A CDC25 family protein phosphatase gates cargo recognition by the Vps26 retromer subunit
- Yale School of Medicine, United States
- University of Iowa, United States
Abstract
We describe a regulatory mechanism that controls the activity of retromer, an evolutionarily conserved sorting device that orchestrates cargo export from the endosome. A spontaneously arising mutation that activates the yeast (Saccharomyces cerevisiae) CDC25 family phosphatase, Mih1, results in accelerated turnover of a subset of endocytosed plasma membrane proteins due to deficient sorting into a retromer-mediated recycling pathway. Mih1 directly modulates the phosphorylation state of the Vps26 retromer subunit; mutations engineered to mimic these states modulate the binding affinities of Vps26 for a retromer cargo, resulting in corresponding changes in cargo sorting at the endosome. The results suggest that a phosphorylation-based gating mechanism controls cargo selection by yeast retromer, and they establish a functional precedent for CDC25 protein phosphatases that lies outside of their canonical role in regulating cell cycle progression.
https://doi.org/10.7554/eLife.24126.001Introduction
Endocytosis mediates internalization of the plasma membrane via vesicles that deliver their content to the endosomal system, a network of related organelles that undergo maturation to generate a terminal endosome that fuses with the lysosome. During endosome maturation, integral membrane proteins are either retained, leading their eventual turnover in the lysosome, or they are exported and delivered to other organelles for re-use. Molecular sorting reactions in the endosome thus play a fundamental role in controlling the composition of the plasma membrane by determining which molecules are recycled back to the plasma membrane and which are degraded.
The retromer protein complex is an evolutionarily conserved sorting device that orchestrates export of integral membrane proteins from the endosome via tubular and vesicular ETCs (Burd and Cullen, 2014; Chi et al., 2015). Retromer is composed of three proteins: Vps35, which resembles an alpha solenoid similar to well characterized vesicle coat proteins such as clathrin; Vps26 (standard name: Pep8), which binds the N-terminal region of Vps35; and Vps29, which binds the C-terminal region of Vps35 (Collins et al., 2008; Hierro et al., 2007; Norwood et al., 2011). The full range of molecular functions that are executed by each retromer subunit have yet to be definitively established, and this is a major obstacle for understanding the mechanisms that underpin retromer-mediated cargo export from the endosome. The helical repeats of Vps35 are thought to provide binding sites for cargo and accessory factors, such as the Snx3 and Snx27 sorting nexins (Harrison et al., 2014; Hierro et al., 2007; Liu et al., 2012) and, in mammals, the WASH complex, an activator of the Arp2/3 actin polymerization complex (Harbour et al., 2012; Jia et al., 2012). Vps26 is structurally related to α- and β-arrestins (Collins et al., 2008; Shi et al., 2006), which function as sorting adapters for G protein-coupled receptors (GPCRs) in clathrin-mediated endocytosis, raising the possibility that it may function in the selection of integral membrane proteins for export. Given the central role that retromer plays in controlling plasma membrane composition and organelle biogenesis, it is logical to expect that retromer activities would be regulated in order to integrate its functions with cell physiology (Seaman, 2012), but examples of such regulation have yet to be identified.
In this study, we used genetic selection in budding yeast (Saccharomyces cerevisiae) to identify gene products that control plasma membrane residence of integral membrane proteins. One mutant obtained displays a loss of retromer-dependent plasma membrane recycling of multiple integral plasma membrane proteins. This phenotype results from a gain-of-function mutation in the dual specificity protein phosphatase (Camps et al., 2000; Patterson et al., 2009), MIH1, encoding the S. cerevisiae homolog of CDC25 (Pal et al., 2008). We show that Vps26 directly recognizes a retromer sorting signal and that the phosphorylation state of Vps26, controlled by Mih1, modulates the affinity of retromer for the Chs3 recycling signal.
Results and discussion
Isolation of yeast mutants deficient in endosomal trafficking
To identify factors that regulate post-Golgi trafficking of integral membrane proteins, we harnessed the activity of yeast chitin synthase 3 (Chs3), an integral membrane enzyme that is trafficked between Golgi and endosomal compartments and the plasma membrane. Delivery of Chs3 to the cell surface is mediated by transport vesicles that are coated with the ‘exomer’ protein complex (Wang et al., 2006). In cells lacking functional exomer, Chs3 is constitutively retained within the cell by a Golgi-endosome trafficking circuit involving a clathrin adapter protein complex 1 (AP1)-dependent retention mechanism (Valdivia et al., 2002; Wang et al., 2006). In cells that lack both functional exomer and AP1 sorting complexes, Chs3 fails to be retained and it is delivered to the plasma membrane via the constitutive secretory pathway (Valdivia et al., 2002) (Figure 1A). Whereas growth of exomer-deficient cells is resistant to calcofluor white (CFW), a cytotoxic molecule that binds chitin in the cell wall (Roncero and Durán, 1985), growth of cells lacking exomer and AP1 is exquisitely sensitive to CFW in the medium (Valdivia et al., 2002) (Figure 1).
Figure 1 with 1 supplement see all
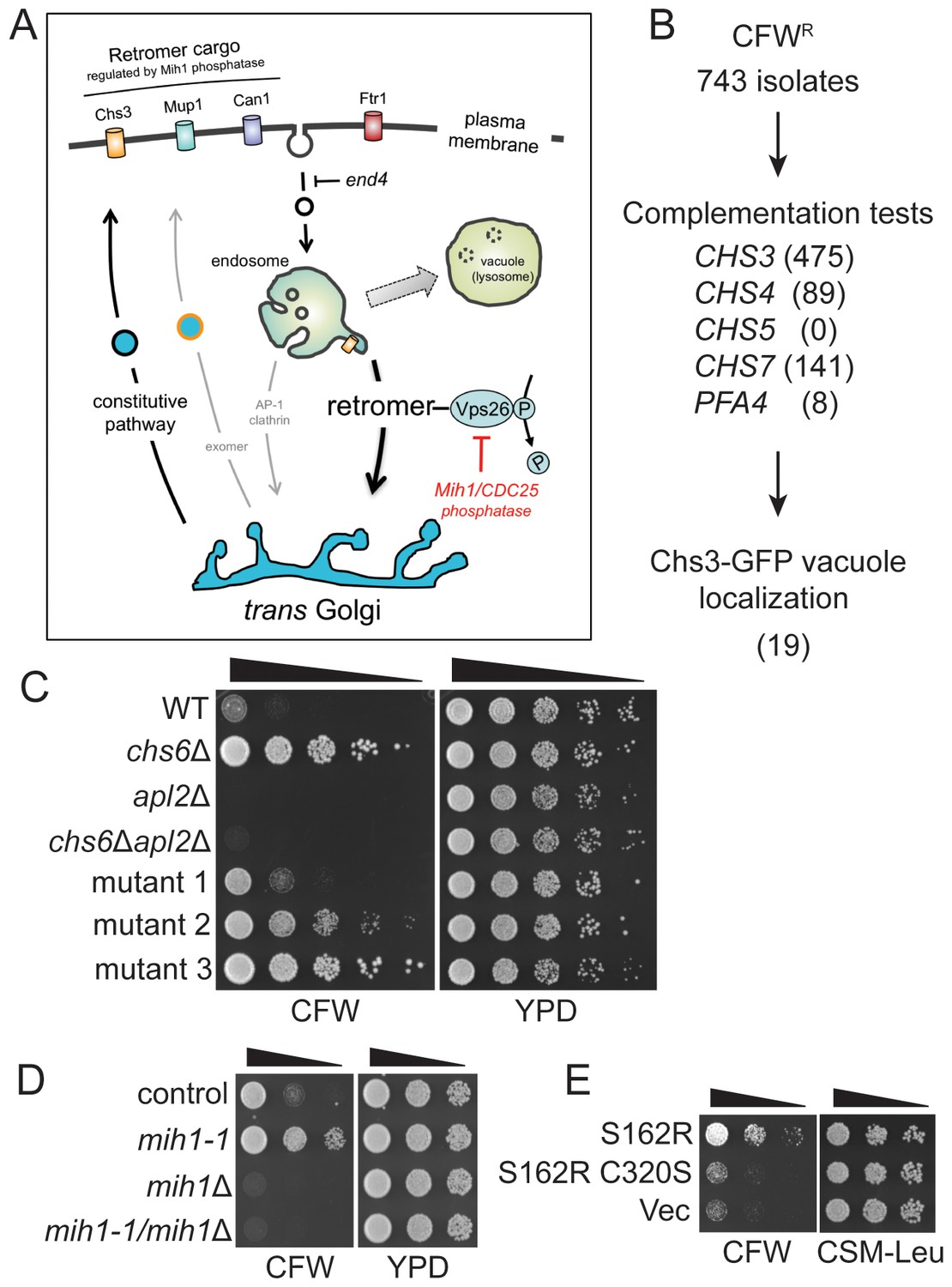
Selection of yeast mutants resistant to calcofluor white.
