

Review Article
Abstract
Pancreatic
ductal adenocarcinoma (PDAC) is one of the fatal cancers among all critical cancers. The progression of the disease is
primarily due to the oncogene activation and
inactivation of tumor suppressor genes causing genome instability and
contributing to this malignancy in human cells.
Somatic mutations drive
cancer progression, and thus identification of such molecular alterations has the potential to deliver a
deeper understanding of the nature of
that tumor. Even though next-generation sequencing has discovered several
functional mutations in KRAS,
TP53, CDNK2A, SMAD4, and BRCA1/2, their clinical effects remain unclear.
Pancreatic cancer remains unmanageable, with a 5-year survival rate of 5-10%.
The biological significance of core
driver genes, the importance of studying somatic mutations leading
to the disease diagnosis their use in clinical
practice and an account
of computational tools and databases that assist in a
detailed mutational analysis have been discussed in this review.
Abstract Keywords
Pancreatic
ductal adenocarcinoma, somatic mutation, KRAS, tumor suppressor, driver
genes, mutational analyses,
diagnosis, computational tools,
databases
1. Introduction
1.1 Pancreatic
cancer
Pancreatic cancer is the eighth leading
cause of cancer-related deaths. In 2020 total of 495,773 cases was reported globally [1].
In 2022, new pancreatic cancer cases reported
in the USA were 62,210 [2]. As per GLOBOCAN, pancreatic cancer is the 24th most
common disease in India, approximately 10,860 new cases have been reported and ranked at 18th
position in terms of the highest
fatality rate [3]. People
diagnosed with pancreatic cancer have a 5-year survival of 5-10%. The survival
rate is affected by several factors.
However, when diagnosed
the specific cancer
stage plays a crucial role [4].
Amongst pancreatic cancer, up to 93% are exocrine adenocarcinoma; the remaining 7% are pancreatic neuroendocrine
tumors. The intraductal papillary mucinous
and pancreatic intraepithelial neoplasia are significant precursors of PDAC [5].
1.2
Molecular Genetics of Pancreatic Cancer
Extensive
research has established that pancreatic cancer is an inherited disease with
various somatic mutations. Analyzing somatic mutations
allows for differentiating pancreatic adenocarcinoma
from other malignant neoplasms of the pancreas [6]. These mutations can be defined as
alterations in the DNA sequence that may arise during replication be repaired incorrectly, or be left unrepaired.
Several exogenous mutagens like chemicals,
UVs, ionizing radiations, and endogenous mutagens like reactive oxygen species, aldehydes, and repairing enzymes, can
cause DNA damage. Different mutational processes have different unique patterns termed mutational signatures.
Analyzing the signature patterns facilitates
quantifying their effect on biological activity in a cancerous and
non-cancerous genome [7]. A germinal mutation
takes place in the germ line.
Germline mutations are also inherited, as the mutant cell participates in
fertilization and passes the mutation to the next generation. Cancer
due to the germline mutation
is inherited or hereditary
cancer. Next-generation analysis has proven promising in identifying germline mutations
in genes, including
CDKN21, TP53, BRCA2, ATM, MLH1, and BRCA1 responsible
for pancreatic cancer progression in 5.5% of the cases. Spontaneous variations occur in somatic cells of a human body
that include Single Nucleotide Variants (SNVs), chromosomal aberrations, Copy Number Variation (CNVs), insertion and deletions, which are known as "somatic mutations” [8]. Somatic
mutations in the early-stage
lead to developmental disorders, whereas intensifying accretion of these
mutations for an extended period can lead to cancer progression.
1.3
Somatic Mutation
Somatic
mutations cannot be passed down to the offspring except for the canine
transmissible venereal tumor. Somatic
mutations influence the antibodies, T cell, and B cell receptors. Some factors such as the environment, often trigger them,
and they build up in any organism's DNA despite effective
DNA repair mechanisms. Somatic mutations occur at a frequency of 2 to 6 mutations per million bases in healthy
tissues [8]. As a result, somatic cells in the same organism may
have different genotypes (somatic mosaicism) in healthy development and ageing. According
to a study [9] point mutations
ranging from 1000 to 20,000 and multiple
insertions, deletions, and rearrangements contribute to cancer development and progression. These figures were
derived from research involving millions of mutations in different cancer forms [10].
