
Comparative genomics of Synechococcus and proposal of the new genus Parasynechococcus
- Published
- Accepted
- Received
- Academic Editor
- Ugo Bastolla
- Subject Areas
- Bioinformatics, Genomics, Microbiology, Taxonomy
- Keywords
- Synechococcus, Phylogenomics, Genomics, Cyanobacteria, Genomic taxonomy
- Copyright
- © 2016 Coutinho et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Comparative genomics of Synechococcus and proposal of the new genus Parasynechococcus. PeerJ 4:e1522 https://doi.org/10.7717/peerj.1522
Abstract
Synechococcus is among the most important contributors to global primary productivity. The genomes of several strains of this taxon have been previously sequenced in an effort to understand the physiology and ecology of these highly diverse microorganisms. Here we present a comparative study of Synechococcus genomes. For that end, we developed GenTaxo, a program written in Perl to perform genomic taxonomy based on average nucleotide identity, average amino acid identity and dinucleotide signatures, which revealed that the analyzed strains are drastically distinct regarding their genomic content. Phylogenomic reconstruction indicated a division of Synechococcus in two clades (i.e. Synechococcus and the new genus Parasynechococcus), corroborating evidences that this is in fact a polyphyletic group. By clustering protein encoding genes into homologue groups we were able to trace the Pangenome and core genome of both marine and freshwater Synechococcus and determine the genotypic traits that differentiate these lineages.
Introduction
Cyanobacteria are unique among prokaryotes due to their ability to perform oxygenic photosynthesis. Members of this phylum are important contributors of global primary production, since they are responsible for a significant fraction of carbon fixation at aquatic habitats (Partensky, Hess & Vaulot, 1999; Richardson & Jackson, 2007). Among the members of this phylum, the sister genera Synechococcus and Prochlorococcus are often the most abundant members of the picophytoplankton (reaching concentrations up to 105 and 106 cells ml−1 respectively), being considered the most important contributors to CO2 fixation taking place in several oceanic regions (Li et al., 1983; Liu et al., 1998; Partensky, Blanchot & Vaulot, 1999). Synechococcus represents a polyphyletic group that encompasses both freshwater, seawater and brackish water lineages. The use of molecular data revealed that marine and brackish water Synechococcus strains are a sister clade to Prochlorococcus, that is distantly related to freshwater Synechococcus strains. Nevertheless, all these organisms are still classified under the same name (Honda, Yokota & Sugiyama, 1999; Robertson, Tezuka & Watanabe, 2001; Shih et al., 2013). Much of the comparative studies regarding Synechococcus strains were focused on marine lineages, and freshwater strains remained poorly characterized. Despite being sister taxa, Prochlorococcus and marine Synechococcus, differ regarding their ecology and biogeographical distribution patterns. Although these organisms frequently co-occur in aquatic environments, Prochlorococcus tends to be more abundant in warm and oligotrophic waters (Partensky, Blanchot & Vaulot, 1999; Bouman et al., 2006). Marine Synechococcus is considered to be ubiquitously distributed, due to its presence in estuarine, coastal and off-shore waters, broader temperature range and high abundance at mesotrophic and eutrophic habitats (Six et al., 2007a).
Genomic studies encompassing both the marine and the distantly related freshwater strains, further expanded the knowledge on the genetic diversity within Synechococcus. Such studies revealed that these organisms developed unique strategies to adapt to their respective environments, that involve several aspects of their metabolism and physiology, e.g. uptake and utilization of nutrients and metals, regulatory systems and motility (Palenik et al., 2006; Six et al., 2007a; Dufresne et al., 2008a). These strains can also be differentiated with regard to their ecology. Several studies have demonstrated differential patterns of biogeographical and seasonal distribution among Synechococcus strains, that are believed to be driven by environmental conditions such as depth, salinity, temperature and nutrient availability (Tai & Palenik, 2009; Paerl et al., 2011; Post et al., 2011).
One of the factors that may have contributed to the remarkable diversity of both freshwater and marine Synechococcus is horizontal gene transfer (HGT). It has been demonstrated that this process plays a significant role into the evolution of Cyanobacterial genomes (Nakamura et al., 2004; Zhaxybayeva et al., 2006). Furthermore, genomic islands have been identified in several genomes of Cyanobacteria, that are thought to have been acquired during infection by Cyanophages, as evidenced by the presence of integrases flanking these regions (Palenik et al., 2003; Palenik et al., 2009). Genes carried by phages, have the potential to be horizontally transferred and may be associated with several metabolic processes, e.g. photosynthesis, carbon and phosphorus metabolism and stress response (Mann et al., 2003; Lindell et al., 2004; Sullivan et al., 2005; Palenik et al., 2003; Dufresne et al., 2008a).
