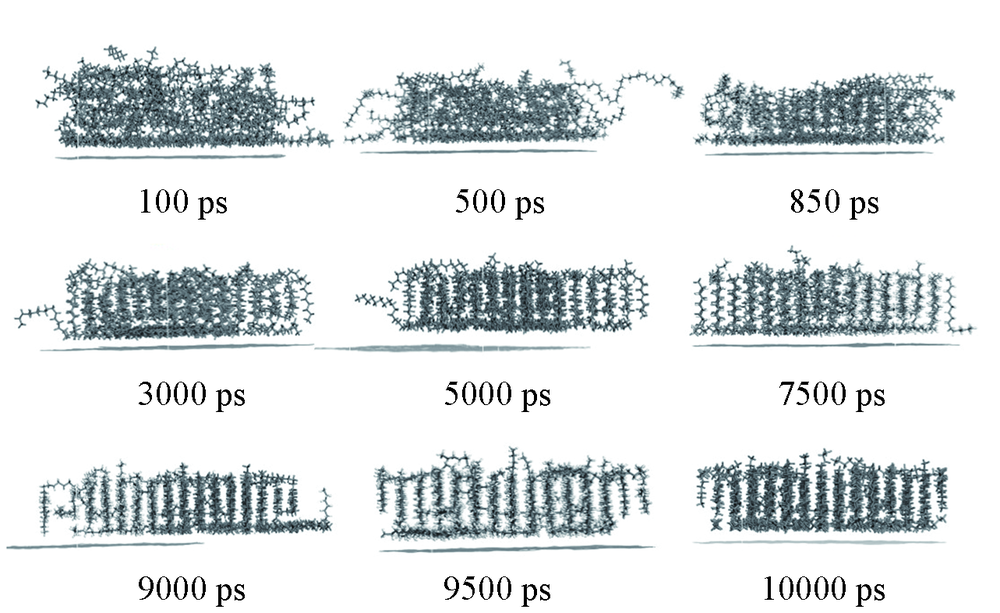
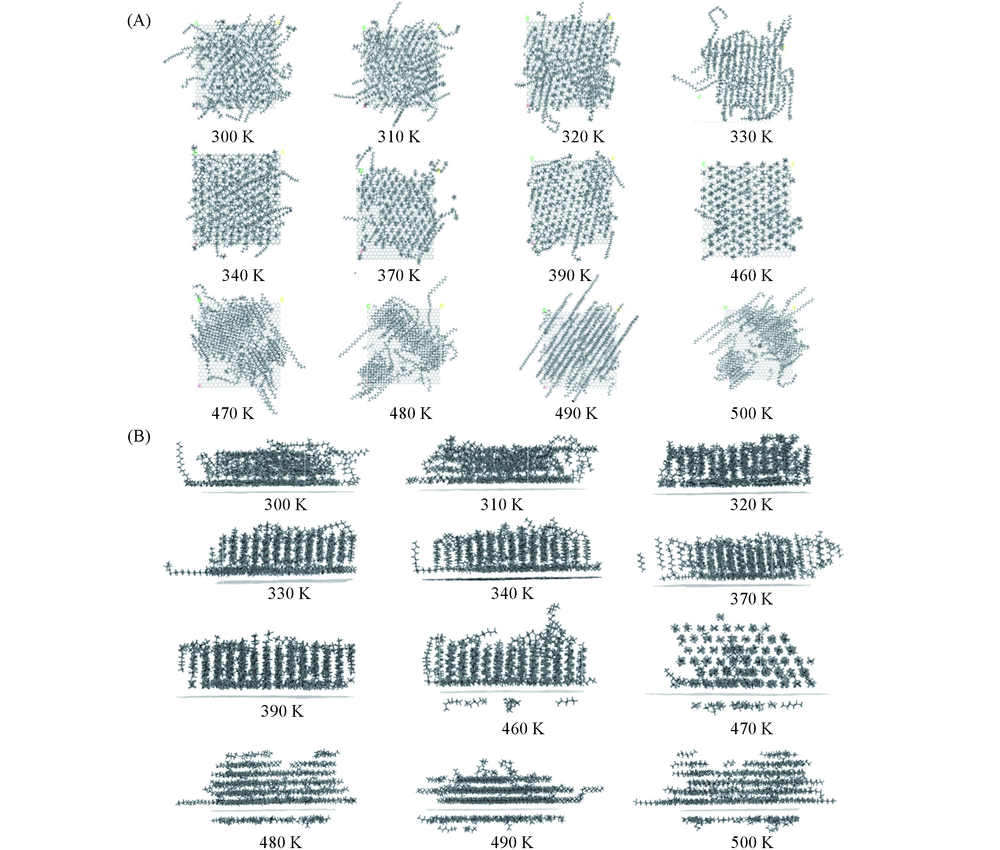
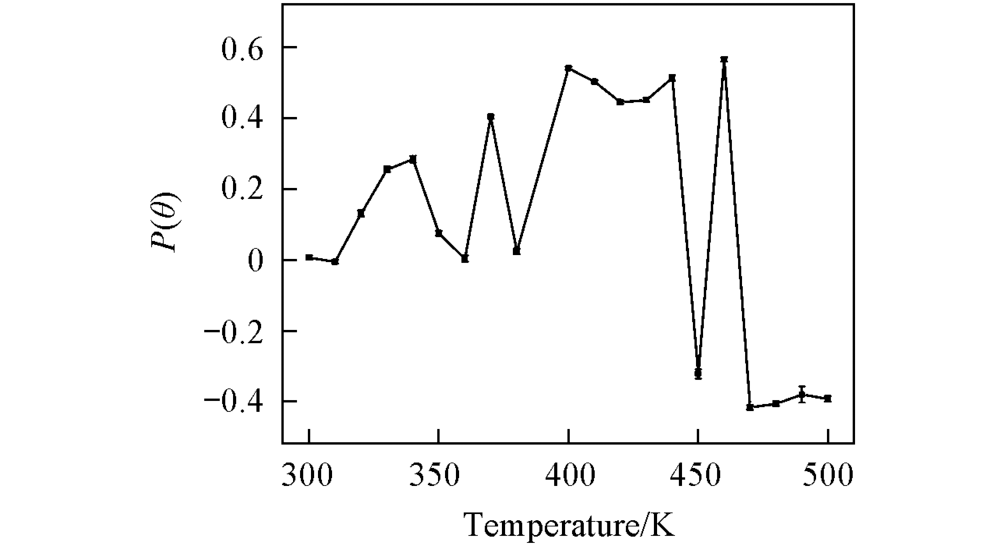
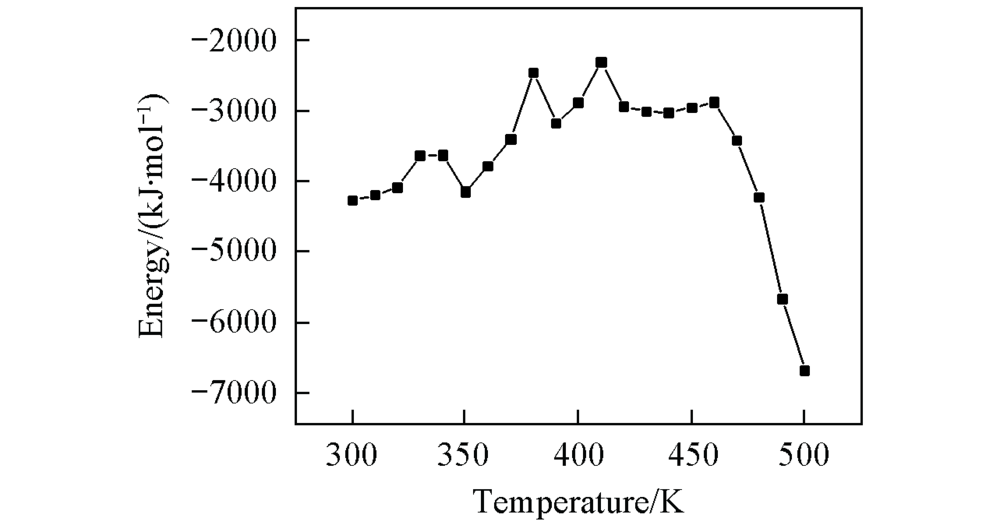
| [1] |
Maldonado E., Roth M.W., Gray P. A., ACS Appl. Mater. Inter., 2009, 1(6), 1211—1217
|
| [2] |
Herwig K.W., Matthies B., Taub H, Phys. Rev. Lett., 1995, 75, 3154—3157
|
| [3] |
Becker K.E., Fichthorn K. A., J. Chem. Phys., 2006, 125, 184706
|
| [4] |
Diama A., Matthies B., Herwig K.W., Hansen F. Y., Criswell L., Mo H., Bai M., Taub H., J. Chem. Phys., 2009, 131, 084707
|
| [5] |
Firlej L., Kuchta B., Roth M.W., Connolly M. J., Wexler C., Langmiur, 2008, 24, 12392—12397
|
| [6] |
Kamiya K., Okada S., Jap. J. Appl. Phys., 2013, 52, 06GD10
|
| [7] |
Liu X.Q., Xue Y., Tian Z. Y., Mo J. J., Qiu N. X., Chu W., Xie H. P, Appl. Surf. Sci., 2013, 19, 190—197
|
| [8] |
Thierfelder C., Witte M., Blankenburg S., Rauls E., Schmidt W.G., Surf. Sci., 2011, 7, 746—749
|
| [9] |
