 (GCA000302655; Islam et al., 2012), Magnaporthe oryzae 70-15 (MG8; Kim et al., 2010), Neurospora crassa (GCA000786625; Baker et al., 2015), Penicillium oxalicum 114-2 (GCA000346795; Liu et al., 2013), Pyrenophora tritici-repentis (GCA000149985; Manning et al., 2013), Sclerotinia sclerotiorum 1980 UF-70 (ASM1469v1; Amselem et al., 2011), Trichoderma reesei RUT C-30 (GCA000513815; Koike et al., 2013), Ustilago maydis 521 (UM1; Kämper et al., 2006), Verticillium dahliae JR2 (GCA000400815; De Jonge et al., 2012) and Yarrowia lipolytica CLIB122 (GCA000002525; Dujon et al., 2004) were downloaded from either the EnsemblFungi database (https://fungi.ensembl.org/index.html) or the NCBI Genbank Genome database (https://www.ncbi.nlm.nih.gov/genbank/). The predicted proteome of D. macrodidyma generated in our study was also included in the analysis. All proteomes were searched against each other using BLASTp (e-value 0.001) and clustered in orthologous gene sets using OrthoMCL v1.4 in the CYVERSE Discovery Environment (https://de.cyverse.org/de/). Single copy genes (clusters with exactly one member per species) found in all 15 fungal proteomes were extracted from the orthologous dataset and the amino acid alignments were performed using MUSCLE v3.8.31 (Edgar, 2004). Gblocks v.0.91b was used to remove ambiguously aligned regions using relaxed selection parameters following Talavera & Castresana (2007). Maximum likelihood (ML) phylogenetic analyses were performed in RAxML (Stamatakis, 2006), using the RAxML GUI v. 1.5b1 (Silvestro & Michalak, 2012). Phylogenetic trees were constructed using the JTT matrix-based model (Jones, Taylor & Thornton, 1992) and 1,000 bootstrap replicates. Ustilago maydis 521 was used as outgroup in the phylogenetic analyses.
Estimation of evolutionary divergence times
To obtain approximate information on divergence events, the phylogenetic tree was timed using RelTime (Tamura et al., 2012) as implemented in MEGA 7 (Kumar, Stecher & Tamura, 2016). RelTime estimated the relative divergence times for each node of the ML tree using the same outgroup taxa. We used seven confidence intervals obtained from the TimeTree database (http://timetree.org; Hedges et al., 2015) as minimum and maximum times to convert the relative times into absolute times (Table S4). Time estimates were performed using maximum-likelihood branch length, local clocks, a JTT matrix-based model (Jones, Taylor & Thornton, 1992) and a discrete Gamma distribution among sites (five categories).
Comparative genomic analyses
The genome sequences of D. macrodidyma JAC15-245 (NCBI GenBank accession JYGD00000000) and F. graminearum PH-1 were downloaded from NCBI GenBank and Ensembl-Fungi database respectively, and used for comparative analysis against the P. foliicola draft genome assembly generated in this study. The predicted proteome generated from this study was used for D. macrodidyma, while the published proteome dataset for F. graminearum was downloaded from the Ensembl-Fungi database. Genome-wide identification, comparison and visualization of orthologous gene clusters among P. foliicola, D. macrodidyma and F. graminearum were performed using the web platform OrthoVenn (http://www.bioinfogenome.net/OrthoVenn/; Wang et al., 2015). The program uses a modified OrthoMCL algorithm to identify orthologous gene clusters from the UniProt/Swiss-Prot database. Proteins potentially involved in carbohydrate metabolism were annotated for each proteome by searching against the database of automated Carbohydrate-active enzyme ANnotation (dbCAN; http://csbl.bmb.uga.edu/dbCAN/annotate.php; Yin et al., 2012). A X2 test of independence, similar to that used in Martinez et al. (2008), was used to identify statistically significant differences across the CAZyme repertoire. Identification of putative enzymes related to the biosynthesis of secondary metabolites was performed from the predicted proteins using the web-based program AntiSMASH (http://antismash.secondarymetabolites.org; Medema et al., 2011; Weber et al., 2015). The secretome was predicted by screening the predicted proteins for different features using a bundle of eight different prediction tools implemented in the web-based program SECRETOOL (Cortázar et al., 2014). Pathogenicity associated genes were identified by performing a local BLASTp search of the predicted proteome against the curated pathogen-host interaction database (PHI-base v.4.0; http://www-phi4.phibase.org/; Winnenburg et al., 2008).
Results
Nuclear genome assembly and annotation
A de novo genome assembly of P. foliicola ATCC13545® was generated from 19.5 million paired end, 300-bp reads, comprising a total of 5.4 Gb of raw sequence data. The resulting genome assembly for P. foliicola was 28.7 Mb, organized in 425 scaffolds (≥500-bp), with an average read depth coverage of 188-fold (Table 1). The N50 scaffold length is 184,987 bp and the three longest scaffolds are 619,696 bp, 551,775 bp, and 524,658 bp. Analysis of the P. foliicola genome assembly using BUSCO identified 420 complete genes out of 429 conserved eukaryotic ortholog dataset and 1,416 complete genes out of 1,438 conserved fungal ortholog dataset (genome completeness scores of 97% and 98%, respectively). Ab initio gene predictions performed with the MAKER2 pipeline for the P. foliicola genome assembly identified 9,272 protein-coding genes with an average predicted protein length of 484 amino acids (Table 1). Meanwhile, gene predictions for the genome of D. macrodidyma identified 16,959 protein-coding genes with an average length of 478 amino acids. Of the three genomes evaluated in this study, gene number predictions were highest for D. macrodidyma, which also had the largest estimated genome size at 58 Mb when compared to P. foliicola and F. graminearum (13,313 genes, 36.1 Mb; Cuomo et al., 2007).