Somatic
mutations include point mutations, repeats, deletions, insertions,
multiplication, copy number loss,
and other genomic variations. When somatic cells split, chromosomal somatic mutations
occur. Chromosome breakages, inappropriate fixing, and unequal material
exchange during chromosome separation cause structural aberrations during this period. These mutations
disrupt genes and their pathways responsible for cell growth and proliferation,
apoptosis, neovascularization, and other cancer hallmarks that lead
to neoplasm development.
1.3.1 Somatic Mutation in Pancreatic Cancer
Somatic mutations
are involved in the progression of cancers, which makes mutational profiling one of the foremost
analyses of the other omics analysis to be considered in clinical practice.
The majority of diagnosis at the clinical
level is based on single-gene mutations. High throughput technologies have underlined
that somatic alterations are a part of the process of growth and development. These somatic alterations may obstruct
gene functions, such as the deactivation of tumor suppressor genes and oncogene
activation and thus disrupt and deregulate
crucial pathways that regulate normal cell growth [11].
Since almost no tumor can form without
somatic mutations, they are essential to oncogenesis [12,13]. Since
the existence or absence of particular mutations
may dictate cancer
therapy, determining a patient's mutational profile is essential in
ensuring successful care. In colorectal,
lung, pancreatic, and other cancer forms specific chemotherapeutics dependent
on mutational status are already part of cancer therapies [14].
The accumulation of somatic
point mutations, also known as single nucleotide variants (SNVs), in the genome
can disrupt cell activity and lead to cancer initiation and progression. The entire repertoire of SNVs across a cancer genome
(which can number in the thousands) can be used to infer clonal populations and research tumor evolution
statistically. As a result, accurate identification
of all somatic SNVs including those with low prevalence is critical since they can identify clones with desirable
phenotypic characteristics. Biomedical investigators researching tumor progression also try to determine
how particular clones are linked to properties like drug resistance, metastatic ability, and fitness under selective therapeutic pressures. Somatic mosaicism
refers to the genetic heterogeneity caused by somatic
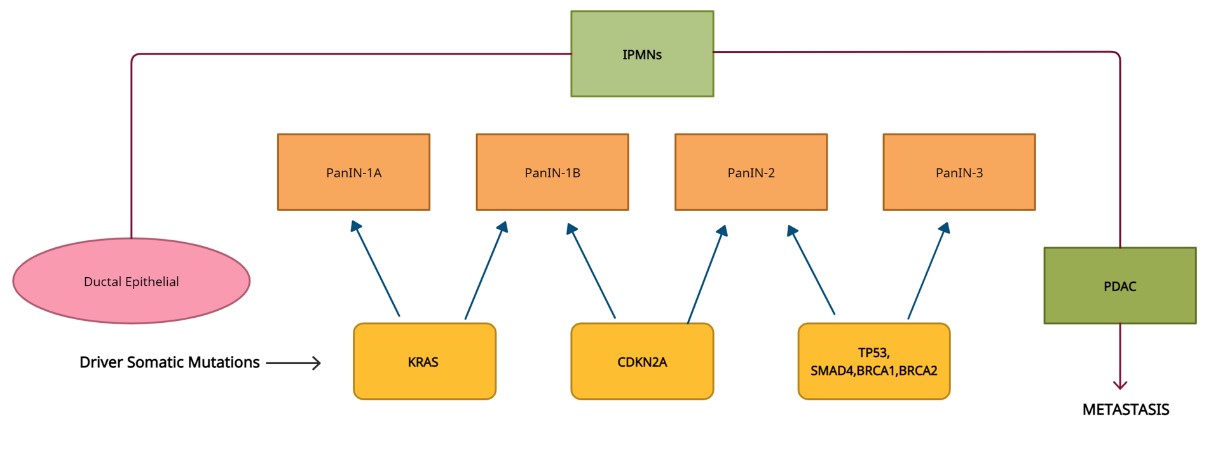
mutations [15]. PDAC is known to arise from PanINlesions (pancreatic intraepithelial neoplasia)
by accumulating somatic
changes in critical
genes over time. (Fig.)
[16].