(A) Schematic diagram of post-Golgi trafficking pathways. In wild-type cells, Chs3 is retained at the Golgi-endosome interface via AP-1 dependent retrieval from the endosome, and is delivered to the plasma membrane via Golgi-derived exomer coated vesicles (gray arrows). In cells used for the genetic selection in this study, chitin synthase 3 (Chs3) is diverted into the constitutive secretory pathway due to deletions of the Chs6 exomer subunit and the Apl2 AP-1 subunit (chs6Δapl2Δ). Endocytosis of Chs3 and other plasma membrane proteins, which is attenuated by the end4 mutation, delivers the internalized proteins to the endosome, where some are sorted by retromer into a recycling pathway, while others are retained in the endosome and delivered to the lysosome-like vacuole and degraded. (B) Strategy to identify novel mutations affecting Chs3 trafficking. The flowchart lists the number of mutants remaining after each step of selection. (C) Representative calcofluor white (CFW) resistant strains. Serial dilutions (1:10; indicated by the black triangle above the photograph) of the indicated strains were spotted onto YPD medium with or without 100 μg/ml CFW. The plates were incubated for three days at 30°C and then photographed. (D) Mutations in MIH1 alter sensitivity of growth to CFW. Serial dilutions (indicated by a triangle above the photograph) of control cells of the indicated genotypes were spotted onto rich medium with or without 50 μg/ml CFW for three days at 30°C. The genotype of the control strain is: chs6Δapl2Δ. The chs6Δapl2Δmih1-1 strain was constructed by introducing the mih1-1 lesion de novo in the original chs6Δapl2Δ parent strain. The mih1Δ designation indicates that the wild-type MIH1 locus was deleted in the original parent strain (chs6Δapl2Δmih1Δ). The mih1-1/mih1Δ designation indicates that the mih1-1 allele was deleted in the original mih1-1 mutant strain. (E) Mih1 phosphatase activity is required for CFW resistance conferred by the mih1-1 mutation. Serial dilutions of chs6Δapl2Δmih1Δ cells containing plasmids expressing the mih1-1 allele (‘S162R’), a double mutant protein (S162R C320S) in which a second mutation was introduced that ablates catalytic activity, or empty vector (‘Vec’), were spotted onto complete synthetic medium with or without 50 μg/ml CFW for three days at 30°C. The host strain is the chs6Δapl2Δmih1Δ strain background.
We used selection on CFW-containing growth medium to obtain CFW-resistant (‘CFWR’) mutants and then identified those exhibiting altered distribution of Chs3 (Figure 1B,C). To bias the selection away from mutations that affect exomer- and AP1-mediated trafficking, we employed a strain deleted for both an exomer subunit (CHS6) and an AP1 subunit (APL2), chs6Δapl2Δ. As previously reported (Valdivia et al., 2002), these cells are sensitive to CFW (Figure 1C), however, we observed spontaneously arising CFWR colonies after several days on solid growth medium containing 100 μg/ml CFW (Figure 1C); we considered these to be good candidates for strains carrying spontaneously arising mutations that cause a reduction in cell wall chitin. Indeed, cells of each of these strains exhibited reduced capacity to bind CFW, as determined by visualizing cells by ultra-violet illumination (not shown).
Complementation tests against a panel of mutants implicated in Chs3-dependent chitin deposition (chs3△, chs4△, chs5△, chs7△, and pfa4△) (Lam et al., 2006; Ono et al., 2000; Trilla et al., 1999; Wang et al., 2006) revealed that, as expected, the majority of CFWR mutants (96%) failed to be complemented (i.e., diploid cells remained CFWR) by any single ‘tester’ strain (Figure 1B), strongly suggesting that lesions in the ‘tester’ genes are responsible for the observed CFW resistance. These mutants were discarded, leaving a subset of 30 strains that were not complemented by any of the tester strains. To determine if Chs3 trafficking is perturbed in cells of any of these strains, we compared localization of a Chs3-GFP fusion protein in each mutant strain to that of the original parent strain. In a population of parental chs6Δapl2Δ cells, Chs3-GFP is observed at the bud neck, intracellular punctae (Golgi and/or endosome compartments), and there is a faint signal in the vacuole lumen of most cells (Figure 2A). In 19 out of 30 CFWR strains examined, there are fewer and less bright punctae, the intensity of Chs3-GFP fluorescence at the bud neck is reduced, and the GFP signal in the vacuole lumen is substantially increased (an example of one such mutant is shown in Figure 2A).
Figure 2 with 1 supplement see all
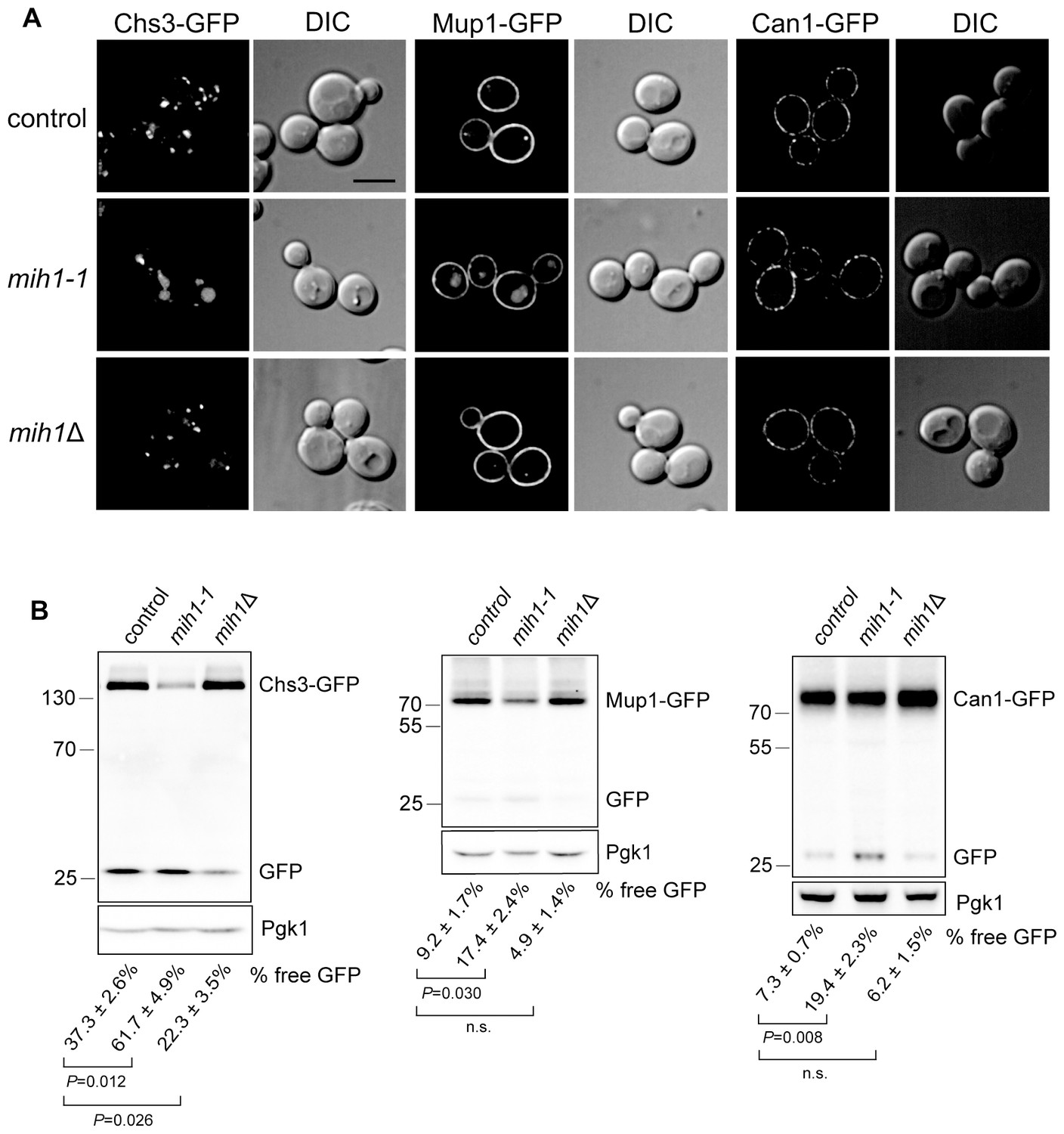
Mih1 regulates turnover of a subset of plasma membrane proteins.
(A) Localization of Chs3-GFP, Mup1-GFP, and Can1-GFP in control cells (chs6Δapl2Δ), mih1-1 (chs6Δapl2Δmih1-1) and mih1Δ (chs6Δapl2Δmih1Δ). A representative medial optical plane of a z-series is shown. Scale bar, 5 µm. (B) Increased proteolysis of Chs3-GFP, Mup1-GFP, and Can1-GFP in mih1-1 (chs6Δapl2Δmih1-1) cells and decreased proteolysis in mih1Δ (chs6Δapl2Δmih1Δ) cells. Mup1-GFP and Can1-GFP expressing cells were grown in methionine- or arginine-deficient medium, respectively, to promote plasma membrane recycling. A representative anti-GFP immunoblot of cell lysates of the indicated strains is shown. The number below each lane indicates the mean proportion (+s.e.m.) of free GFP to the total GFP signal in the lane calculated from a minimum of three independent measurements. Anti 3-phosphoglycerate kinase (Pgk1) blotting was used to control for loading. Statistical significance for pairwise comparisons is indicated; n.s., not statistically significant. The positions of molecular weight (kDa) protein standards is indicated on the left side of the gel.
Whole-genome sequencing of the most severe vacuolar localization mutant led to the identification of a cytosine-to-adenine mutation in the MIH1 ORF that causes a S162R substitution in the Mih1 protein. That this mutation is responsible for the phenotypes that we selected was confirmed by the identical phenotypes of cells in which this mutation was constructed de novo in the parent strain, and by deletion of the MIH1 locus in the original mutant strain, which reverted the enhanced vacuolar targeting of Chs3 (Figure 2) and conferred enhanced sensitivity to CFW (Figure 1D). We termed this allele mih1-1.