| Genome features | Pseudonectria foliicola | Fusarium graminearum | Dactylonectria macrodidyma |
|---|---|---|---|
| Genome size (Mb) | 28.7 | 38.1 | 58.0 |
| Average sequence coverage | 188 | 10 | 46 |
| Total number of scaffolds | 425 | 5 | 850 |
| GC content (%) | 54.3 | 48.2 | 49.9 |
| Transposable elements (%) | 0.7 | 0.06 | 6.5 |
| Predicted proteins | 9,272 | 14,164 | 16,454 |
| NCBI accession | LMTV00000000 | AACM00000000 | JYGD00000000 |
Transposable elements, repetitive DNA and repeat induced mutations
Only 0.7% (196,205-bp) of the P. foliicola nuclear genome assembly contained TEs based on the REPET pipeline analysis (Table 1). Annotation of TEs across the P. foliicola genome assembly identified 191 TE matches. Of these, 141 were Class I (retrotransposons) TEs comprising ∼0.4% of genome and 45 were Class II (DNA) TEs making up ∼0.3% of the genome. Long terminal-repeats (LTRs) were found to be the most abundant Class I order (∼0.3% of genome) and included matches in the Copia, Gypsy and BEL/Pao retrotransposon superfamilies. Almost all Class II TEs were identified as Helitrons, with only ∼0.001% of the genome identified with TIR or “unknown” TEs (Wicker code: DXX). Evidence of RIP mutation affecting P. foliicola TEs was only found for Helitrons based on alignment of 37 sequences (3.7-kb in length) and di-nucleotide indexes: TpA/ApT = 1.8 and (CpA + TpG)/(ApC + GpT) = 0.3.
The draft genome of D. macrodidyma genome as originally published did not include information about TEs. Here, we also analyzed that assembly for the presence of TEs and RIP mutations using the same parameters used for P. foliicola for comparative purposes. As REPET identified over 2,000 D. macrodidyma TEs of “unknown” superfamily, we performed an additional tblastx search (e-value: 0.01, percent query coverage per hsp: ≥70%, minimum percent identity of hits: ≥70%) against Repbase and reclassified five TEs based on the top match if it shared the same class and order. REPET analysis identified ∼6.5% (3,794,887-bp) of the D. macrodidyma genome to be made up of TEs (2,178 elements). Of these, 1,489 elements were Class I TEs and comprised ∼5.3% of the genome and 689 were Class II TEs and comprised ∼1.2% of the genome. The most common Class I retrotransposon orders were: LTRs (∼3.2%), “Unknown RXX” (∼1.1%), DIRS-like (∼0.9%), LINE (∼0.1%) and SINE (0.001%). For Class II DNA elements, the most common orders were: TIR (∼0.7%), Helitron (∼0.4%) and “Unknown DXX” (∼0.2%). Evidence for RIP mutation affecting D. macrodidyma TEs using alignment and di-nucleotide counts was identified in the sets of 91 Helitron and 146 DIRS TEs. For the Helitrons, RIP indexes were calculated as: TpA/ApT = 1.3 and (CpA + TpG)/(ApC + GpT) = 0.9, while for DIRS: TpA/ApT = 1.4 and (CpA + TpG)/(ApC + GpT) = 0.3.
Mitochondrial genome size and gene content
The mitogenomes of P. foliicola and D. macrodidyma were each contained within a single scaffold, measuring 58.3 kb and 44.2 kb respectively (scaffold 56, scaffold 68). Although the mitogenome-containing scaffolds for these two organisms were considerably smaller than that of F. graminearum (95.7 kb), a full complement of protein-coding genes was contained: apocytochrome b (cob), ATP-synthase subunits (atp6, atp8, atp9), cytochrome oxidase subunits (cox1, cox2, cox3), and NADH subunits (nad1, nad2, nad3, nad4, nad4L, nad5, nad6). This set of 14 protein-coding mitochondrial genes is highly conserved among fungi, and shared with animal mtDNA (Bullerwell & Lang, 2005). A full complement of tRNAs necessary for translation was encoded in the mitogenomes (25 total), as were small and large subunit rRNAs (one and three copies, respectively). Neither P. foliicola nor D. macrodidyma mitogenomes contained copies of the four large unidentified open-reading frames that are encoded by the F. graminearum mitogenome (Al-Reedy et al., 2012), accounting for the observed difference in sizes between these species.
Mitochondrial genome synteny
The organization of the mitogenomes of P. foliicola, D. macrodidyma and F. graminearum was highly conserved at the gene level. As observed in the mitochondria of most ascomycete fungi, all genes, rRNAs and tRNAs were oriented in the same direction. The three mitogenomes contained a nearly identical ordering of shared genes (Fig. 2). Only a single tRNA (tRNA-G) was positioned differently in the P. foliicola and D. macrodidyma mitogenomes relative to F. graminearum. Two tRNAs (tRNA-R and tRNA-L) were positioned differently between D. macrodidyma and P. foliicola/F. graminearum. This conserved ordering of the three mitogenomes was consistent with previous observations that the mitochondrial DNA of fungi in the order Hypocreales is highly conserved (Al-Reedy et al., 2012; Pantou, Kouvelis & Typas, 2008).

Figure 2: Comparisons of the mitogenomes of Pseudonectria foliicola (Pf), Dactylonectria macrodidyma (Dm) and Fusarium graminearum (Fg).