Comparative genomics has successfully been applied to several groups of organisms, allowing for the identification of new species, reconstruction of phylogenies and definition of genomic traits that are responsible for the metabolic and ecological differences observed between these organisms (Chen et al., 2006; Makarova et al., 2006; Thompson et al., 2009; Thompson et al., 2013; Thompson et al., 2014). Bacterial taxonomy applies a polyphasic approach, i.e. integration of phenotypic, genotypic and phylogenetic data, for the classification of microorganisms, based on traits that range from the molecular to the ecological level (Colwell, 1970; Vandamme et al., 1996). Microbial taxonomy has come to incorporate whole genome information, giving rise the field of genomic taxonomy, that uses the massive amounts of information contained in complete genome sequences, for the classification and differentiation between microbial lineages (Coenye et al., 2005). Nevertheless, the very concept of bacterial species remains elusive, and therefore the classification of bacteria into species, based on genomic or any other type of feature, remains challenging.
A total of 24 complete Synechococcus genomes have been sequenced until the year of 2013. These sequences were obtained from strains isolated from several habitats throughout the globe, each possessing unique genetic, metabolic and ecologic traits. Despite all being named Synechococcus, these genomes represent a polyphyletic group and the genetic similarities, and differences between these strains have not been well characterized in a broad-scale comparative genomic analysis. By identifying groups of homologous genes shared between these genomes, we were able to trace the core-genome and the pan-genome of Synechococcus and Prochlorococcus. Based on these results and on phylogenomic reconstruction, we propose the creation of the genus Parasynechococus, a sister clade to Prochlorococcus.
Materials and Methods
Samples
A total of 24 complete Synechococcus genomes publicly available as of August 2013 were retrieved from Genbank for analysis (Table 1). Only two genomes were classified at the species level: Synechococcus elongatus strain PCC6301 and Synechococcus elongatus strain PCC7942. The genomes were sequenced from isolates obtained from several aquatic environments around the globe, including freshwater, coastal and open water marine environments and covering a depth range from 0 to 1,800 meters. These genomes show marked variation regarding their size (2.12–5.97 Mb), G+C content (48.20%–65.40%) and amount of protein encoding genes (2,510–5,702) (Table 1), suggesting that they may have originated from organisms with long divergence times.
| Organism/Strain | Source | NCBI Accession number | Scaffolds | Lenght (Mbp) | GC(%) | CDS |
|---|---|---|---|---|---|---|
| Synechococcus elongatus PCC 6301 | Freshwater | NC_006576.1 | 1 | 2.7 | 55.50 | 2901 |
| Synechococcus elongatus PCC 7942 | Freshwater | NC_007604.1 | 2 | 2.74 | 55.46 | 2882 |
| BL107 | Blanes Bay, Mediterranean Sea, 1,800 m | NZ_DS022298.1 | 1 | 2.29 | 54.20 | 2655 |
| CB0101 | Chesapeake Bay | NZ_ADXL00000000.1 | 94 | 2.69 | 64.20 | 2881 |
| CB0205 | Chesapeake Bay | NZ_ADXM00000000.1 | 78 | 2.43 | 63.00 | 2661 |
| CC9311 | California current, Pacific (coastal), 95 m | NC_008319.1 | 1 | 2.61 | 52.40 | 3164 |
| CC9605 | California current, Pacific (oligotrophic), 51 m | NC_007516 | 1 | 2.51 | 59.20 | 3016 |
| CC9902 | California current, Pacific (oligotrophic), 5 m | NC_007513 | 1 | 2.23 | 54.20 | 2635 |
| JA23Ba213 | Octopus Spring, Yellowstone Park | NC_007776 | 1 | 3.05 | 58.50 | 3064 |
| JA33Ab | Octopus Spring, Yellowstone Park | NC_007775.1 | 1 | 2.93 | 60.20 | 3036 |
| PCC 6312 | Freshwater, California | CP003558.1 | 2 | 3.72 | 48.49 | 3795 |
| PCC 7002 | Unknown | NC_010475.1 | 7 | 3.41 | 49.16 | 3008 |
| PCC 7335 | Snail shell, intertidal zone, Puerto Penasco, Mexico | ABRV00000000.1 | 11 | 5.97 | 48.20 | 5702 |
| PCC 7336 | Sea Water Tank, Berkey University | ALWC00000000.1 | 1 | 5.07 | 53.70 | 5093 |
| PCC 7502 | Sphagnum bog | CP003594.1 | 3 | 3.58 | 40.60 | 3703 |
| RCC307 | Mediterranean Sea, 15 m | NC_009482.1 | 1 | 2.22 | 60.80 | 2571 |
| RS9916 | Gulf of Aqaba, Red Sea, 10 m | NZ_DS022299.1 | 1 | 2.66 | 59.80 | 2927 |
| RS9917 | Gulf of Aqaba, Red Sea, 10 m | NZ_CH724158.1 | 1 | 2.58 | 64.40 | 2719 |
| WH 5701 | Long Island Sound, USA | NZ_CH724159–NZ_CH724167 | 116 | 3.28 | 65.40 | 3185 |