MIH1 is homologous to CDC25, first discovered in Schizosaccharomyces pombe, which encodes a dual-specificity protein phosphatase that dephosphorylates the Cdc2 cyclin-dependent kinase (CDK; Cdc28 in S. cerevisiae) to activate it (Pal et al., 2008; Patterson et al., 2009; Russell and Nurse, 1986). The substitution caused by the mih1-1 mutation lies outside of the phosphatase motif, however, the phosphatase activity of Mih1 is necessary for the observed phenotypes of the mih1-1 mutant, because introduction of a second mutation, cysteine(320) to serine, which changes an active site residue required for catalysis (Yano et al., 2013), ablates CFWR caused by the mih1-1 mutation (Figure 1E). Mih1 undergoes cell cycle regulated phosphorylation at numerous sites and this is correlated with inhibition of its activity toward Cdc28/CDK (Pal et al., 2008). While S162 is not a known site of phosphorylation, several residues in the vicinity of S162 are proposed to be phosphorylated by protein kinase C (PKC) (Yano et al., 2013), a protein kinase shown to regulate trafficking of Chs3 (Valdivia and Schekman, 2003). Thus, we speculate that the S162R mutation may impinge on regulation of Mih1 activity.
As the proposed function of Mih1 is to activate Cdc28/CDK by dephosphorylating an active site tyrosine residue (Pal et al., 2008), we tested the possibility that the mih1-1 associated trafficking phenotypes are due to hyper-activation of Cdc28/CDK. Immunoblotting with anti-phospho-CDK antiserum did not reveal a change in the amount of phosphorylated Cdc28/CDK in lysates from cultures of chs6Δapl2Δ versus chs6Δapl2Δmih1-1 cells (Figure 1—figure supplement 1), probably because multiple phosphatases act redundantly to activate Cdc28/CDK (Kennedy et al., 2016). Accordingly, this hypothesis was further tested by deleting the gene encoding the Swe1 kinase that phosphorylates Cdc28/CDK to inhibit its activity (Lianga et al., 2013; Pal et al., 2008); if the mih1-1 allele exerts its effect via activated Cdc28/CDK, deletion of SWE1 should revert the CFWR and Chs3-GFP trafficking phenotypes. However, we observed that CFW resistance and Chs3-GFP localization are unaffected by the swe1Δ mutation (Figure 1—figure supplement 1). In addition, deletion of SWE1 in the original chs6Δapl2Δmih1-1 mutant strain (chs6Δapl2Δmih1-1swe1Δ) has no effect on CFW sensitivity or Chs3-GFP localization (Figure 1—figure supplement 1). These data indicate that the consequences of the mih1-1 mutation on Chs3 trafficking are not exerted via activated Cdc28/CDK.
Mih1 regulates recycling of plasma membrane proteins
The data suggest that CFW resistance is caused by an increased rate of turnover of Chs3 in the vacuole of chs6Δapl2Δmih1-1 cells. Anti-GFP immunoblotting of cell extracts confirmed this; in lysates of chs6Δapl2Δmih1-1 cells expressing Chs3-GFP, GFP is cleaved from ~62% of the Chs3-GFP fusion protein, but just ~37% is processed to GFP in lysates from the chs6Δapl2Δ parent (Figure 2). Importantly, in parental cells lacking MIH1 (chs6Δapl2Δmih1Δ), GFP is cleaved from just ~22% of the Chs3-GFP fusion protein with a concomitant accumulation of full length Chs3-GFP relative to parental cells (Figure 2). Thus, the mih1-1 and mih1Δ alleles have opposing consequences, indicating that native Mih1 is a physiological regulator of Chs3 trafficking.
In order to determine if the mih1-1 mutation uniquely affects trafficking of Chs3, we examined localization and processing of GFP-tagged forms of several nutrient transporters that are maintained at the plasma membrane via endocytic recycling in chs6Δapl2Δmih1-1 and chs6Δapl2Δmih1Δ cells. As observed for Chs3-GFP in the original mih1-1 mutant, increased vacuole localization and proteolytic processing of Mup1-GFP (a GFP tagged methionine transporter) and Can1-GFP (a GFP tagged arginine transporter) is observed (Figure 2). In addition, deletion of MIH1 (i.e., chs6Δapl2Δmih1Δ cells) results in an increase in the levels of full-length Mup1-GFP and Can1-GFP (Figure 2), while the localization, levels, and proteolytic processing of Ftr1-GFP (a GFP tagged iron transporter), Vps10-GFP (a GFP-tagged integral membrane protein that is trafficked from the endosome to the Golgi by retromer), and GFP-Snc1 (a GFP tagged v-SNARE of secretory vesicles) are not affected by mutations in MIH1 (Figure 2—figure supplement 1). These results indicate that Mih1 regulates recycling of a subset of proteins that transit the plasma membrane.
In principle, the mih1-1 mutation could affect sorting upon export from the Golgi or sorting at the endosome, after cargo has been internalized by endocytosis. To distinguish between these modes, we monitored localization and processing of Chs3-GFP and Mup1-GFP in a strain in which the end4-1 mutation was introduced into the chs6Δapl2Δmih1-1 strain (chs6Δapl2Δmih1-1 end4-1). This mutation substantially limits endocytosis via the clathrin-dependent pathway (Munn and Riezman, 1994), resulting in the accumulation of recycling proteins on the plasma membrane (Strochlic et al., 2007). In cells carrying the end4-1 mutation, the Chs3-GFP and Mup1-GFP (Figure 3A,C) fluorescence signals in the vacuole is decreased, and the proportion of GFP cleaved from the fusion proteins also decreases (Figure 3B,D). These data indicate that Mih1 acts downstream of endocytosis to regulate the abundance of Chs3 and Mup1 on the plasma membrane, strongly suggestive of a role in endocytic recycling.
Figure 3
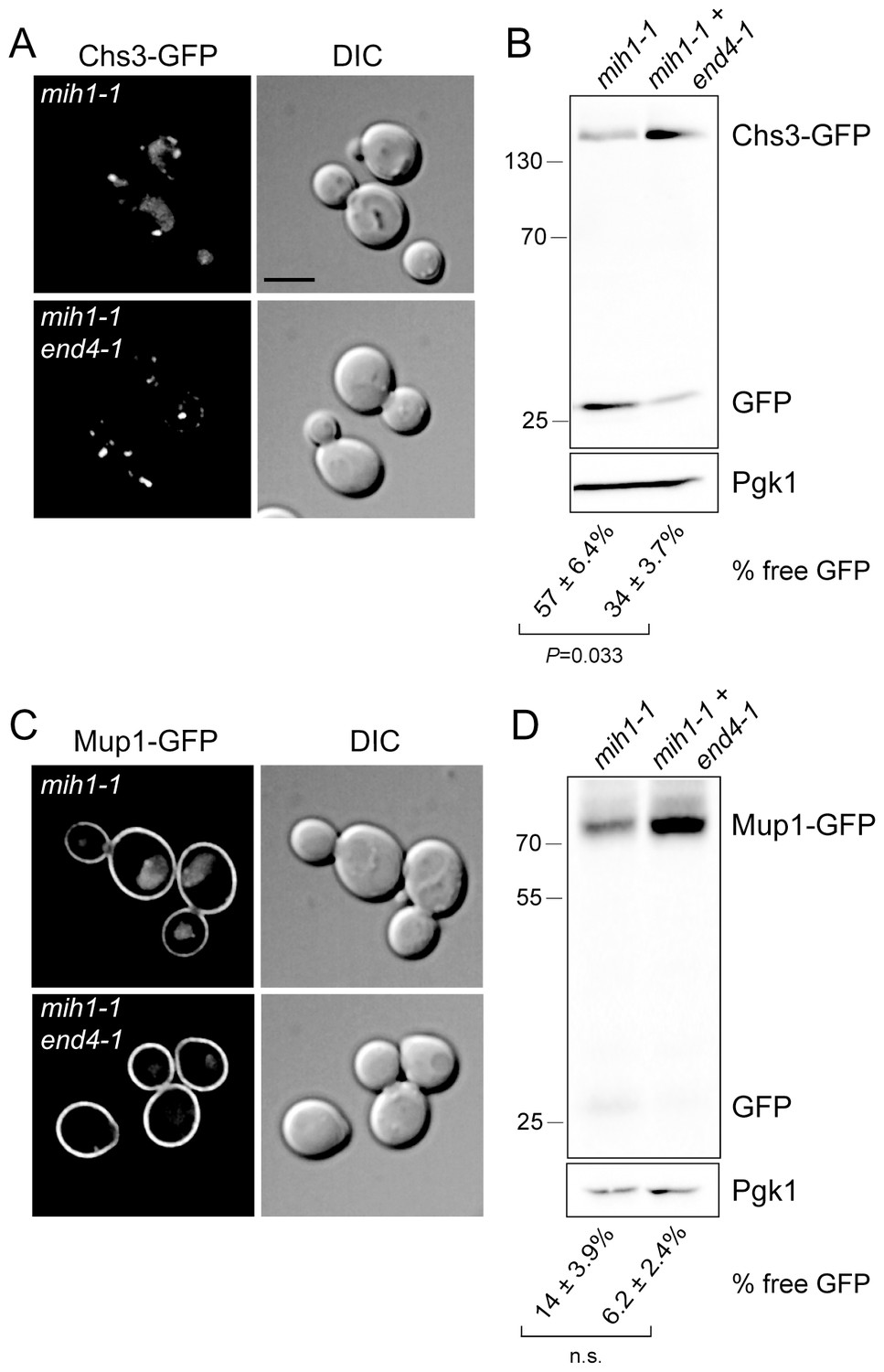
Mih1 regulates protein sorting at the endosome.