Shared areas contain intergenic sequence that has been collapsed to improve diagram readability; the starting and ending coordinates are plotted at the beginning and end of these regions.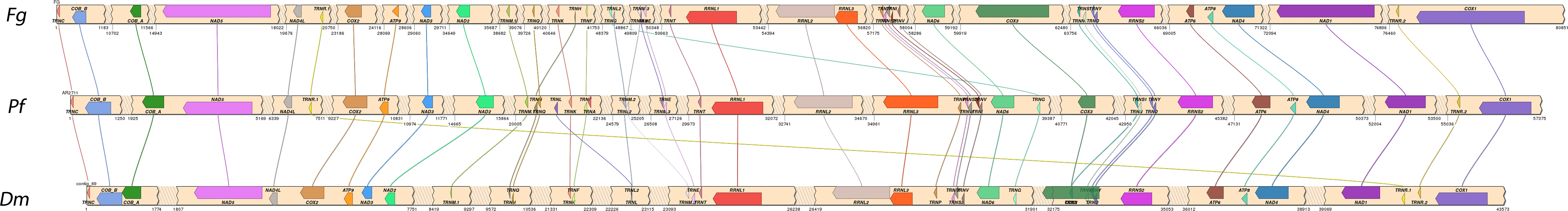{kind=link}
Identification of mating type idiomorphs in P. foliicola
The presence of the conserved alpha domain indicative of the MAT1-1 mating type idiomorph was identified in P. foliicola scaffold 102. In the same scaffold, the APN2 (encoding DNA lyase) and SLA2 (encoding cytoskeletal protein) genes were found flanking the mating type idiomorph, as well as the MAT-locus associated gene COX13 (cytochrome c oxidase subunit VIa homolog). The HMG-box region indicative of the MAT1-2 mating type idiomorph was not found within the P. foliicola genome. Based on these data, P. foliicola appears to be a heterothallic fungus, requiring a partner of the alternate mating type in order to initiate the sexual life cycle.
Phylogeny and divergence estimation
The phylogenetic relationships and divergence times among the fungal genomes studied is displayed in Fig. 3. Fourteen publicly available fungal genomes were used to examine the phylogenetic placement of P. foliicola through the analysis of single copy orthologous genes. The program OrthoMCL identified 16,356 gene clusters, from which 1,884 orthologous genes were shared across all 15 fungal species. From these shared gene clusters, 1,511 orthologous genes present as single copies were used for the phylogenetic analysis. The final dataset after removal of ambiguously aligned regions consisted of 388.7 Mb. The ML analysis identified, with high bootstrap support (>70%), five major clusters representing the ascomycete classes Sordariomycetes, Leotiomycetes, Eurotiomycetes, Dothideomycetes, and Saccharomycetes (Fig. 3). Within the Sordariomycetes, P. foliicola was more closely related, albeit appearing basal, to D. macrodidyma and F. graminearum, all of which belonged to the Nectriaceae in the order Hypocreales. These phylogenetic relationships were consistent with previous reports (Lombard et al., 2015; Yin et al., 2015). Using RelTime methods and a JTT matrix-based model, the estimated log likelihood value was −142633.0660. The divergence of P. foliicola from the Nectriaceae species D. macrodidyma and F. graminearum was estimated to have occurred ∼132 Mya. The overall estimates of divergence times in the tree are in agreement with divergence times reported for the order Hypocreales (Sung, Poinar & Spatafora, 2008).
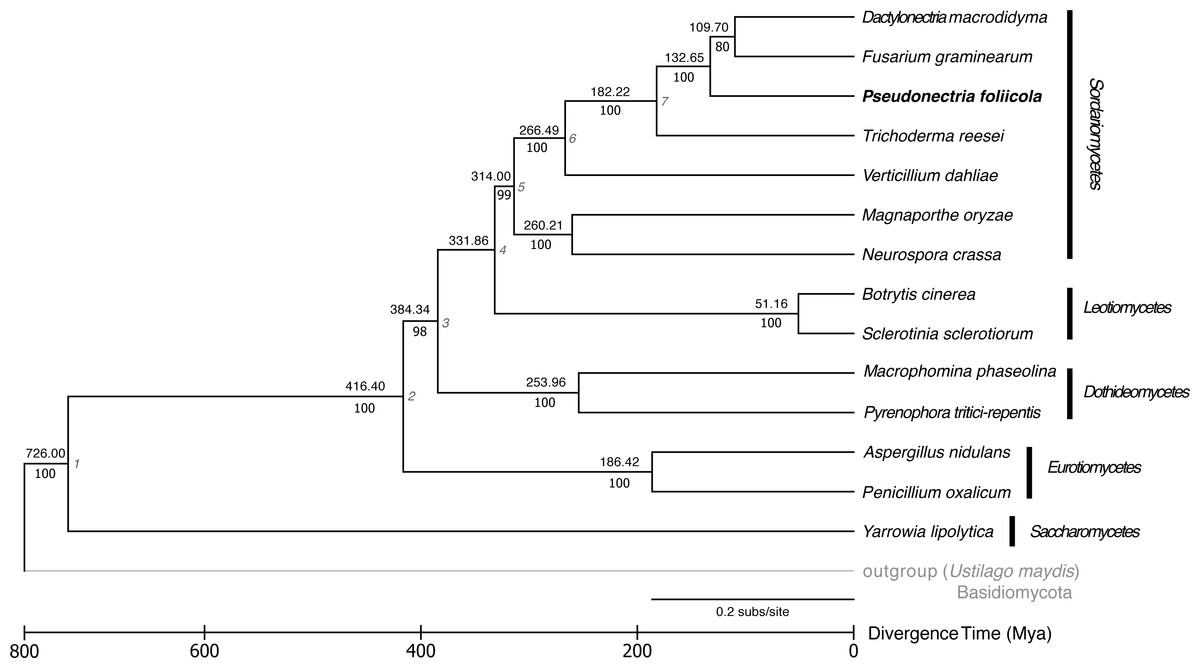
Figure 3: Reconstruction of the phylogenetic relationships and divergence times of Pseudonectria foliicola relative to other fungal species.