| WH 7803 | Sargasso Sea, 25 m | NC_009481 | 1 | 2.37 | 60.20 | 2660 |
| WH 7805 | Sargasso Sea | NZ_CH724168.1 | 3 | 2.63 | 57.60 | 2931 |
| WH 8016 | Woods Hole, MA, USA | AGIK00000000.1 | 16 | 2.69 | 54.10 | 3184 |
| WH 8102 | Sargasso Sea | NC_005070.1 | 1 | 2.43 | 59.40 | 2752 |
| WH 8109 | Sargasso Sea | ACNY00000000.1 | 1 | 2.12 | 60.10 | 2510 |
Genomic taxonomy
The genomes of the 24 strains were compared through six methods: I) Average Nucleotide Identity (Konstantinidis & Tiedje, 2005a; Konstantinidis & Tiedje, 2005b), II) Average Amino acid Identity (Konstantinidis & Tiedje, 2005a) III) Dinucleotide signature (Karlin, Mrazek & Campbel, 1997), IV) in silico DNA-DNA hybridization (Meier-Kolthoff et al., 2013), V) Identity of rrsA gene sequences (16S rRNA gene) and VI) Multi-locus sequence analysis (MLSA) of genes rrsA, gyrB, pyrH, recA and rpoB. The Cgview comparison tool (Grant, Arantes & Stothard, 2012) was used to create a genome-wide homology map, based on protein identity using S. elongatus PCC 7942 as the reference genome. Synteny between Prochlorococcus and Synechococcus genomes was analyzed through whole-genome alignments performed in progressiveMauve (Darling, Mau & Perna, 2010).
To facilitate analyzes for genomic taxonomy we developed a program written in Perl named GenTaxo, freely available at sourceforge.net/projects/gentaxo/. This tool receives as input genome sequences in FASTA format to calculate three metrics for genome comparison: Average Nucleotide Identity (ANI), Average Amino acid Identity (AAI) and distances between genomes based on dinucleotide signature.
Homologue identification
The 82,703 proteins encoded in 24 Synechococcus and 13 Prochlorococcus genomes (Thompson et al., 2013) were analyzed through OrthoMCL v1.4 (Li, Stoeckert & Roos, 2003), allowing for the identification of both orthologous and paralogous genes shared between these taxa. Homologue identification was also performed between Synechococcus OrthoMCL was run using the following parameters: inflation factor of 1.25 and e-value ≤10−05.
Phylogenomic reconstruction
Orthologous groups identified by OrthoMCL were used to reconstruct the phylogeny of Synechococcus and Prochlorococcus genomes. Protein sequences of 607 orthologous genes shared between Synechococcus and Prochlorococcus (with no identified paralogs) were aligned through MUSCLE (Edgar, 2004). Next, protein alignments were converted to nucleotide alignments through pal2nal (Suyama, Torrents & Bork, 2006) and each of the 607 alignments were concatenated. Distances between taxa were calculated through the Tajima-Nei method using the concatenated alignment. The phylogenomic tree was reconstructed through the Neighbor-joining algorithm using MEGA5. Bootstrap tests were performed in 1,000 replicates.
Results
Genomic taxonomy
The results from the different methods of comparative genomic analysis all indicated the same pattern: the analyzed strains were extremely distinct regarding genomic content. With the exception of Synechococcus elongatus PCC6301 and Synechococcus elongatus PCC7942, the remaining pairs of strains presented values for ANI and AAI drastically below the species cutoff (95% for both methods). The results obtained from DDH and dinucleotide signatures corroborated these patterns as all the strains had an estimated level of DNA-DNA hybridization below 70% (expect for the aforementioned pair) and the majority of genome pairs yielded distances based on dinucleotide content above the 0.01 cutoff. The same trend was observed through MLSA and 16S rRNA gene comparisons.
Phylogenomic reconstruction
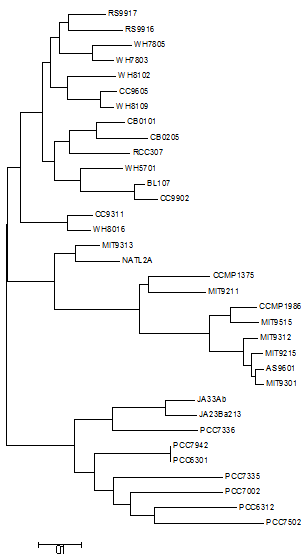
The phylogeny of Synechococcus was reconstructed based on the sequences of 607 orthologous genes, with no paralogs, shared between Synechococcus and Prochlorococcus genomes (Fig. 1). Bootstrap values were above 70% for the majority of nodes, indicating very high consistency of topology. Two major clades of Synechococcus could be identified. The first, made up mostly of marine strains: CB0101, CB0205, WH5701, RCC307, RS9917, RS9916, WH7805, WH7803, BL107, CC9902, WH8102, CC9605, WH8109, WH8016 and CC9311. The second clade is made up mostly of freshwater strains: JA23Ba213, JA33AB, PCC7336, PCC7942, PCC6301, PCC7335, PCC7002, PCC7502 and PCC6312. Tree topology suggested that the 24 genomes represent a polyphyletic group that can be divided in marine strains, a sister taxa of Prochlorococcus, and freshwater and inter-tidal strains.