(A) Endocytosis promotes vacuolar localization of Chs3-GFP in mih1-1 cells. Micrographs showing Chs3-GFP in mih1-1 (chs6Δapl2Δmih1-1) and mih1-1end4-1 (chs6Δapl2Δmih1-1end4-1) cells are shown. A representative medial optical plane of a z-series is shown. Scale bar, 5 µm. (B) Cleavage of GFP from Chs3-GFP is attenuated by the end4-1 mutation. Anti-GFP blotting of cell lysates was used to quantify the pools of full length Chs3-GFP and free GFP. The number below each lane indicates the mean (+s.e.m.) proportion of free GFP to the total GFP signal in the lane calculated from a minimum of three independent measurements. Statistical significance is indicated. Anti-Pgk1 blotting was used to control for loading. The positions of molecular weight (kDa) of protein standards are indicated on the left side of the gel. (C) Vacuolar localization of Mup1-GFP requires endocytosis. Micrographs showing Mup1-GFP in mih1-1 (chs6Δapl2Δmih1-1) and mih1-1end4-1 (chs6Δapl2Δmih1-1end4-1) cells cultured in medium lacking methionine are shown. A representative medial optical plane of a z-series is shown. Scale bar, 5 µm. (D) Cleavage of GFP from Mup1-GFP is attenuated by end4-1 mutation. The data are presented as described in the legend to panel B. Due to the small amount of free GFP, the difference is not statistically significant (n.s.).
Plasma membrane recycling of Chs3 requires retromer
Sorting nexins are components of endosomal recycling/retrograde sorting devices (Cullen and Korswagen, 2012) and we therefore speculated that one or more sorting nexins are required for Chs3 recycling in the absence of AP1-mediated recycling (i.e., as in chs6Δapl2Δ parental cells). To test this, CFW resistance of cells deleted of a sorting nexin with an established role in endocytic recycling introduced into the parental chs6Δapl2Δ Chs3-GFP was examined (Figure 4A). This screen revealed that deletion of vps5△ and vps17△, but no other sorting nexin deletion, including snx3Δ, which has been shown to ablate plasma membrane recycling of Ftr1-GFP (Strochlic et al., 2007), conferred resistance to CFW comparable to the chs6Δapl2Δmih1-1 strain. It is noteworthy that Vps5 and Vps17 form a SNX-BAR heterodimer that functions as a component of the retromer sorting complex (Horazdovsky et al., 1997; Seaman et al., 1998) and, indeed, deletion of the gene encoding each retromer subunit also confers resistance to CFW (Figure 4A) and Chs3-GFP is localized prominently to the vacuole lumen (Figure 4B). These results indicate that Chs3 is exported from the endosome via a retromer-dependent recycling pathway.
Figure 4

Mih1 regulates the phosphorylation state of Vps26 retromer subunit.
(A) Growth of strains lacking genes encoding endosomal sorting nexins and retromer deletions in the presence of CFW. Serial dilutions of control (chs6Δapl2Δ), mih1-1 (chs6Δapl2Δmih1-1), snx3Δ (chs6Δapl2Δsnx3Δ), snx4Δ (chs6Δapl2Δsnx4Δ), vps5Δ (chs6Δapl2Δvps5Δ), vps17Δ (chs6Δapl2Δvps17Δ), mvp1Δ (chs6Δapl2Δmvp1Δ), vps26Δ (chs6Δapl2Δvps26Δ), vps29Δ (chs6Δapl2Δvps29Δ) and vps35Δ (chs6Δapl2Δvps35Δ) cells were spotted onto rich medium with or without 50 μg/ml CFW for three days at 30°C. (B) Localization of Chs3-GFP in control (chs6Δapl2Δ), mih1-1 (chs6Δapl2Δmih1-1) and vps29Δ (chs6Δapl2Δvps29Δ) (as a representative retromer deletion). A representative medial optical plane of a z-series is shown. Scale bar, 5 µm. (C) Endogenously expressed Vps35-GFP, Vps29-GFP, Vps26-GFP, Vps17-GFP or Vps5-GFP fusion proteins were immunopurified from chs6Δapl2Δmih1Δ cells and samples of the collected material were analyzed by immunoblotting with a pan phosphoryl amino acid antiserum (top) or anti-GFP (bottom). An amount of cell lysate equivalent to 0.5% of the starting material for the purifications was run in the ‘load’ lanes. The positions of molecular weight (kDa) protein standards are indicated on the left side of the gels. (D) Vps26 phosphorylation status. Endogenously expressed Vps26-GFP fusion protein was immunopurified from chs6Δapl2Δ (control), chs6Δapl2Δmih1-1 (mih1-1) and chs6Δapl2Δmih1Δ (mih1Δ) cells and subjected to immunoblot assay with a pan phosphoryl amino acid antiserum (top) or anti-GFP (bottom). The molecular weight (kDa) of protein standards is indicated on the left side of the gels. (E) Purified Mih1 proteins. The indicated Mih1 proteins were purified from yeast cell lysates. A coomassie blue stained gel of aliquots of the eluted fractions used for activity assays is shown. (F) In vitro assay of Vps26 dephosphorylation by Mih1. Purified Mih1 proteins or lambda protein phosphatase were incubated with immunopurified Vps26-GFP. Equivalent portions of each reaction were examined by immunoblot with a pan phosphoryl amino acid antiserum (top) or anti-GFP (bottom). ‘Mock’ indicates a reaction that received no purified phosphatase.
Regulation of retromer by Mih1
Deficient trafficking of multiple proteins (Chs3, Mup1, and Can1) in the endosomal system of mih1-1, mih1Δ, and retromer null cells leads us to speculate that Mih1 regulates the activity of a general endosomal sorting and trafficking device – retromer – via dephosphorylation of one or more retromer subunits. To test this, we immunopurified GFP-tagged forms of each retromer subunit from chs6△apl2△ and chs6△apl2△mih1△ cells, and then probed the precipitates with a pan phosphoryl–amino acid antiserum (Figure 4C). We observe an accumulation of phosphorylated Vps26-GFP in cells lacking Mih1 (chs6△apl2△mih1△); we also note that a phosphorylated form of Vps35-GFP was detected, but only in a subset of these experiments (not shown). Phosphorylated Vps26-GFP could also be detected after GFP immuno-purification from wild-type cell lysate, but not in lysate prepared from mih1-1 cells (Figure 4D), consistent with the mih1-1 allele conferring enhanced activity, as predicted from the genetic analyses.
Changes in the phosphorylation status of Vps26 in mih1-1 and mih1Δ cells raise the possibility that phosphorylated Vps26 is a substrate of Mih1, though this is unexpected, as CDC25 family phosphatases are thought to be dedicated toward CDKs (Boutros et al., 2007). We tested this by assaying the activities of purified Mih1, Mih1-1, and the catalytically inactive C320S form of Mih1, toward phosphorylated Vps26. Mih1 proteins were purified from yeast strains that overexpress each protein (Figure 4E), and Vps26-GFP was purified from a mih1Δ strain. The results clearly show that the wild-type and Mih1-1 phosphatases, but not Mih1(C320S), dephosphorylate Vps26, which was confirmed by comparing to the products of a reaction using pure protein phosphatase derived from bacteriophage lambda (Figure 4F). Accumulation of phosphorylated Vps26 in mih1Δ cells, decreased phosphorylation of Vps26-GFP in mih1-1 cells, and the demonstration that phosphorylated Vps26 is a substrate for purified Mih1 in vitro, lead us to conclude that Vps26 is a bona fide substrate of Mih1 in vivo.
Ten phosphorylated residues in Vps26 have been identified (Stark et al., 2010) (Bodenmiller et al., 2008), notably including five serine residues located within a loop that connects two β strands (corresponding to ‘loop 6’ in the structure of mammalian Vps26), as well as several positions corresponding to structurally significant features of arrestin family proteins, to which Vps26 is structurally related. To address the significance of Vps26 phosphorylation status controlled by Mih1, we first compared localizations and steady state abundances of GFP- and/or FLAG epitope-tagged Vps26 and Vps35 in wild-type, mih1-1, and mih1Δ cells; no differences were noted between these strains, suggesting that Mih1 does not control organelle targeting or turnover of retromer proteins (not shown). Therefore, as an indicator of retromer function, we assessed CFW resistance of mutant strains in which annotated phosphorylated residues in Vps26 were changed to alanine (Ala), and observed that no single point mutation affected CFW resistance of the cells (not shown). We therefore removed most of loop 6 (22 of 33 amino acids) including the five annotated phosphorylated serine and threonine residues, using the structure of human Vps26A (Shi et al., 2006) as a guide to avoid distorting the global fold of the mutant protein. Deletion of loop 6 (‘Vps26ΔL6’) confers increased sensitivity to CFW (Figure 5A), suggesting that this enhances plasma membrane localization of Chs3-GFP. That is, deletion of this loop appears to potentiate retromer-mediated recycling of Chs3. Consistent with residues of loop 6 being targets of Mih1 activity, the amount of phosphorylated Vps26ΔL6 detected by immunoblotting was reduced by ~50% compared to native Vps26 (Figure 5B). Next, each of the known and potential sites of phosphorylation of loop 6 were substituted with a phosphomimetic or non-phosphoryl residues and growth on CFW medium was scored (Figure 5C). Similar to cells with the loop 6 deletion, substitution of all serine (Ser) and threonine (Thr) residues with glutamate (Glu) results in enhanced CFW sensitivity (Figure 5C, rows 1, 5), suggesting that phosphorylation of these residues potentiates retromer-mediated recycling. Importantly, these substitutions exerted this effect in the context of the mih1-1 mutation (Figure 5C, compare rows 2 and 3), correlating the charge on these residues with the CFWR phenotype of mih1-1 cells. In contrast, substitution of these residues with non-phosphoryl alanine residues conferred modest resistance CFW (Figure 5C, compare rows 1 and 8), indicating that these substitutions diminish retromer function. Thus, the data argue that Mih1 regulates retromer-dependent recycling in part by controlling the phosphorylation status of Vps26 loop 6 residues. We note that the data also suggest Mih1 has additional targets that affect the growth response to CFW, as deletion of mih1 is epistatic to the Vps26 loop 6 (A) substitutions (Figure 5C, compare rows 7 and 8).