The maximum likelihood (ML) tree analysis and time tree (RelTime method) were conducted on the concatenated dataset of 1,511 single copy orthologous genes. Numbers above branches indicate the approximate relative times of divergence (Mya) between two lineages. The seven calibration points used are indicated at the nodes and listed in Table S4. Scale representation under the tree demonstrate divergence times of genes. Statistical support values corresponding to ML are indicated below the branches. The basidiomycete Ustilago maydis was used as the outgroup.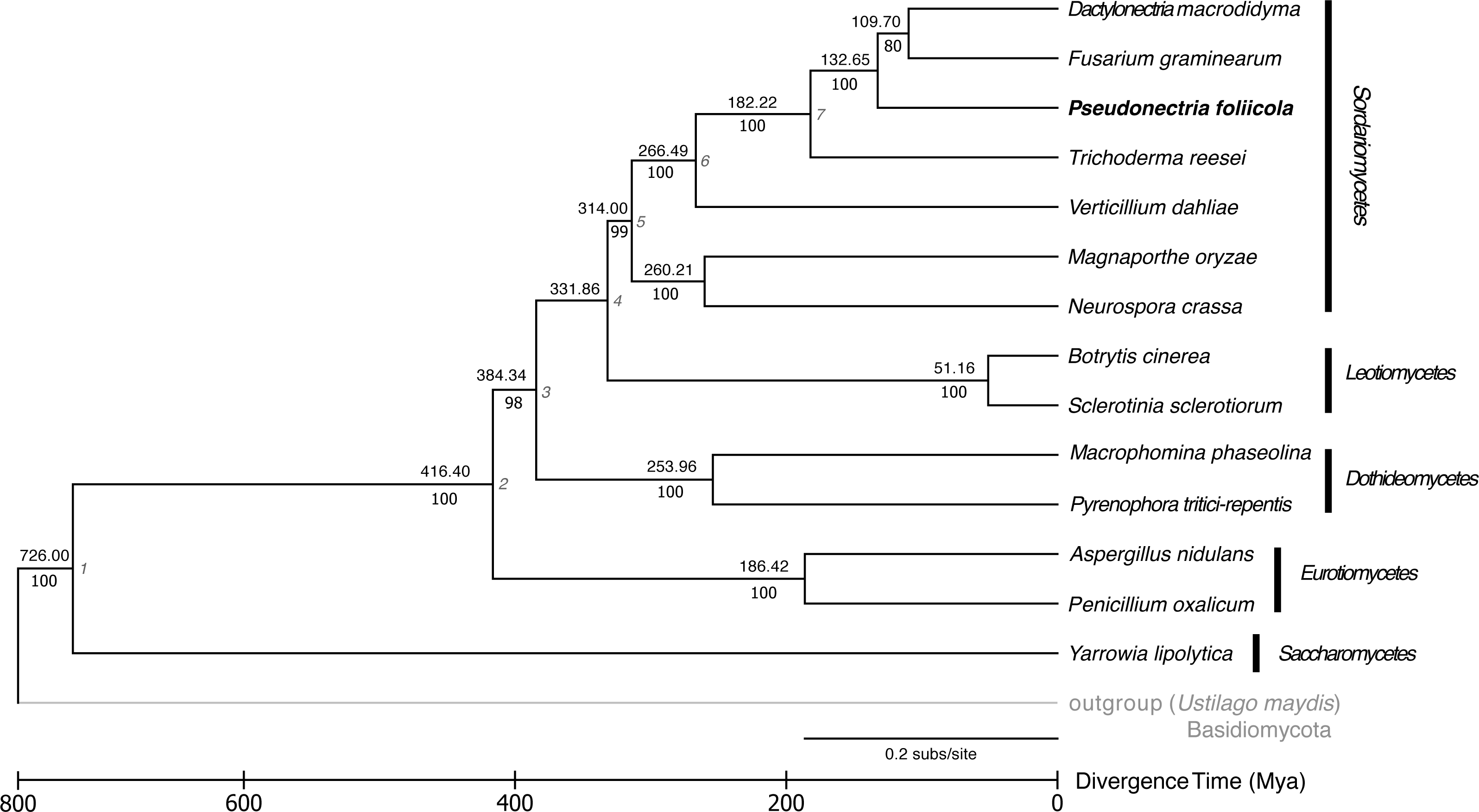{kind=link}
Comparative genomic analysis of P. foliicola and other fungi in the Nectriaceae
Gene orthology
Orthologous gene clusters were identified for P. foliicola, D. macrodidyma and F. graminearum using OrthoVenn (Fig. 4). Proteins from these three fungal species formed 10,403 orthologous clusters, of which 7,135 clusters were shared among all three species. The top three Swiss-Prot annotations among the core shared clusters were the Acyl-CoA-binding domain-containing protein (44 proteins), pleiotropic drug resistance protein 4 (30 proteins) and a short-chain dehydrogenase TIC 32, chloroplastic (12 proteins). Sixteen clusters representing 48 predicted proteins were unique to P. foliicola, while 610 and 124 species-specific protein clusters were identified in D. macrodidyma and F. graminearum, respectively. Only 0.2% of the P. foliicola proteome was unique, a low percentage relative to 1.3% for the F. graminearum and 6.0% for the D. macrodydima proteomes. Although the majority of the 16 clusters unique to P. foliicola had no annotations based on the UniProt/Swiss-Prot and Gene Ontology (GO) databases, three of the unique clusters were identified as (1) vegetative cell wall protein Gp1/structural constituent of cell wall (GO:0005199), (2) thioredoxin/protein disulfide oxidoreductase activity (GO:0009507), and (3) leucine-rich repeat extensin-like protein 3/structural constituent of cell wall (GO:0005618). Biological processes and molecular functions annotated by the GO database for the species-unique gene clusters were most abundant in D. macrodidyma (66 biological processes, 28 molecular functions), and least abundant in P. foliicola (five biological processes, two molecular functions; Table S1). Three GO categories were found enriched (hypergeometric test on OrthoVenn, p-value <0.05) in the P. foliicola unique clusters: (1) a glycerol ether metabolic process, (2) structural constituent of cell wall and, (3) a protein disulfide oxidoreductase activity. Despite the large number of unique gene clusters found in D. macrodidyma, only one GO category, an oxidoreductase activity acting on single donors with incorporation of molecular oxygen, was found to be enriched.