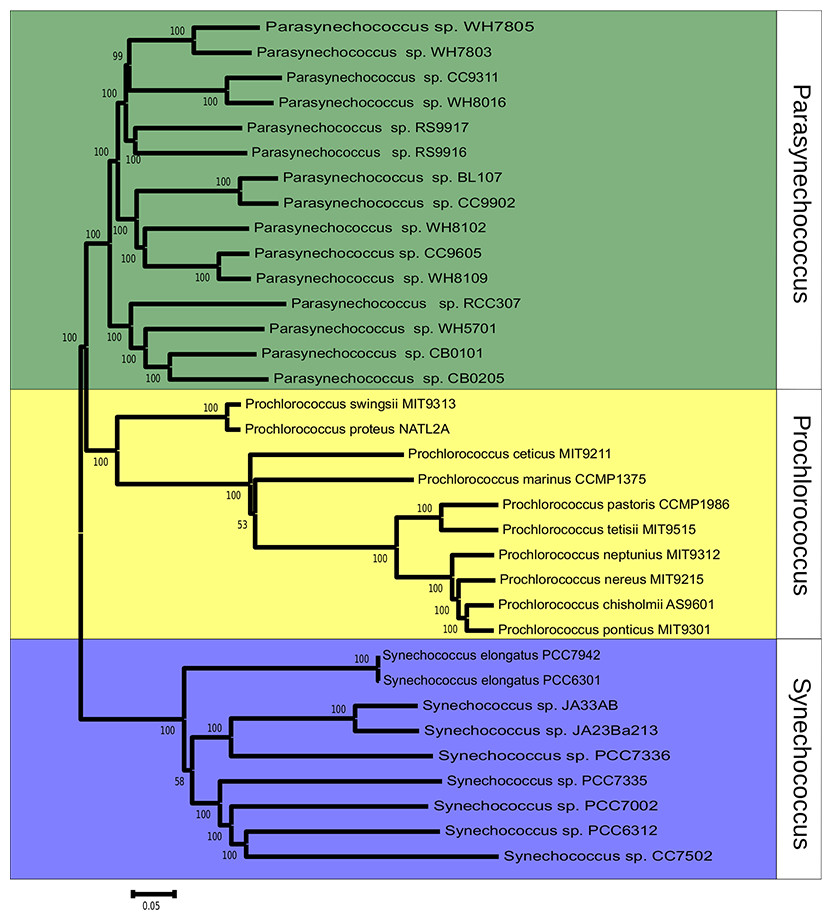
Figure 1: Phylogenomic reconstruction 37 Synechococcus and Prochlorococcus strains based on the concatenated alignment of 607 ortholog genes.
The tree was constructed through neighbor-joining using the Tajima-Nei method. Bootstrap tests were conducted with 1000 replications.{kind=link}
Homologue identification
The 82,703 protein encoding genes analyzed represented the pan-genome of the Synechococcus and Prochlorococcus, the diversity of all protein encoding genes of these genomes. This analysis identified 8,167 homologous groups (Table S1), of those, 744 were shared between all lineages, thus representing the core-genome of Synechococcus and Prochlorococcus. Out of all genes, 15,724 are exclusive of a single genome, of which 577 have a paralog within the same genome and 15,147 are orphans, i.e. are exclusive of a single genome and have no identified orthologs or paralogs (Table S2).
Genomic map and synteny
A whole-genome identity map was created by comparing all the genomes against that of Synechococcus elongatus PCC7942 through blastx (Altschul et al., 1990). This analysis (Fig. 2) revealed low identity levels (<80%) across the extension of the genomes, and indicated several sites of potential insertion/deletion events at the genome of Synechococcus elongatus PCC7942. Even though S. elongatus PCC6301 and S. elongatus PCC7942 are very closely related, the map illustrated that their genomes were not completely identical. While the majority of their genomes shows very high identity levels (>90%), as demonstrated by the dominance of black and dark red colors of the outermost circle, several segments appeared to be exclusive of PCC7942 and not present in PCC6301, which are examples of variation of genomic content between organisms that have very short divergence times.