Figure 5
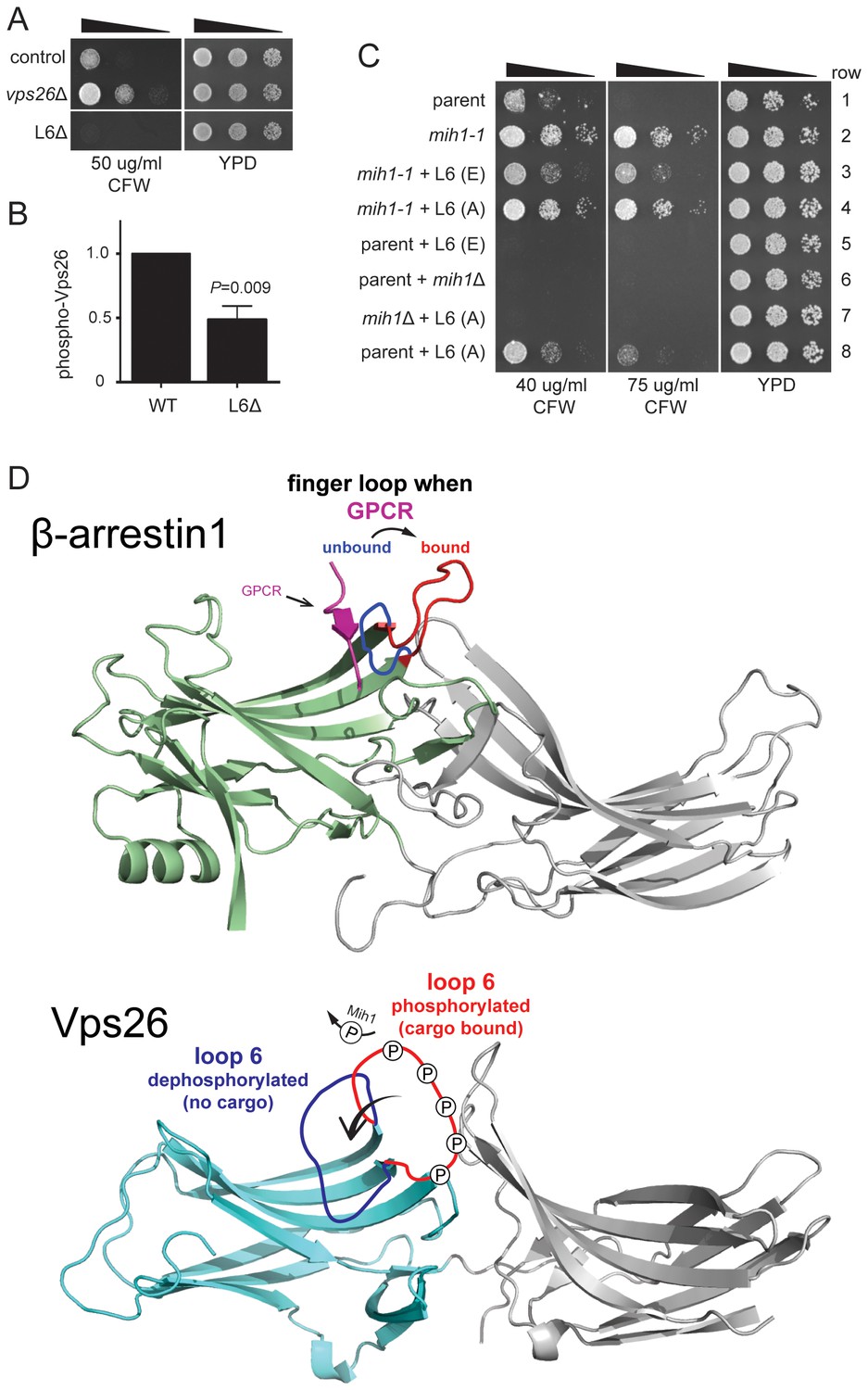
Substitutions within Vps26 loop 6 influence cargo recycling.
(A) Deletion of loop 6 of Vps26 confers CFW sensitive growth. Serial dilutions of control (chs6Δapl2Δ), vps26Δ (chs6Δapl2Δvps26Δ) and L6Δ (chs6Δapl2Δvps26ΔL6) cells were spotted on YPD medium with or without CFW at the indicated concentration for three days at 30°C. (B) Vps26 loop 6 is de-phosphorylated by Mih1 in vivo. The amount of phosphorylated Vps26ΔL6 -GFP relative to native Vps26-GFP is shown. The mean (+s.e.m.) of three independent determinations is plotted. Statistical significance is indicated. (C) Phospho-mimetic substitutions in loop 6 of Vps26 confer sensitivity to CFW. Serial dilutions of control (chs6Δapl2Δ), mih1-1 (chs6Δapl2Δmih1-1), mih1−1 + L6 (E) (chs6Δapl2Δmih1-1vps26-loop 6 (E)), mih1−1 + L6 (A) (chs6Δapl2Δmih1-1vps26-loop 6 (A)), L6 (E) (chs6Δapl2Δvps26-loop 6 (E)), mih1Δ (chs6Δapl2Δmih1Δ), mih1Δ + L6 (A) (chs6Δapl2Δmih1Δvps26-loop 6 (A)) and L6 (A) (chs6Δapl2Δvps26-loop 6 (A)) cells were spotted on YPD medium with or without CFW at the indicated concentration for three days at 30°C. (D) Top: Structure of human β-arrrestin1 bound to a peptide derived from the sequence of the V2 vasopressin receptor (Shukla et al., 2013) (PDB: 4JQI). The ‘finger loop’, which corresponds to loop 6 of Vps26, is shown in the inactive (i.e., GPCR unbound) and active (GPCR bound) conformations to illustrate the manner in which it occludes GPCR binding in the inactive state. Bottom: Structure of human Vps26a (Shi et al., 2006) (PDB: 2FAU) with the extended loop 6 of yeast modeled in the dephosphorylated and phosphorylated conformations. We suggest that phosphorylation relieves an auto-inhibited conformation of the loop that antagonizes cargo recognition and that dephosphorylation by Mih1 restores the auto-inhibited state.
Retromer contributes to Chs3 recycling in wild-type cells (Arcones et al., 2016), and especially in chs6Δapl2Δ cells (Figure 4A). Thus, Chs3 should possess a recycling signal that is recognized by retromer. As previously noted (Weiskoff and Fromme, 2014), a ‘YYL’ sequence (amino acids 12–14) located within the N-terminal cytoplasmic segment of Chs3 is a candidate and the results of three experiments confirmed this. First, substitution of these three residues with Ala results in CFW resistance (Figure 6A). Second, an immobilized fusion protein composed of the first 52 amino acids of Chs3 fused to glutathione S transferase (GST) captures both Vps26 and Vps35 retromer subunits from cell lysates (Figure 6B). We note that a previous report found that isolated human Vps26 recognizes the retromer sorting motif of the SorLA protein (Fjorback et al., 2012), and that in our experiments Vps26 was slightly, but reproducibly enriched on this matrix relative to Vps35. As Vps26 is the least tightly associated yeast retromer subunit (Seaman et al., 1998), these data suggest that Vps26 may directly recognize GST-Chs3(1-52) while Vps35 capture is indirect. Third, ligand-binding assays employing pure recombinant Vps26 and GST-Chs3(1-52) shows that Vps26 does indeed recognize GST-Chs3(1-52) (Figure 6C), while there is no detectable binding to GST with an irrelevant C-terminal sequence (Figure 6C). Binding of Vps26 to GST-Chs3(1-52) is saturable, with an estimated equilibrium dissociation constant, KD, of 19 μM (Figure 6D). In contrast, binding of Vps26 to a mutant GST-Chs3 fusion protein in which the YYL sequence was changed to AAA was not saturated, even at 100 μM Vps26 (KD >40 μM). These results confirm that Vps26 recognizes this sequence and, in light of the data demonstrating retromer-dependent trafficking of Chs3, indicate that this sequence constitutes a retromer-dependent sorting signal.