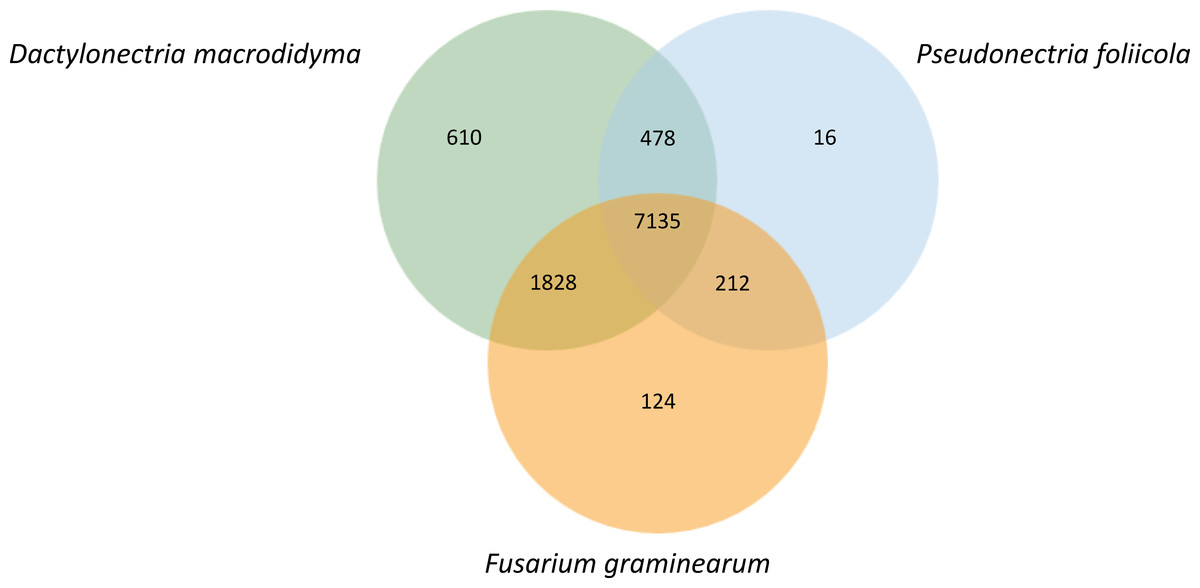
Figure 4: Orthologous genes shared between Pseudonectria foliicola, Dactylonectria macrodidyma and Fusarium graminearum.
{kind=link}
The CAZyme repertoire
Annotation of the P. foliicola proteome identified 448 CAZyme modules, including domains encoding 179 glycoside hydrolases (GH), 88 glycosyl-transferases (GT), 18 polysaccharide lyases (PL), 64 carbohydrate esterases (CE), 46 carbohydrate-binding modules (CBM), and 53 enzymes with auxiliary activities (AA) (summarized in Table 2, Fig. 5 and Table S2). The D. macrodidyma and F. graminearum genomes encoded 1,086 and 707 CAZyme modules, respectively. The CAZyme encoding genes represented between 4.8 and 6.6% of the predicted proteome for the three fungal species (Table 2).
| Name | Pseudonectria foliicola | RF% | Dactylonectria macrodidyma | RF% | Fusarium graminearum | RF% |
|---|---|---|---|---|---|---|
| AA | 53 | 0.52 | 147 | 0.84 | 110 | 0.79 |
| CBM | 46 | 0.53 | 116 | 0.73 | 80 | 0.65 |
| CE | 64 | 0.83 | 210 | 1.37 | 127 | 1.08 |
| GH | 179 | 1.94 | 447 | 2.59 | 268 | 2.01 |
| GT | 88 | 1.01 | 125 | 0.78 | 100 | 0.82 |
| PL | 18 | 0.17 | 41 | 0.20 | 22 | 0.16 |
| Total | 448 | 4.83% | 1,086 | 6.40% | 707 | 5.31% |
Notes:
- AA
-
Auxiliary activity families
- CBM
-
Carbohydrate-binding modules
- CE
-
Carbohydrate esterase families
- GH
-
Glycoside hydrolase families
- GT
-
Glycosyltransferase families
- PL
-
Polysaccharide lyase families
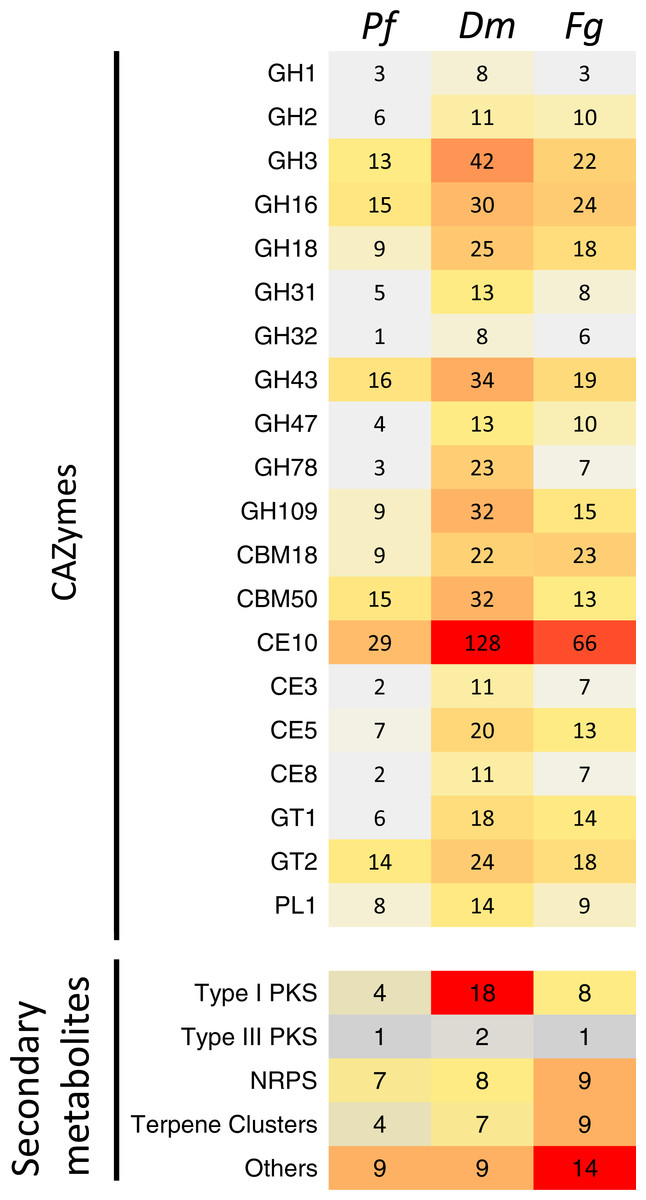
Figure 5: Comparison of the predicted CAZymes and secondary metabolite clusters identified from the genome assemblies of Pseudonectria foliicola (Pf), Dactylonectria macrodidyma(Dm) and Fusarium graminearum (Fg).