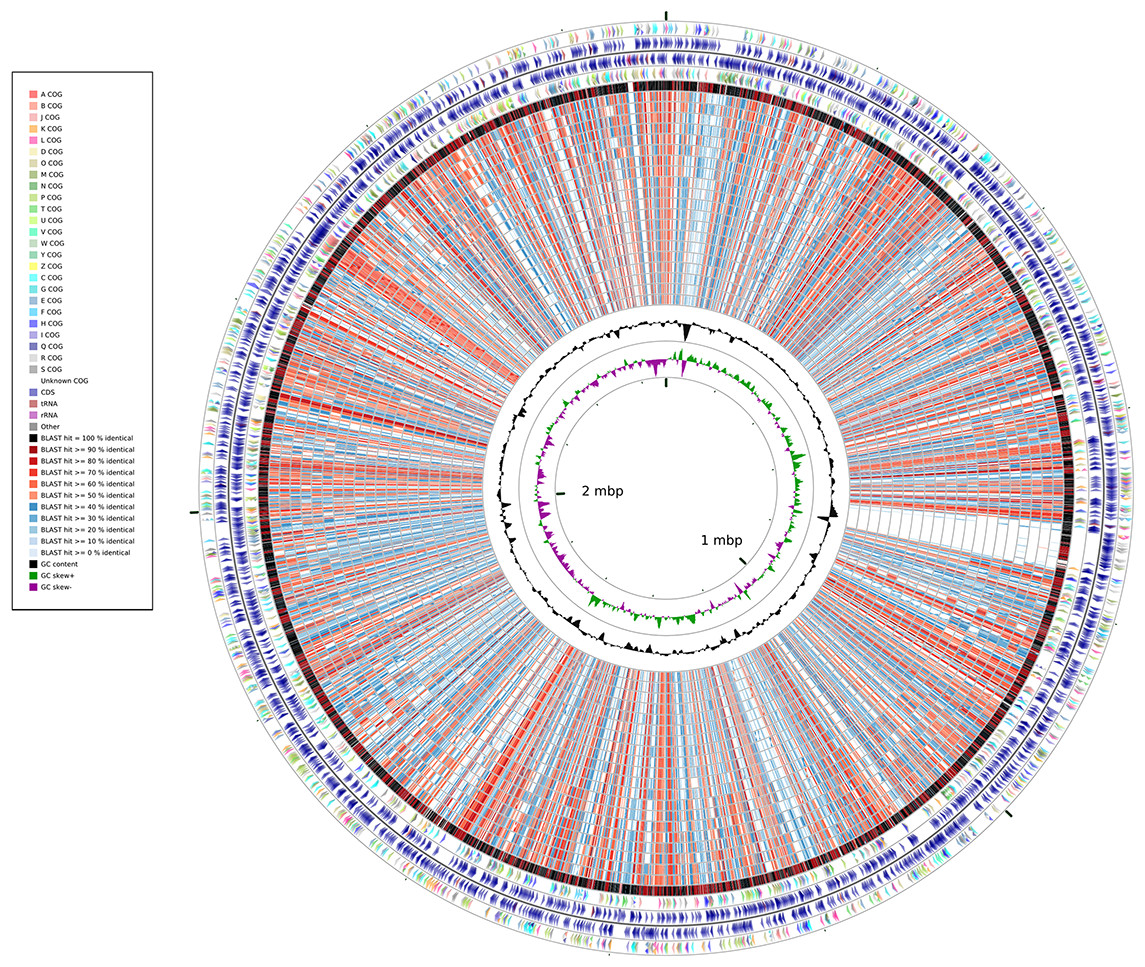
Figure 2: Whole-genome identity map generated through Cgview comparison tool.
Protein sequences of each strain were compared against the genome o PCC7942 through blastx. Rings represent, from outermost to innermost: 1) Genes of the plus strand color coded by COG category; 2) Protein encoding and RNA genes at the plus strand; 3) Genes of the minus strand color coded by COG category; 4) Protein encoding and RNA genes at the minus strand; 5) PCC6301; 6) PCC7335; 7) PCC6312; 8) PCC7002; 9) PCC7336; 10) PCC7502; 11) WH5701; 12) JA23Ba213; 13) JA33Ab; 14) WH7805; 15) RS9917; 16) WH7803; 17) RCC307; 18) RS9916; 19) WH8016; 20) CC9605; 21) CC9311; 22) WH8102; 23) BL107; 24) WH8109; 25) CC9902; 26) G+C Content; 27) G+C Skew.{kind=link}
Whole-genome alignments between marine and freshwater Synechococcus revealed little to no synteny between the genomes of these lineages and a significant amount of genome rearrangement events occurring between strains (Figs. S2A and S2B). This pattern was not observed when analyzing Prochlorococcus genomes, which showed a somewhat syntenic genome organization (Fig. S2C).
Discussion
Phylogenomics of Synechococcus and Prochlorococcus
The phylogenomic reconstruction based on the concatenated alignments of 607 orthologs separated the 24 Synechococcus genomes into two major groups, roughly segregating seawater and freshwater strains, and is supported by high bootstrap values (Fig. 1). This pattern points toward a division between these two lifestyles early on the evolutionary history of these organisms. Previous studies that investigated the evolution of Synechococcus suggested that marine strains are closely related to Prochlorococcus and distant from members of freshwater strains, which would make Synechococcus a polyphyletic group that encompasses at least two genera (Honda, Yokota & Sugiyama, 1999; Robertson, Tezuka & Watanabe, 2001; Fuller et al., 2003; Shih et al., 2013), that can be further divided into several subclades (Fuller et al., 2003; Shih et al., 2013; Matzke, Shih & Kerfeld, 2014). The phylogeny was reconstructed based on the genes of the core-genome of Prochlorococcus and of both marine and freshwater Synechococcus strains. These 607 ortholog groups encode constitutive functions and genes that are very unlikely to undergo HGT. Nevertheless, to rule out the possibility that the segregation between the two lineages emerges from environment specific HGT, a second tree was constructed, using concatenated alignments of pyrH, recA and gyrB genes (Fig. S1). This tree further corroborates the consistency of the obtained topology.