Figure 1. Driver gene
mutations in pancreatic adenocarcinoma carcinogenesis, classification of
pancreatic intraepithelial neoplasia (PanIN) precursor of pancreatic cancer, at
its different stages (1A, 1B, 2, 3) due to the somatic mutations occurring in
driver genes leading to cancer metastasis.
The
occurrence of pancreatic cancer begins with precursor lesions such as intraductal papillary mucinous neoplasms
(IPMN), pancreatic intraepithelial neoplasia (PanIN) and mucinous cystic
neoplasms (MCN). One of the most common and well-described PDAC
precursor lesions is PanIN. Gene mutations occurring at the Pancreatic
intraepithelial neoplasia stage in due course advanced dysplastic condition.
According to the dysplasia state precursor lesions exhibit varying levels of
mucin, varied architectural patterns and different proliferation rates which
eventually results in changes in gene functionality and cancer cell
progression.
Isolation might
occur in copy number alteration in PDAC. These events are similar to structural alterations in which a chromosome substitutes
for another. Chromothripsis is one of the standard
techniques through which various structural alterations occur in one
cataclysmic mitotic incident.
Detection of chromothripsis through susceptible techniques shows that it can be located in 65% of PDACs,
in many cases, before polyploidization. Chromothripsis can occur
separately or with the additional complex genomic incident
and involve multiple
chromosomes resulting in gene amplification, deletions, or double minutes formation
in either case. Alternatively, structural rearrangements could result from continuous genomic damage caused
by a lack of DNA repair. Advance extensive studies of DNA copy-number
changes (CNAs) led to the discovery
of WGD in human tumors. WGD was more than twice as prevalent (13per cent each) as TERT promoter and oncogenic KRAS mutations.
In
a comprehensive series of pancreatic cancers, whole-genome sequencing revealed
2.64 Mb of a mutational burden on
an average per somatic mutation [17]. The
four most frequently mutated tumor suppressor genes are The Kirsten rat sarcoma (KRAS) oncogene, the
tumor suppressor protein 53 (TP53), SMAD family member 4 (SMAD4), and the
cyclin-dependent kinase inhibitor 2A (CDKN2A). All these mutations dysregulate signaling pathways, thereby affecting the proliferation of tumor
cells and crosstalk with the desmoplastic TNM (tumor, nodes,
and metastasis) surrounding them [18].
Next-generation sequencing is an excellent technique for classifying and systematizing the full spectrum
of somatic alterations and their characteristics. In sporadic pancreatic cancer studies whole-exome sequencing and whole-genome sequencing have
led to identifying genes that, when
mutated, can induce tumorigenesis [19].
KRAS, TP53, CDKN2A, and SMAD4 are the
four main genetic alterations identified in PDAC and most mutations are point mutations [20].
The most prevalent KRAS and TP53 mutations are seen in early-stage intraepithelial neoplasia implying that they
have a role in tumor
initiation [12].
2.
Mutations in
pancreatic cancer
The
classification of PDAC with the understanding of molecular, genetic, and
morphological details will be beneficial in developing targeted
and potent therapeutics in clinical practice. The detailed analysis
of somatic variants
will bring out essential findings. Other than top genes like KRAS,
TP53, SMAD4, CDKN2A, and BRCA1/2studies
have reported somatic mutations in various
genes (Fig. 2) such as ATM, TGFBR2,
ARID2A, SF3B1, GNAS, EGFR, ERBB3, GAT6
that are involved in [20-23] crucial
biological pathways causing
PDAC. Driver genes in PDAC are
listed below, and a few frequently reported
mutations according to COSMIC and TCGA
are mentioned in Table 1.

Figure 2. Gene mutation frequency
of top 20 mutated genes in pancreatic cancer.