GH, glycoside hydrolases; CBM, Carbohydrate-binding modules; CE, Carbohydrate esterases; GT, Glycosyl transferases; PL, Polysaccharide lyases; PKS, Polyketides; NRPS, Non-ribosomal peptide synthase.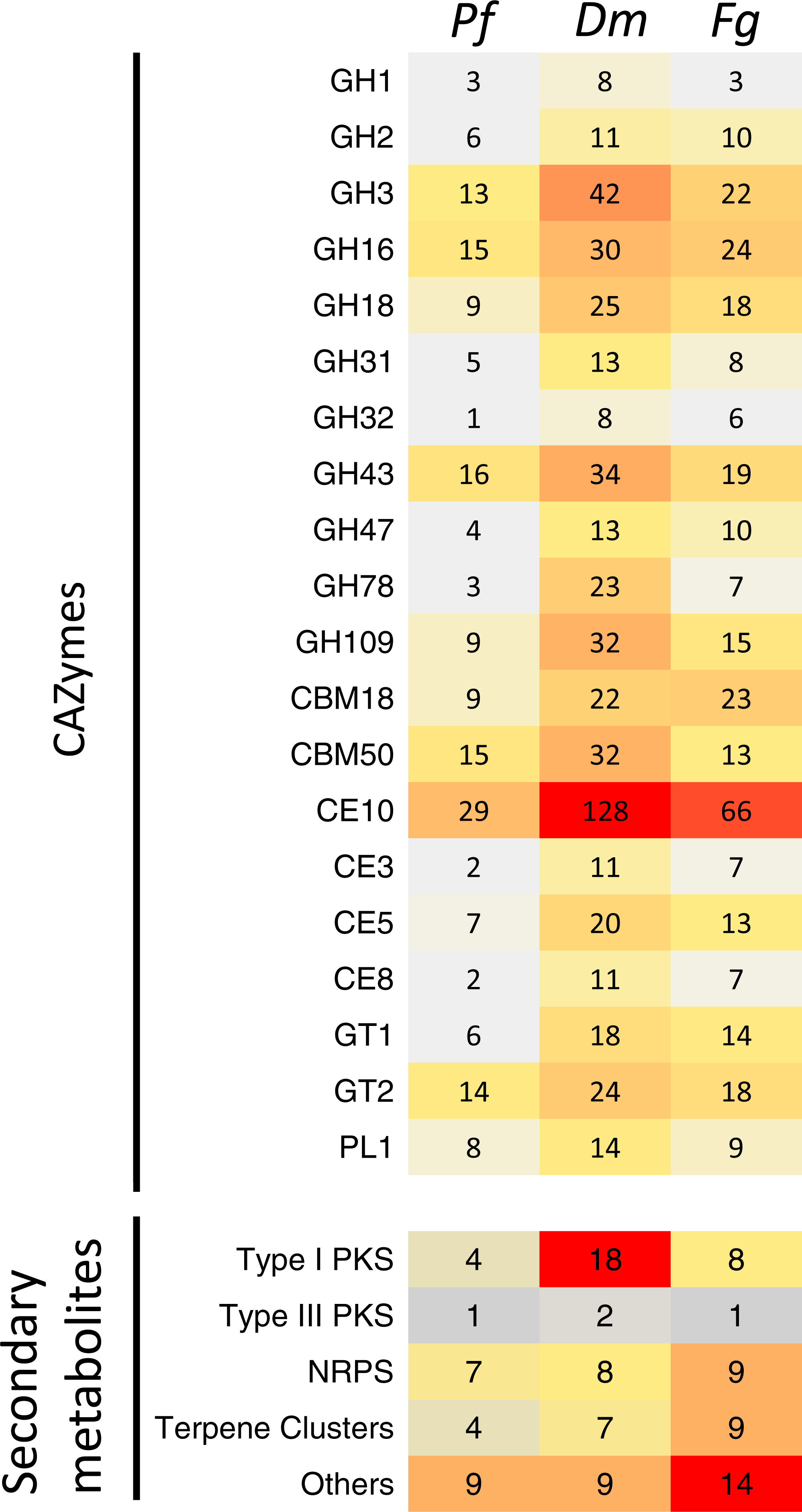{kind=link}
An increased number of CAZyme modules was identified from the D. macrodidyma proteome when compared to P. foliicola, F. graminearum and a database of 91 other plant pathogenic, facultative pathogenic, saprophytic and symbiotic fungi (Table S2; Zhao et al., 2013). Although not significant in X2 tests against P. foliicola and F. graminearum, D. macrodidyma has an increased number of GH and CE enzymes. This is largely due to the GH3 family, which is associated with cellulose degrading activities, the GH28 family, with pectinase activities, as well as the less characterized GH78 and GH109 families. Within the CE and PL module groups, increased numbers in the D. macrodidyma genome were observed in the families CE3, CE5, CE10 and PL1 (Fig. 5). The CE3 and CE5 have acetyl xylan esterase and cutinase activities and are common modules with a higher representation in Ascomycetes relative to Basidiomycetes (Zhao et al., 2013). CE10 families have carboxylesterases activities but can also act on non-carbohydrate substrates (Cantarel et al., 2009). The PL1 family is the most commonly found among fungi, particularly in plant pathogenic species (Zhao et al., 2013). The number of CE families in D. macrodidyma (233) is comparable to that of the pea root pathogen Fusarium solani, previously regarded as having the most CEs with 223 (Zhao et al., 2013).