Pan and core genome composition of Prochlorococcus and Synechococcus
Previous studies have investigated the pangenome of marine Synechoccocus and of Cyanobacteria as a whole. These analyzes estimated a core-genome of 1,228 orthologue groups for marine Synechococcus and Prochlorococcus (Dufresne et al., 2008b) and of 892 orthologue groups for Cyanobacteria (Mulkidjanian et al., 2006). Our results point to a comparable figure, i.e. a core genome for marine and freshwater Synechococcus of 866 orthologue groups that drops to 744 when Prochlorococcus is included. The somewhat smaller core-genome obtained here is a consequence of using a larger number of genomes that belong to a broader range of phylogenetic lineages (Shih et al., 2013). These studies also came to similar conclusions regarding the functional roles of the orthologue groups which make up the core-genome, many of which encode genes associated to essential physiologic functions, e.g. photosynthesis and DNA metabolism, cell division, circadian cycle and ribosomal proteins. Meanwhile the non core orthologue groups are usually involved in habitat adaptation and nutrient uptake (Mulkidjanian et al., 2006; Dufresne et al., 2008b).
Genetic distinctions between marine Synechococcus/Prochlorococcus and freshwater Synechococcus
A total of 310 orthologue groups are present in all marine Synechococcus and Prochlorococcus but absent from at least one freshwater Synechococcus genome (Table S1). Among the traits that differentiate these two clades, their carbon concentration mechanisms are among the most relevant. Cyanobacteria have carboxysomes, intracellular microcompartments formed by a protein shell that encapsulates the enzymes carbonic anhydrase and ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO). Carbonic anhydrase converts to CO2 around RuBisCO that in turn utilizes this CO2 molecule to synthesize phosphoglycerate. The compartmentalization brought by the carboxysome, enhances the efficiency of carbon fixation by elevating the levels of intracellular CO2 around RuBisCO (Rae et al., 2013). Two forms of carboxysome have been identified that differ regarding their enzymes, transporters, structural proteins and the RuBisCO isoform within them. Marine Cyanobacteria have α-Carboxysomes while coastal and freshwater species harbor β-Carboxysomes (Yeates et al., 2008). Genes encoding Carboxysome shell proteins CsoS2 and CsoS3 and Carboxysome peptides A and B are present in all the genomes marine Synechococcus and Prochlorococcus, these proteins are characteristic of α-Carboxysomes. Meanwhile proteins of the ccm operon (L, M, N and O), which are characteristic of β-Carboxysomes were found exclusively and in all of the members freshwater Synechococcus clade. These findings indicate that marine and freshwater Synechococcus use distinguished apparatus for carbon concentration, meaning that these groups could have differential contributions regarding their roles as primary producers and to the global biogeochemical carbon cycle. Although, despite the different protein composition of α and β carboxysomes, functional differences between them are still poorly characterized (Yeates et al., 2008).
A total of 46 orthologue groups are found exclusively on genomes of Prochlorococcus and marine Synechococcus clade and absent from all freshwater Synechococcus lineages. Among those, the ones with well characterized functions include proteins such as: a Fructokinase (carbohydrate metabolism), an inorganic pyrophosphatase (phosphorus metabolism), a carboxypeptidase (protein degradation and maturation), an aspartocyalase (aspartic acid biosynthesis), GMC oxidoreductase (carbohydrate metabolism), a Ribonuclease (tRNA maturation), and an RpoD like sigma factor (transcriptional regulation). A total of 71 orthologue groups are found exclusively and in all genomes of freshwater Synechococcus, the ones with well characterized functions include: a Lycopene cyclase (carotenoid biosynthesis), a RecJ exonuclease (DNA repair), an oligopeptide permease (peptide transport), a citramalate synthase (aminoacid biosynthesis), a folate transporter and several ABC transporters.
Genetic distinctions between marine and freshwater Synechococcus
Besides carboxysome structure, marine Synechococcus can be differentiated from freshwater Synechococcus by: 16S rRNA gene homology to marine strains of Synechococcus, absence of Fructose-1,6-bisphosphatase and 6-phosphofructokinase and presence of DNA polymerase II (not detected in any of the other Synechococcus genomes) and also by the presence of 101 identified homologs groups that are specific to either one of these two lineages. Many of these genes are involved in essential metabolic processes, i.e. carbohydrate metabolism, nutrient uptake, transcriptional regulation and DNA replication. Nevertheless, molecular assays would be required to describe how the physiology of these organisms is affected by the presence/absence of these proteins, which unfortunately is outside of our scope. However it is reasonable to assume that these organisms make use of alternative proteins to fulfill those roles, which could belong to the many poorly characterized and hypothetical proteins that are present in these genomes (Tables S1 and S2).