Ricca A., Bauschlicher C.W., Chem. Phys., 2016, 324, 455—458
|
| [10] |
Liu X.Q., Tian Z. Y., Chu W., Xue Y, Acta Phys.-Chim. Sin., 2014, 30(2), 251—256
|
|
(刘晓强, 田之悦, 储伟, 薛英. 物理化学学报, 2014, 30(2), 251—256)
|
| [11] |
Gutierrez-Maldonado S.E., Garate J. A., Retamal M. J., Cisternas M. A., Volkmann U. G., Perez-Acle T, Chem. Phys. Lett., 2017, 674, 64—70
|
| [12] |
Gao J.P., Luedtke W. D., Landman U., J. Chem. Phys., 1997, 106, 4309—4318
|
| [13] |
Enevoldsen A.D., Hansen F. Y., Diama A., Taub H., Dimeo R. M., Neumann D. A., Copley J. R. D., J. Chem. Phys., 2007, 126, 104704
|
| [14] |
Hansen F.Y., Criswell L., Fuhrmann D., Herwig K. W., Diama A., Dimeo R. M., Neumann D. A, Phys. Rev. Lett., 2004, 92, 046103
|
| [15] |
Klein J., Kumacheva E., Science, 1995, 269, 816—819
|
| [16] |
Zhao L., Yang H., Li Z., Li Z.S., Sun J. Z., Chem. J. Chinese Universities, 2006, 27(7), 1340—1342
|
|
(赵莉, 杨华, 李卓, 李泽生, 孙家锺. 高等学校化学学报, 2006, 27(7), 1340—1342)
|
| [17] |