Figure 6
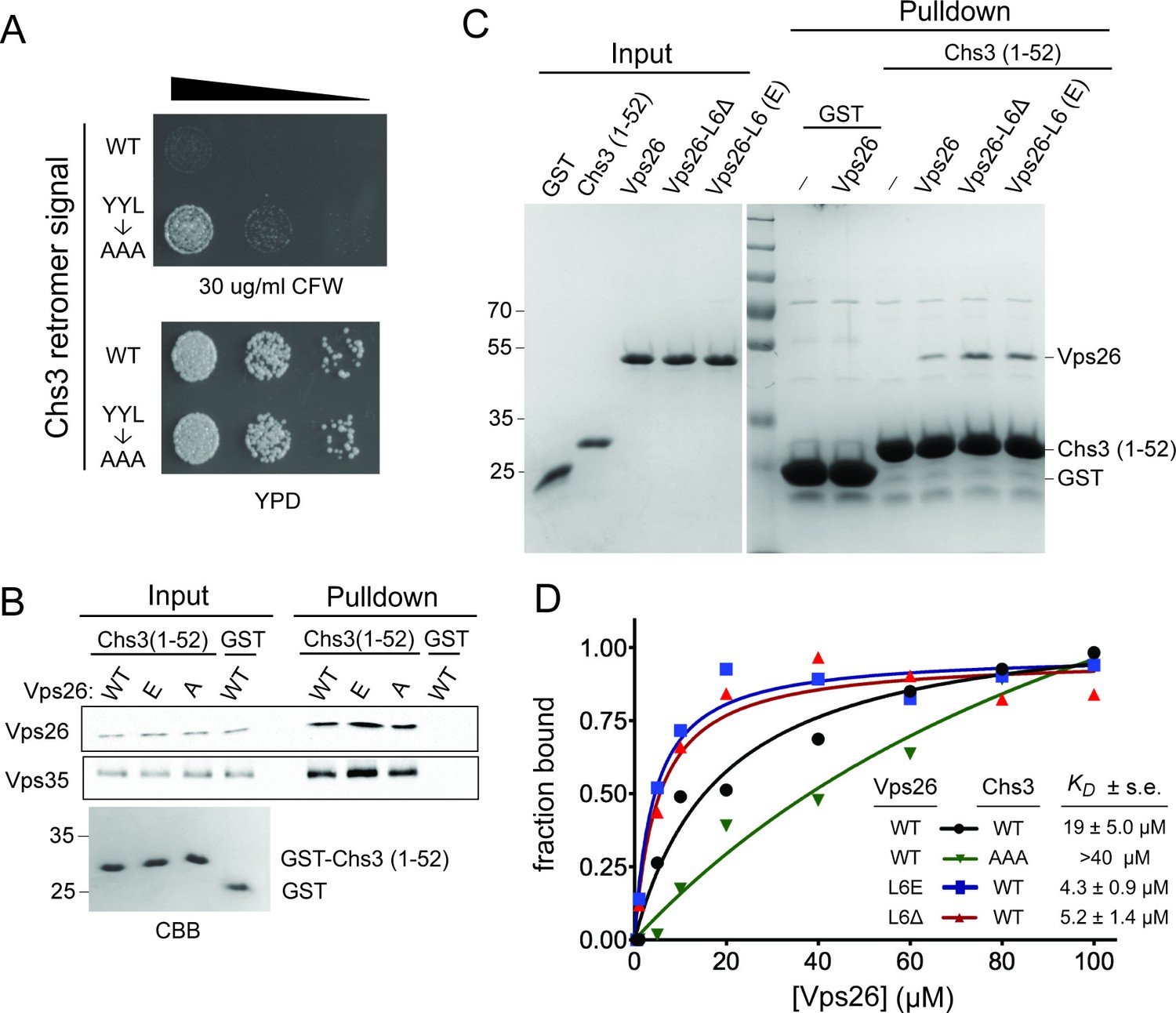
Vps26 loop 6 controls affinity for a retromer sorting signal.
(A) Substitution of a putative retromer sorting motif in Chs3 confers resistance CFW. Chs3 residues Y12YLwere changed to alanine in chs6Δapl2Δ cells. Serial dilutions of control (chs6Δapl2Δ) and mutant cells were spotted on calcofluor plates (30 μg/ml) and grown for three days at 30°C. (B) The Vps26 and Vps35 retromer subunits are captured by beads presenting Chs3 residues 1–52. Lysates from cells expressing GFP tagged Vps26 (or its variants) or Vps35-FLAG were incubated with immobilized GST-Chs3 (amino acids 1–52; ‘GST-1–52’) or GST. An amount of cell lysate equivalent to 0.5% of the starting material for the purifications was run in the ‘load’ lanes. The relative enrichment of each protein in the pulldown fraction (means and standard deviations of three independent experiments) is indicated. A coomassie blue-stained gel of 3% of each starting affinity resins is shown below the immunoblots. The molecular weight (kDa) of protein standards is indicated on the left side of the gels. (C) Vps26 binds directly to the YYL Chs3 sorting signal. Binding assays of pure, 6xHis-tagged wild-type Vps26, Vps26 loop 6 deletion (Vps26ΔL6) and loop 6 phosphomimetic Vps26 (Vps26-L6(E) to GST-Chs3(1-52) or the same portion of Chs3 with alanines substituted for the YYL sequence (GST-Chs3(YYL->AAA)) are shown. The input fractions show one microgram of the starting material used for each assay. The positions of molecular weight (kDa) protein standards are indicated on the left side of the gel. (D) Loop 6 controls affinity of Vps26 for a retromer sorting signal. Equilibrium binding measurements of pure, recombinant wild-type Vps26, Vps26L6Δ, and Vps26-L6(E) for the GST-Chs3(1-52) fusion protein, and wild-type Vps26 for the GST-Chs3(YYL->AAA) fusion protein, are shown. The points plotted in the graphs represent the mean fraction of bound Vps26 proteins, calculated from single measurements using three independent protein preparations of each Vps26 protein. The calculated KD values (±s.e.) are listed. Saturation binding to the GST-Chs3(YYL->AAA) fusion protein was not observed, so a dissociation constant could not be calculated.
As the results of the cell-based experiments suggest that loop 6 impedes retromer-dependent recycling of Chs3, we measured the affinity of Vps26 lacking this loop (Vps26ΔL6) to GST-Chs3(1-52). This form of Vps26 exhibits a nearly 4-fold higher affinity for GST-Chs3(1-52) (KD of 5.2 μM) (Figure 6D). In close agreement with this measurement, the loop 6 phosphomimetic form of Vps26 (Vps26(L6E) binds GST-Chs3(1-52) with a KD of 4.3 μM (Figure 6D). Taken together, the data correlate an increase in the affinities with which the Chs3 recycling signal is recognized by Vps26 with sorting outcome; an increase in the affinity of cargo resulting from phosphorylation of loop 6 results in enhanced recycling of Chs3.
How does phosphorylation of Vps26 control the affinity with which Vps26 recognizes cargo? A role for Vps26 in cargo recognition had been raised previously on the basis of its structural homology to arrestin proteins (Collins et al., 2008; Shi et al., 2006), which serve as cargo adapters for clathrin-mediated endocytosis of GPCRs (Kang et al., 2014; Moore et al., 2007), and the finding that Vps26 binds the retromer sorting motif of SorLA (Fjorback et al., 2012). Atomic structures of activated β-arrestin1 (Kim et al., 2013), especially that of β-arrestin1 bound to a phosphorylated peptide derived from the human V2 vasopressin receptor (termed ‘V2Rpp’) (Shukla et al., 2013) (Figure 5D), potentially provide remarkable insight into this question. Arrestins are composed of two tandem β sandwich domains whose relative orientation toggles between an inactive conformation and an active conformation that binds ligand-activated, phosphorylated GPCR (Kim et al., 2013; Shukla et al., 2013, 2014). The phosphorylated C-terminal segment of GPCR binds to the N-terminal domain in a concave ‘cup’ that is partly occluded in the inactive conformation by a loop, termed the ‘finger loop,’ that connects two strands within a β sheet (Figure 5D) (Shukla et al., 2013). In the active, receptor-bound conformation of β-arrestin1, the finger loop extends from the domain, allowing it to contact the membrane core of the GPCR and exposing the binding site of the GPCR C-terminal segment (Figure 5D) (Shukla et al., 2013; Szczepek et al., 2014).
The finger loop of β-arrestin1 corresponds to the phosphorylated loop 6 of Vps26 (Figure 5D). It is unlikely that loop 6 directly recognizes the retromer sorting signal. Rather, we suggest that it functions as a gate that controls access of the β sheet of N-terminal domain to cargo (Figure 5D). In its unphosphorylated state, the extended loop would antagonize cargo engagement via an auto-inhibited conformation, akin to the conformation of the finger loop of β-arrestin1 in the inactive state, and phosphorylation of loop residues would displace it from occluding access to the surface of the β sheet, akin to the displacement of the β-arrestin-1 finger loop in the active conformation. This mechanism provides for a tunable system for adjusting the abundance of proteins on the cell surface via recycling regulated by kinase-phosphatase networks that are responsive to metabolic and environmental cues. Surprisingly, neither the mih1-1 or mih1Δ mutations affect the distribution of Vps10, a well characterized retromer cargo that cycles between the Golgi apparatus and endosome (Figure 2—figure supplement 1), indicating the existence of at least one additional mode of cargo recognition by retromer, speculated to be mediated by the Vps35 retromer subunit (Nothwehr et al., 1999, 2000). Consistent with this, a recently published structure of a Vps35-Vps26-Snx3 complex bound to a peptide containing a retromer sorting signal revealed an additional mode of cargo recognition involving the Snx3-Vps26 interface (Lucas et al., 2016). Multiple modes of cargo recognition by retromer provide a mechanistic explanation for the diversity of cargo-specific sorting factors that function with retromer, such as the Snx3, SNX-BAR, and Snx27 sorting nexins (Cullen, 2008; Cullen and Korswagen, 2012; Strochlic et al., 2007). As cargo recognition plays a key role in targeting soluble retromer to the endosome membrane (Harrison et al., 2014; Lucas et al., 2016), an important implication of our findings is that distinct cargo recognition modes could drive the formation of distinct retromer-containing complexes on the endosome membrane.