Two-component response regulators are among the mechanisms used by bacteria to sense environmental stimulus and adapt accordingly by modulating protein activity through alteration of transcriptional patterns (West & Stock, 2001; Laub & Goulian, 2007). Freshwater Synechococcus and marine Synechococcus strains differ regarding the presence and abundance of proteins of two-component response regulators, histidine kinases and transcriptional regulators (e.g. sigma factors). These orthologue groups are indicated by stars on column 4 of Tables S1 and S2. Unfortunately, the specific roles of the majority of these proteins remains unknown. Nevertheless, the fact that each strain harbors a distinctive set of these regulators points to unique responses to environmental stimuli and mechanisms for transcriptional regulation for each of them.
Phenotypic and ecological traits of marine and freshwater Synechococcus
An extensive body of knowledge has been focused on characterizing differences and similarities between Synechococcus strains based on phenotypical and ecological traits. Most of this work has been focused on marine and coastal strains, and unfortunately freshwater strains remain poorly characterized. Nevertheless, those phenotypic analyzes corroborate our results, as they all point to significant distinction between the Synechococcus lineages. A time-series study of the abundance patterns of the clades that include lineages CC9311, CC9605, WH8102 and CC9902 at a coastal Pacific ocean environment revealed that these organisms have distinctive temporal distribution patterns throughout the year, with the clades that include strains CC9311 and CC902 emerging as dominant members of the community while the clades of WH8102 and CC9605 appear as low-abundance members throughout most of the time series (Tai & Palenik, 2009). Another time series study performed at the Gulf of Aqaba also reported uneven distribution of Synechococcus clades throughout the year, and linked fluctuations in the abundance of these lineages to their preferences regarding nutrient utilization strategies (Post et al., 2011). A study focused on the spatial distribution of several clades of marine Synechococcus (which include some of the strains analyzed here, namely WH8016, CC9311, WH8109, RS9917 and WH8102) across oceanic provinces provided strong evidence for distinctive distribution signatures across the oceans for each one of the analyzed clades, which could be driven by differences in the capacities of these organisms to adapt to nutrient availability and temperature (Zwirglmaier et al., 2008).
Phycobilisomes are light-harvesting complexes present in Cyanobacteria. These structures are formed by a Phicocyanin core that can be linked phycobiliproteins and phicoerythrin. These structures are responsible for absorbing energy from light that is transferred to Chlorophyll molecules (Glazer, 1985; Mullineaux, 2008). Therefore, phycobilisome structure determines the light spectra that can be used by a given organism, and consequently its capacity to photosynthesize at different environments. Eleven marine Synechococcus strains have had their phycobilisome structures analyzed and compared, revealing that even within this group of closely related organisms there is a remarkable diversity regarding their light-absorption apparatus (Six et al., 2007b). The functioning and tolerance of fluctuations in irradiance of the light harvesting apparatus of lineages WH8102, RS9917 and RCC307 has been shown to be distinct between them and also different from that of Prochlorococcus. These differences are thought to be associated with niche-partitioning between these organisms, that make use of distinct light spectra for photosynthesis (Six et al., 2007a).
Horizontal gene transfer influences the evolution of Synechococcus
Mobile genetic elements and horizontal gene transfer play a significant role at the evolution of Cyanobacteria (Zhaxybayeva et al., 2006; Palenik et al., 2009). Among the homologous groups identified, many of those that show drastic differences in their number of copies at each genome are associated with mobile genetic elements. As an example, an homologue group encoding for a transposase was detected exclusively in the genome of strain PCC 7335. Thirty-five copies of this gene were detected, making it the protein with most copies in this genome. Three other homologue groups encoding integrases were more abundant in the genome of PCC 7335 than any of the others analyzed genomes. Interestingly, PCC 7335 has the largest genome among the analyzed strains followed by PCC 7336, whose genome if also filled with multiple copies of transposases. This pattern suggests that these transposases may be responsible for the increased genome of these strains, as these elements mediate acquisition of exogenous DNA (Ochman, Lawrence & Groisman, 2000; Juhas et al., 2009). Besides these integrases, many homologous groups identified are also involved in gene transfer events (e.g. plasmid proteins, CRISPR, transposons and phage proteins). Altogether, these results provide evidence that horizontal gene transfer agents are important drivers of the evolution of these genomes, contributing to the diversification of the group, these elements may be one of the sources of the extensive genomic plasticity found among marine and freshwater Synechococcus.