Yu X., Han M., Yang X.Z., Chem. J. Chinese Universities, 2011, 32(1), 180—184
|
|
(余翔, 韩铭, 杨小震. 高等学校化学学报, 2011, 32(1), 180—184)
|
| [18] |
Xia T.K., Landman U., Science, 1993, 261, 1310—1312
|
| [19] |
Roth M.W., Firlej L., Kuchta B., Connolly M. J., Maldonado E. E., Wexler C., J. Phys. Chem. C, 2016, 120(2), 984—994
|
| [20] |
Yang J.S., Yang C. L., Wang M. S., Chen B. D., Ma X. G, Phys. Chem. Chem. Phys., 2011, 34, 15476—15482
|
| [21] |
Liu J., Zhao L., Lv Z.Y., Li Z. S., Chem. J. Chinese Universities, 2008, 29(12), 2389—2392
|
|
(刘佳, 赵莉, 吕中元, 李泽生. 高等学校化学学报, 2008, 29(12), 2389—2392)
|
| [22] |
Sütay1 B., Yurtsever M., J. Mol. Model., 2017, 23, 150
|
| [23] |
Sun H., J. Phys. Chem. B, 1998, 102, 7338—7364
|
| [24] |
Sun H., Ren P., Frie J.R., Comput. Theor. Polym. Sci., 1998, 8, 229—246
|
| [25] |
Nose S., J. Chem. Phys., 1984, 81, 511—519
|
| [26] |
Hoover W.G., Phys. Rev. A, 1985, 31, 1695—1697
|
| [27] |
Yang H., Zhao X.J., Sun M., Phys. Rev. E, 2011, 84, 011803
|
 ), 张辉2
), 张辉2