Secondary metabolite clusters
Annotation of the P. foliicola, F. graminearum and D. macrodidyma proteomes identified key enzyme clusters for the biosynthesis of secondary metabolites such as non-ribosomal peptide synthases (NRPS), polyketide synthases (PKS), terpene synthases (TS), among others (summarized in Fig. 5). The genome of P. foliicola contained 25 secondary metabolite clusters, in contrast with F. graminearum and D. macrodidyma with a total of 41 and 44 clusters, respectively (Fig. 5).
Secretome
The predicted secretome for P. foliicola was also relatively small, comprising just 346 proteins. In comparison, the genomes of D. macrodidyma and F. graminearum contained 607 and 457 predicted secreted proteins (Table S3 ). However, for all three species, the secretome made up between 3.4 to 3.7% of the predicted proteome.
Virulence associated genes
The genomes of P. foliicola, D. macrodidyma and F. graminearum were screened against PHI-base, a curated database that contains pathogenicity, virulence and effector genes from fungi, oomycete and bacterial pathogens (Winnenburg et al., 2008). Relative to the total proteome, the frequency of virulence-associated genes was highest in P. foliicola (14.5%) compared to F. graminearum (12.2%) and D. macrodidyma (9.5%) (Table 3). Genes associated with the loss of pathogenicity, reduced virulence and those with mixed outcomes were identified in higher frequencies in the P. foliicola proteome than in the D. macrodidyma and F. graminearum proteomes (Table 3). Genes associated with loss of pathogenicity and with reduced virulence have been identified by PHI-base from transgenic strains of fungal, oomycete and bacterial pathogens that either fail to cause disease or that cause quantitatively lower degrees of disease than the wild-type strains (Winnenburg et al., 2008; Urban et al., 2015).
| Name | Pseudonectria foliicola | RF% | Dactylonectria macrodidyma | RF% | Fusarium graminearum | RF% |
|---|---|---|---|---|---|---|
| Chemistry target | 7 | 0.08 | 8 | 0.05 | 9 | 0.07 |
| Effector | 15 | 0.16 | 22 | 0.13 | 19 | 0.14 |
| Enhanced antagonism | 2 | 0.02 | 2 | 0.01 | 2 | 0.02 |
| Increased virulence | 7 | 0.08 | 7 | 0.04 | 7 | 0.05 |
| Increased virulence (Hypervirulence) | 13 | 0.14 | 20 | 0.12 | 18 | 0.14 |
| Lethal | 65 | 0.70 | 80 | 0.47 | 87 | 0.65 |
| Loss of pathogenicity | 103 | 1.11 | 116 | 0.68 | 99 | 0.74 |
| Mixed outcome | 112 | 1.21 | 124 | 0.73 | 114 | 0.86 |
| Reduced virulence | 459 | 4.95 | 510 | 3.01 | 490 | 3.68 |
| Unaffected pathogenicity | 558 | 6.02 | 712 | 4.2 | 782 | 5.87 |
| Other | 2 | 0.02 | 2 | 0.01 | 2 | 0.02 |
| Total | 1,343 | 14.5% | 1,603 | 9.45% | 1,629 | 12.24% |
Discussion
Pseudonectria species, P. foliicola and P. buxi, are economically important fungal pathogens responsible for increased costs in foliar disease management of boxwood plants worldwide. Here, we present a draft genome sequence for P. foliicola, including a comparative analysis of this genome against two other plant pathogens in the Nectriaceae. The 28.7 Mb P. foliicola draft genome assembly is smaller than the reported size of other fungi in the Ascomycota (average genome size 36.9 Mb; Mohanta & Bae, 2015). This assembly represents the smallest genome known from the Nectriaceae, in which genomes range from 36.1 to 58.1 Mb: D. macrodidyma (58.0 Mb; Malapi-Wight et al., 2015), Neonectria ditissima (44.9 Mb; Gomez-Cortecero, Harrison & Armitage, 2015) and F. graminearum (36.1 Mb; Cuomo et al., 2007). Consistent with genome size, the number of predicted gene models from the P. foliicola assembly is reduced but comparable to the number of gene models predicted in other Ascomycota fungi with similar genome size (e.g., Patellaria atrata, 28.7 Mb, 7,794 gene models (JGI); Mohanta & Bae, 2015). Pseudonectria foliicola also contains one of the smallest cohorts of TEs reported for filamentous fungi, similar to the genomes of Trichoderma atroviridae, T. reesei, and T. virens (ranging from 0.48 to 0.57%; Kubicek et al., 2011; Martinez et al., 2008) as well as F. graminearum (<1% of repetitive DNA; Cuomo et al., 2007). A strong correlation between genome size and repeat content was reported in a study of 18 Dothidiomycete genomes (Ohm et al., 2012) however, based on the low percentage of TEs found in F. graminearum and D. macrodidyma, this correlation may not be sustained within the Nectriaceae.