Bacterial genomes are dynamic, constantly undergoing contraction through gene loss and expansion mediated by horizontal gene transfer (Puigbò et al., 2014). Recent studies have explored the drastic genome reduction that occurred during the evolution of Prochlorococcus (Kuo, Moran & Ochman, 2009; Batut et al., 2014). However, no evidence for such process has been observed for either clade of Synechococcus. Instead, our data points to an opposite trend among these taxa, acquisition of new genes leading to enlargement of genomes driven by invasion of exogenous DNA through horizontal gene transfer. This invasive DNA molecules are able to fixate in species with small effective population sizes, in which genetic drift is more relevant than natural selection for genome evolution (Batut et al., 2014). Considering the superior cell densities of Prochlorococcocus compared to Synechococcus, genetic drift is expected to be less influential over the first than the letter, thus favoring a reduced genome size as consequence of strong natural selection. These distinctions of genome size between Synechococcus and Prochlorococcus may be associated with the environmental distribution of these organisms. The first are typical of freshwater and coastal environments, that are richer in nutrients than the oligotrophic waters occupied by the latter. Therefore, the selective pressure towards a reduced genome may be more pronounced over Prochlorococcus since it thrives in nutrient deprived environments.
A new taxonomic classification for Synechococcus
The results from ANI, AAI, and in silico DDH analyzes indicate that, with the exception of strains PCC7942 and PCC6301, the level of dissimilarity found between these genomes suggests very long diverge times. Such a trend is also corroborated by the 13,511 identified orthologous groups that can differentiate these lineages. It is therefore likely that these genomes represent different species, which according to the phylogenomic reconstruction can be segregated into two different genera: a sister clade to Prochlorococcus formed by the marine strains with the proposed name Parasynechococcus, and a second clade formed by freshwater Synechococcus strains.
A formal description of Parasynechococcus following the criteria established by the International Journal of Systematic and Evolutionary Microbiology follows:
List of the strains included in the taxon: Based on the phylogenomic reconstruction, the following strains are assigned to Parasynechococcus: CB0101, CB0205, WH5701, RCC307, RS9917, RS9916, WH7805, WH7803, BL107, CC9902, WH8102, CC9605, WH8109, WH8016 and CC9311.
Tabulation of the characteristics of each strain: Tables S1 and S2 list the collection of orthologous groups that can be used to differentiate between these strains based on their genomic content.
List of characteristics considered essential for membership in the taxon: Characteristic traits of Parasynechococcus are: 16S rRNA gene closely related to marine strains of Synechococcus and Prochlorococcus, α-Carboxysomes, absence of Fructose-1, 6-bisphosphatase and 6-phosphofructokinase and presence of DNA polymerase II. Also the orthologue groups described in Tables S1 and S2 can be used to differentiate this genera from Prochlorococcus and freshwater Synechococcus and also between the Parasynechococcus 15 strains.
-
List of characteristics which qualify the taxon for membership in the next higher taxon:
Both Prochlorococcus and Parasynechococcus belong to the Synechococcaceae family, order Chroococcales. These organisms are grouped together on the basis of phylogenetic reconstruction, nevertheless the phenotypic and genotypic traits that distinguish this family remain poorly characterized.
List of diagnostic characteristics: Tables S1 and S2 also list the genomic traits that distinguish Paraynechoccus from the sister taxa Prochlorococcus.
Designation of the type for that taxon: Strain WH8102 was chosen as the type strain of Parasynechococcus. This strain represents the first complete genome of the genus to be sequenced, has reasonable amount of descriptive data encompassing several aspects of its biology, such as carboxysome structure (Iancu et al., 2007), the light-harvesting apparatus (Six et al., 2007b), seasonal abundance patterns (Tai & Palenik, 2009; Post et al., 2011), and nutrient uptake and utilization (Moore et al., 2005; Su et al., 2006; Tetu et al., 2009). Also, as required, this strain is available in two international culture collections (Roscoff Culturing Collection, France and NCMA, USA).
Reactions of the type strain: To our knowledge, there is no large scale dataset that consistently assessed phenotypic traits concerning metabolic reactions performed by these strains. Therefore, we limited our description to the genotypic traits that differentiate this lineage.
Conclusions
Comparative genomics and phylogenomic reconstruction allowed the identification of two genera: Synechococcus and Parasynechococcus. The two clades and their individual members have marked differences regarding their genetic content, including taxa-specific homologues. This genetic variability pertains to central aspects of the physiology of these organisms and to their interactions with their environment. Future studies should strive to establish how the differences in the genetic content of these taxa affect their lifestyle, specifically with regard to nutritional demands, metabolism, carbon fixation methods and light-utilization strategies.
Supplemental Information
Orthologous groups.
Ortholog composition across the 24 analyzed genomes. Only orthologs present in at least 2 genomes are shown.
Oprhan groups.
Orphan composition. List of genes present in only one of each of the 24 genomes with no identified homologs in other genomes.
Phylogenetic reconstruction.
Phylogeny of Synechococcus and Prochlorococcus reconstructed based on the concatenated alignments of genes pyrH, recA and gyrB.
{kind=link}
Synteny maps based on whole-genome alignments.
(A) Marine Synechococcus (B) Freshwater Synechococcus (C) Prochlorococcus.
{kind=link}