A future challenge is to identify the signaling pathways and regulatory inputs that modulate plasma membrane composition via retromer-mediated recycling. Importantly, the identification of Mih1 as a physiological regulator of retromer function reveals a new facet of function for CDC25 family phosphatases, which are currently thought to act solely on cyclin-dependent kinases to control cell cycle progression (Boutros et al., 2007). The activities of CDC25 family phosphatases are subject to intricate regulation via kinases and phosphatases that respond to a variety of cellular stimuli and impinge on various cell cycle checkpoints. One possible regulator of Mih1 function with regard to Chs3 recycling is PKC, which is has been shown to control Mih1 signaling (Yano et al., 2013) and to promote Chs3 trafficking from the TGN to the plasma membrane via the exomer pathway (Valdivia and Schekman, 2003). As PKC is a core component of the cell wall integrity pathway, Mih1-mediated regulation of Chs3 recycling by retromer might reflect a stress response to defects in the cell wall.
The results presented here indicate that, in addition to the role that yeast Mih1 plays in cell cycle progression, it also controls trafficking of plasma membrane proteins that impact nutrient homeostasis and cell wall remodeling – key axes of cell growth control. Over-expression of CDC25 proteins is observed in some human cancers, leading to speculation that gain-of-function dysregulation, akin to the Mih1-1 protein characterized in this study, contributes to cell transformation. However, it has not been possible to attribute the effects of increased CDC25 expression levels solely to inappropriate progression through the cell cycle and our study raises the possibility that this may be due, in part, to unappreciated activities of CDC25 proteins on non-CDK substrates.
Materials and methods
Yeast strains and culture conditions
Request a detailed protocolAll yeast strains were constructed in BY4742 (MATα his3-1, leu2-0, met15-0, and ura3-0) (Open Biosystems/Thermo Scientific, Waltham, MA) by homologous recombination of gene-targeted, polymerase chain reaction (PCR)-generated DNAs using the method of Longtine et al (Longtine et al., 1998). Mutant strains were derived either from the EUROSCARF KANMX deletion collection (Open Biosystems/Thermo Scientific, Waltham, MA) or by replacement of the complete reading frame with a HIS3MX6, NATMX6 or URA3 cassette. The introduction of point or truncation mutations into the genome was performed according to the established methods (Gray et al., 2004, 2005; Toulmay and Schneiter, 2006). To make the Vps26-E mutant, all serine and threonine codons in loop 6 region of Vps26 (87–113 aa) were replaced by glutamate codons; To make the Vps26-A mutant, all serine and threonine codons in loop 6 region of Vps26 were replaced by alanine codons. To express TAP-tagged Mih1, a TAP tag DNA construct was integrated immediately upstream of the native stop codon of strain TVY614 (MATα, ura3–52, his3-Δ200, trp1-Δ 901, lys2–801, suc2-Δ9, leu2–3, 11, pep4Δ::LEU2, prb1Δ::HISG, prc1Δ::HIS3). Next, DNAs generated by PCR using pFA-kanMX6-PGAL1 and pBS1539 as templates were used to integrate the GAL1 promoter immediately upstream of the native MIH1 start codon and a TAP tag immediately upstream of the native stop codon, respectively. To construct similar strains expressing the mih1-1 or C320S alleles, the MIH1 locus of TVY614 was first replaced with the URA3 gene. These strains were then transformed with DNA generated by PCR of the mih1 locus using genomic DNA from strains containing the mih1-1 or C320S alleles as template. Transformants were selected on plates containing 5-fluoroorotic acid and replacement of the URA3 locus by the mih1 allele was confirmed by DNA sequencing. Next, a TAP tag and the GAL1 promoter were integrated as described for the native MIH1 locus. All newly constructed strains were confirmed to present relevant autotrophic/drug resistance markers (indicating that they were derived from the BY4742 parent) and chromosomal manipulations were confirmed by PCR amplification and DNA sequencing of the relevant locus.
Cells were grown in rich YP medium (2% bacto-peptone, 1% yeast extract, 2% glucose), or complete synthetic medium (Sunrise Science Product, San Diego, CA). Corresponding nutrient(s) were omitted from media to maintain selection for auxotrophic markers and/or plasmids (Sherman et al., 1979). Cells used to produce TAP-tagged Mih1 were grown at 23°C in YP +2% raffinose+0.1% glucose until an OD600 ≈ 0.6, when galactose was added to 2% and growth continued overnight. To obtain calcofluor-resistant mutants, chs6Δapl2Δ cells were spread onto YPD plates containing 100 μg/mL CFW, and the plates were incubated at 30°C for three days. Colonies were picked and re-streaked on a CFW plate. To identify mutant strains with CFWR-conferring mutations in expected genes, each mutant strain was mated to chs3Δ, chs4Δ, chs5Δ, chs7Δ and pfa4Δ strains (in the BY4741 background) and CFWR was evaluated. If the resulting diploid cells were CFWR, the candidate was determined to have a recessive mutation in the gene which is lacking in the mating partner and the strain was discarded. Candidates that were not complemented by any ‘tester’ strain were retained. Whole genome DNA sequencing of the parent and mih1-1 strains was used to identify the lesion in the mih1-1 strain. DNA sequencing was performed by the Iowa Institute of Human Genetics, University of Iowa using the Illumina HiSeq platform. Genome assembly was done using Lasergene software (DNASTAR, Madison, WI).
For spot growth assays, cells from cultures were grown to OD600 ≈ 0.5 in liquid YPD medium were washed with water and fresh YPD medium, and then a 10-fold dilution series was spotted onto plates with a series of concentrations of CFW (0–150 μg/mL). The plates were assessed after 2–3 days of incubation at 30°C.
Immunoblot and immunoprecipitation
Request a detailed protocolCells from cultures were grown to OD600 ≈ 0.5 in liquid complete synthetic medium or YPD medium. Equal amounts of cells were collected and TCA-precipitated, and then proteins were resolved by SDS-PAGE. Primary antibodies in these studies include: anti-GFP (1:2000; Roche), anti-Pgk1 (3-phosphoglcerate kinase) (1:10,000; Invitrogen), anti-phosphoserine/threonine/tyrosine antibody (1:500; Abcam), anti-FLAG (1:5000; sigma), anti-His (1:1000; Biolegend) and anti-Phospho-cdc2 (Tyr15) (1:1000; Cell Signaling Technology).
For immunoprecipitation of GFP tagged retromer subunits, cells were grown to OD600 ≈ 0.5 in complete synthetic medium, and then were washed and frozen in liquid nitrogen prior to disruption by bead beater in lysis buffer (20 mM HEPES pH = 7.4, 150 mM KOAc, 5% glycerol, 1% NP40, proteinase inhibitor cocktail (Roche), PhosSTOP (Roche)). GFP-nAb magnetic beads (Allele Biotech) were used to capture GFP tagged proteins. After binding, the beads were extensively washed and then eluted with SDS-PAGE sample buffer.
Light microscopy and image analysis
Request a detailed protocolCells were grown to OD600 ≈ 0.5 in liquid medium were mounted in growth medium on a microscope slide. Image stacks were collected at 0.3-µm z increments on a DeltaVision workstation (Applied Precision) based on an inverted microscope (IX-70; Olympus) using a 100 × 1.4 NA oil immersion lens. Images were captured at 24°C with a front illuminated sCMOS, 2560 × 2160 pixels camera and deconvolved using the iterative-constrained algorithm and the measured point spread function. Image analysis and preparation were done using Softworx 6.1 (Applied Precision Instruments) and ImageJ v1.50d (Rasband, 2015).
Protein expression and purification
Request a detailed protocolPlasmids, pET28a-Vps26 and pGST-Chs3 (1-52 amino acid) (Weiskoff and Fromme, 2014), were used to express 6XHis-Vps26 and GST-Chs3 (1-52) fusion proteins in Escherichia coli. Cells were grown in LB media containing the appropriate antibiotics in baffled flasks filled to 20% of the total volume to OD600 ≈ 0.5–0.7 and IPTG was added to a final concentration of 0.5 mM and growth was continued for 3 hr at 37°C. Proteins were purified in batch using GSH beads (GE Healthcare Life Sciences) or Ni-NTA Agarose (Qiagen) and confirmed by SDS-PAGE and immunoblot with corresponding antibodies against each tag. Purified protein concentration was quantified by BCA assay (Pierce).
Frozen Mih1-TAP cells were thawed and then lysed (50 mM Tris, pH 7.4, 300 mM NaCl2, 1 mM DTT, proteinase inhibitor (Roche), 0.5%Triton) using an EmulsiFlex-C3 system (Avestin). Lysates were clarified by ultracentrifugation at 100,000 x g for 1 hr at 4°C. IgG agarose beads (GE Healthcare) were rinsed three times in lysis buffer. Beads were added to cleared lysate and incubated 4 hr at 4°C with rotation. Beads were washed (10 × 1 ml) and suspended in tobacco etch virus (TEV) protease buffer (50 mM Tris, pH 7.4, 150 mM NaCl2, 0.5 mM EDTA, 1 mM DTT). TEV protease was added to the buffer and incubated overnight at 4°C. To reduce the association of Mih1-TAP with IgG beads, IgG beads were precipitated and rinsed in calmodulin binding buffer (20 mM Tris, pH 7.4, 300 mM NaCl2, 4 mM CaCl2, 1 mM magnesium acetate, 1 mM imidazole, 10 mM 2-mercaptoethonal). Calmodulin magnetic beads were rinsed, suspended in calmodulin binding buffer, and added directly to precipitated IgG beads. The mixed beads were incubated at 4°C with rotation for 4 hr. Calmodulin magnetic beads were isolated magnetically and washed extensively. Mih1-TAP proteins were eluted with elution buffer (20 mM Tris, pH 8.0, 150 mM NaCl2, 10 mM EGTA and 10 mM 2-mercaptoethanol).