The uniquely small genome size of P. foliicola is similar to the genome assembly recently reported from Escovopsis weberi (29.5 Mb), a highly specialized mycoparasitic fungus that also belongs to the order Hypocreales (De Man et al., 2016). Specialized pathogens or those with a narrow host range are predicted to maintain only the essential cohort of genes, and lose those no longer needed in their particular niche (e.g., De Man et al., 2016; Lee & Marx, 2012) and while there are exceptions (e.g., Spanu et al., 2010), the comparative genome analysis of the three Nectriaceae genomes in this study follows that prediction. From the three fungal species analyzed in this study, only P. foliicola has a narrow host range (only known from Buxus spp.) while F. graminearum (reported frequently in a large number of hosts in Poaceae) and D. macrodidyma (reported on grapevine, avocado and olive trees) have increasingly broader host ranges and larger genomes and proteomes. Divergence time estimates indicated that P. foliicola, D. macrodidyma and F. graminearum diverged from their common ancestral organism ca. 132 Mya. This relatively distant split may account for the differences observed in pathogenicity and host range between these species, and indicate that ongoing gene loss resulting in a reduced genome size is a major contributor to the genome evolution of P. foliicola.
Enzymes that degrade plant cell wall carbohydrates can be essential during the infection and decomposition of host plant tissue, particularly for necrotrophic and hemibiotrophic fungi (Gibson et al., 2011; Zhao et al., 2013). Consequently, CAZyme profiles can be used as indicators of the fungal lifestyle. In previous studies, necrotrophic and hemibiotrophic fungal plant pathogens have been reported to produce a large repertoire of these enzymes (Gibson et al., 2011; Knogge, 1996), and typically exhibit expanded arsenals of CAZymes in their genomes, relative to biotrophic and obligate fungi that typically exhibit the lowest numbers (Zhao et al., 2013). Similarly, the fungal secretome and secondary metabolites, both involved in the host-pathogen interaction process (Van den Burg et al., 2006; Yu & Keller, 2005), can correlate with the lifestyle of a fungal pathogen (Lowe & Howlett, 2012; Ohm et al., 2012). Consistent with its small genome and proteome size, P. foliicola has the smallest cohort of CAZymes, SM clusters and secreted proteins relative to D. macrodidyma and F. graminearum. Despite the reduced number of total CAZyme clusters in P. foliicola, all clusters are well represented and comparable to those found in the genomes of fungi with different lifestyles (comparison among the 91 fungal CAZyme profiles by Zhao et al., 2013). Within the Nectriaceae, comparisons among P. foliicola, D. macrodidyma and F. graminearum show increased numbers of CAZymes in several clusters for D. macrodidyma relative to the other two fungi, although not significant. The glycosyl hydrolases (GH), enzymes with an important role in the complete breakdown of the plant cell wall for successful infection (Cantarel et al., 2009), were the most abundant type of secreted protein and CAZymes found across all three species compared.
The pathogenicity profile of P. foliicola as predicted by comparisons against the PHI-base database shows a higher relative frequency of genes associated with loss of pathogenicity and reduced virulence, when compared to D. macrodidyma and F. graminearum. The genome characteristics of P. foliicola described in our analyses may help explain the apparent inability of this fungus to penetrate host plant tissue and its dependence on wounding or winter damage for successful infections. Shi & Hsiang (2014) reported primary infection by P. buxi on leaves and stems of various Buxus species through wounded tissue resulting in general plant decline. A similar strategy may be employed by the closely related species P. foliicola; however, due to its recent taxonomic placement, it remains uncertain whether previous disease reports and epidemiology studies correspond to either Pseudonectria species.
Conclusions
Despite the economic importance of fungi in the Nectriaceae family, only a small number of genome resources are currently available. A survey of public databases shows that less than 5% of the estimated 900 fungal species in this family have been sequenced on the whole genome scale (NCBI-GenBank, the Joint Genome Institute Mycocosm and Ensembl databanks). To our knowledge, the P. foliicola genome is the smallest known genome in the Nectriaceae. Currently, it is unknown if the genome characteristics of other fungal pathogens in the Nectriaceae are similar to those of P. foliicola, D. macrodidyma or F. graminearum, or if these genomes represent the extremes. With the advent and accessibility of next generation sequencing technologies we expect that more in depth comparative genomics studies will characterize fungal groups of great economic and ecological importance. The quality of microbial draft genomes and consequently the predicted size of the associated proteome can also be influenced by the next generation sequencing platform and assembly software (Mavromatis et al., 2012). Even though improvements on sequencing chemistry and better assembly algorithms have reduced the chance of errors, further sampling of genomes of fungi in the Nectriaceae and other families, ideally with a wide range of life styles, would help determine if the differences observed in annotation rates for some protein classes in our study is due to real biological differences or if they might be an artifact of the technology used to generate these draft genomes. Furthermore, the availability of fungal genomes will aid in the resolution of important fungal lineages and explore beyond the commonly used standard molecular markers for taxonomic classification.
Supplemental Information
Molecular functions, biological processes, and cellular components annotated by the Gene Ontology (GO) database using OrthoVenn (http://www.bioinfogenome.net/OrthoVenn/, on November 22, 2017)
Comparative analysis of fungal CAZymes (from Zhao et al., 2013 and this study)
CBM, carbohydrate binding module; CE, carbohydrate esterase; GH, glycoside hydrolases; GT, glycosyltransferase; PL, polysaccharide lyase.