Protein phosphatase assay
Request a detailed protocolMih1, Mih1-1, and Mih1(C320S) were purified as described above. Vps26-GFP was immunoprecipitated from chs6Δapl2Δmih1Δ cells. The immobilized Vps26-GFP was incubated with purified Mih1 proteins in the phosphatase assay buffer (40 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 5 mM DTT) at 30°C for 1 hr. The immobilized Vps26-GFP was rinsed, boiled and subjected to immunoblotting with a pan phosphoryl amino acid antiserum.
In vitro binding assays
Request a detailed protocolPurified GST-Chs3 (1-52) or GST was immobilized to glutathione sepharose. Immobilized GST-Chs3 (1-52) or GST (1 μM) was then mixed with purified WT or mutant 6XHis-Vps26 proteins at the indicated concentration. After overnight incubation, the beads were washed (10 × 1 ml) with PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM TCEP, pH = 7.4). The bound 6XHis-Vps26 proteins were extracted from the beads with SDS-PAGE sample buffer and detected by immunoblot with an anti-His antibody (Biolegend). Immunoblot signals were quantified using Image Lab software (Biorad).
Statistical analyses
Request a detailed protocolPrism 7 software (GraphPad, La Jolla, CA) was used to calculate means, standard deviations and standard errors of mean values. The Student’s unpaired t test was used for statistical analyses. A p<0.05 value was considered to indicate statistical significance.
References
-
PhosphoPep--a database of protein phosphorylation sites in model organismsNature Biotechnology 26:1339–1340.https://doi.org/10.1038/nbt1208-1339
-
CDC25 phosphatases in cancer cells: key players? good targets?Nature Reviews Cancer 7:495–507.https://doi.org/10.1038/nrc2169
-
Retromer: a master conductor of endosome sortingCold Spring Harbor Perspectives in Biology 6:a016774.https://doi.org/10.1101/cshperspect.a016774
-
Dual specificity phosphatases: a gene family for control of MAP kinase functionFASEB Journal 14:6–16.
-
Biogenesis of endosome-derived transport carriersCellular and Molecular Life Sciences 72:3441–3455.https://doi.org/10.1007/s00018-015-1935-x
-
Endosomal sorting and signalling: an emerging role for sorting nexinsNature Reviews Molecular Cell Biology 9:574–582.https://doi.org/10.1038/nrm2427
-
Sorting nexins provide diversity for retromer-dependent trafficking eventsNature Cell Biology 14:29–37.https://doi.org/10.1038/ncb2374
-
Multiple repeat elements within the FAM21 tail link the WASH actin regulatory complex to the retromerMolecular Biology of the Cell 23:2352–2361.https://doi.org/10.1091/mbc.E11-12-1059
-
Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor traffickingCurrent Opinion in Cell Biology 27:63–71.https://doi.org/10.1016/j.ceb.2013.11.005
-
Palmitoylation by the DHHC protein Pfa4 regulates the ER exit of Chs3The Journal of Cell Biology 174:19–25.https://doi.org/10.1083/jcb.200602049
-
A Wee1 checkpoint inhibits anaphase onsetThe Journal of Cell Biology 201:843–862.https://doi.org/10.1083/jcb.201212038
-
Rab GTPase regulation of retromer-mediated cargo export during endosome maturationMolecular Biology of the Cell 23:2505–2515.https://doi.org/10.1091/mbc.E11-11-0915
-
Regulation of receptor trafficking by GRKs and arrestinsAnnual Review of Physiology 69:451–482.https://doi.org/10.1146/annurev.physiol.69.022405.154712
-
Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeast: identification of six new END genesThe Journal of Cell Biology 127:373–386.https://doi.org/10.1083/jcb.127.2.373
-
Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1The Journal of Cell Biology 180:931–945.https://doi.org/10.1083/jcb.200711014
-
Dual-specificity phosphatases: critical regulators with diverse cellular targetsBiochemical Journal 418:475–489.https://doi.org/10.1042/BJ20082234
-
Effect of Calcofluor white and Congo red on fungal cell wall morphogenesis: in vivo activation of chitin polymerizationJournal of Bacteriology 163:1180–1185.
-
A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeastThe Journal of Cell Biology 142:665–681.https://doi.org/10.1083/jcb.142.3.665
-
The retromer complex - endosomal protein recycling and beyondJournal of Cell Science 125:4693–4702.https://doi.org/10.1242/jcs.103440
-
BookMethods in Yeast Genetics: A Laboratory ManualCold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
-
The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domainNature Structural & Molecular Biology 13:540–548.https://doi.org/10.1038/nsmb1103
-
Grd19/Snx3p functions as a cargo-specific adapter for retromer-dependent endocytic recyclingThe Journal of Cell Biology 177:115–125.https://doi.org/10.1083/jcb.200609161
-
Distinct N-terminal regions of the exomer secretory vesicle cargo Chs3 regulate its trafficking itineraryFrontiers in Cell and Developmental Biology 2:47.https://doi.org/10.3389/fcell.2014.00047
Article and author information
Author details
Tabitha A Peterson

Christopher G Burd
Funding
National Institutes of Health (GM061221)
- Christopher G Burd
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Rob Piper (University of Iowa) for assistance with genome sequencing and analysis, Doug Kellogg (University of California, Santa Cruz) for discussions regarding Mih1, Chris Fromme (Cornell University) for the GST-Chs3(1-52) expression plasmid, and colleagues for critical comments on the manuscript. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number GM060221.
Version history
- Received: December 9, 2016
- Accepted: March 30, 2017
- Accepted Manuscript published: March 31, 2017 (version 1)
- Version of Record published: April 28, 2017 (version 2)
Copyright
© 2017, Cui et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,463
- Page views
-
- 445
- Downloads
-
- 11
- Citations
Article citation count generated by polling the highest count across the following sources: Crossref, PubMed Central, Scopus.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A CDC25 family protein phosphatase gates cargo recognition by the Vps26 retromer subunit
eLife 6:e24126.
https://doi.org/10.7554/eLife.24126
Further reading
-
- Biochemistry and Chemical Biology
- Medicine
Type 2 diabetes (T2D) is associated with higher fracture risk, despite normal or high bone mineral density. We reported that bone formation genes (SOST and RUNX2) and advanced glycation end-products (AGEs) were impaired in T2D. We investigated Wnt signaling regulation and its association with AGEs accumulation and bone strength in T2D from bone tissue of 15 T2D and 21 non-diabetic postmenopausal women undergoing hip arthroplasty. Bone histomorphometry revealed a trend of low mineralized volume in T2D (T2D 0.249% [0.156–0.366]) vs non-diabetic subjects 0.352% [0.269–0.454]; p=0.053, as well as reduced bone strength (T2D 21.60 MPa [13.46–30.10] vs non-diabetic subjects 76.24 MPa [26.81–132.9]; p=0.002). We also showed that gene expression of Wnt agonists LEF-1 (p=0.0136) and WNT10B (p=0.0302) were lower in T2D. Conversely, gene expression of WNT5A (p=0.0232), SOST (p<0.0001), and GSK3B (p=0.0456) were higher, while collagen (COL1A1) was lower in T2D (p=0.0482). AGEs content was associated with SOST and WNT5A (r=0.9231, p<0.0001; r=0.6751, p=0.0322), but inversely correlated with LEF-1 and COL1A1 (r=–0.7500, p=0.0255; r=–0.9762, p=0.0004). SOST was associated with glycemic control and disease duration (r=0.4846, p=0.0043; r=0.7107, p=0.00174), whereas WNT5A and GSK3B were only correlated with glycemic control (r=0.5589, p=0.0037; r=0.4901, p=0.0051). Finally, Young’s modulus was negatively correlated with SOST (r=−0.5675, p=0.0011), AXIN2 (r=−0.5523, p=0.0042), and SFRP5 (r=−0.4442, p=0.0437), while positively correlated with LEF-1 (r=0.4116, p=0.0295) and WNT10B (r=0.6697, p=0.0001). These findings suggest that Wnt signaling and AGEs could be the main determinants of bone fragility in T2D.
-
- Biochemistry and Chemical Biology
Heat stress can cause cell death by triggering the aggregation of essential proteins. In bacteria, aggregated proteins are rescued by the canonical Hsp70/AAA+ (ClpB) bi-chaperone disaggregase. Man-made, severe stress conditions applied during, e.g., food processing represent a novel threat for bacteria by exceeding the capacity of the Hsp70/ClpB system. Here, we report on the potent autonomous AAA+ disaggregase ClpL from Listeria monocytogenes that provides enhanced heat resistance to the food-borne pathogen enabling persistence in adverse environments. ClpL shows increased thermal stability and enhanced disaggregation power compared to Hsp70/ClpB, enabling it to withstand severe heat stress and to solubilize tight aggregates. ClpL binds to protein aggregates via aromatic residues present in its N-terminal domain (NTD) that adopts a partially folded and dynamic conformation. Target specificity is achieved by simultaneous interactions of multiple NTDs with the aggregate surface. ClpL shows remarkable structural plasticity by forming diverse higher assembly states through interacting ClpL rings. NTDs become largely sequestered upon ClpL ring interactions. Stabilizing ring assemblies by engineered disulfide bonds strongly reduces disaggregation activity, suggesting that they represent storage states.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}