Table 1. List of genes and mutations predominantly involved in pancreatic cancer.
|
S.NO |
Gene name |
Mutation type |
Position |
|
1. |
KRAS |
MISSENSE |
G12D,
G12V, G12C, G13D, Q61H, G12R, G12A, G12S, A146T |
|
2. |
PIK3CA |
MISSENSE |
E545K,
H1047R, E542K, R88Q, H1047L, N345K, E726K, G118D |
|
3. |
NRAS |
MISSENSE |
Q61R,
Q61K, Q13R, G12D, Q61L |
|
4. |
FBXW7 |
MISSENSE |
R465H,
R505G, R465C |
|
5. |
BRAF |
MISSENSE |
V600E,
V600M |
|
6. |
CDKN2A |
STOP
GAINED |
R80*,
R58* |
|
7. |
APC
|
STOP
GAINED |
R1450*,
R876*, R1114* |
|
FRAMESHIFT |
T1556NFs3* |
||
|
8. |
3'UTR PTEN
|
MISSENSE |
R130Q,
R130G |
|
FRAMESHIFT |
K267RFs*9,
T319* |
||
|
STOP
GAINED |
R233*,
R130* |
||
|
9. |
TP53
|
MISSENSE
|
R175H,
R248Q, R273C, R273H, R248W, R282W, Y220C, G245S, H179R, H193R, V157F, Y163C,
R273L, C176F, I195T, R249S, E285K, C176Y |
|
STOP
GAINED |
R213*,
R196*, R342*, R306*, Q192* |
||
|
SPLICE
REGION |
T125T |
||
|
10. |
ARID1A
|
FRAMESHIFT |
D1850Tfs*33 |
|
STOP
GAINED |
R1989* |
||
|
11. |
IDH1 |
MISSENSE |
R132H,
R132C |
|
12. |
FGFR2 |
MISSENSE |
S252W |
|
13. |
FGFR3 |
MISSENSE |
S249C |
|
14. |
CTNN31 |
MISSENSE |
S37F |
|
15. |
EGFR |
MISSENSE |
L858R |
|
16. |
GNAQ |
MISSENSE |
Q209F |
|
17. |
AKT1 |
MISSENSE |
E17K |
|
18. |
ERBB2 |
MISSENSE |
S310F |
|
19. |
GNA11 |
MISSENSE |
Q209L |
|
20. |
PPP2R1A |
MISSENSE |
P179R |
|
21. |
BCOR |
MISSENSE |
N1459S |
|
22. |
HRAS |
MISSENSE |
Q61R |
|
23. |
POLE |
MISSENSE |
P286R |
|
24. |
SPECC1 |
FRAMESHIFT |
N303TFs*63 |
|
25. |
JAK1 |
FRAMESHIFT |
K860NFs*16 |
|
26. |
RPL22 |
FRAMESHIFT |
K15RFs*5 |
|
27. |
UBR5 |
FRAMESHIFT |
E2121KFs*28 |
|
28. |
CTCF |
FRAMESHIFT |
T204NFs*26 |
|
29. |
KMT2D |
FRAMESHIFT |
F2354LFs*17 |
|
30. |
AKAP9 |
FRAMESHIFT |
K39RFs*17 |
2.1 KRAS Mutation
In
90 per cent to 93 per cent of pancreatic tumors oncogenic KRAS mutations are detected. KRAS is a GTPase
of size 21kDa, which gets activated on binding to GTP and deactivated upon binding with GDP. As KRAS gets
activated, it further activates RAF family kinases RAF-1, BRAF, and ARAF.
RAF family members then get phosphorylated and activate MEK-1
and MEK-2. These MEK-1 and MEK-2
further activate the extracellular
regulatory kinases ERK-1 and ERK-2. These cause cell proliferations by bringing cytosolic and nuclear proteins
like transcription factors
ELK-1 and c-Jun [24].
The mutations cause constitutive activation of KRAS, resulting in various processes like uncontrolled
proliferation, which causes cancer to develop and spread across the cells and tissues.
KRAS is also responsible for regulating multiple
signaling pathways which are reportedly involved in cancer progressions,
such as PI3K-AKT, PLC- PKC, and RAL. Mutations
of the codons G12, G13, or Q61, by and large, correspond to constitutively active KRAS, activated KRAS. The periodic
mutations in K117 and A146 are also
known to occur. Activating mutations in KRAS are reported in ninety-five per
cent of pancreatic cancer cases. Of
these ninety-nine per cent of all mutations occur in G12 (G12D- 50%) [25].
Mutations
in KRAS highly contribute to the initiation and progression of pancreatic
cancer. KRAS mutations alter RAS proteins. Practically every mutation in KRAS is SNVs in PDACs, appearing in codons 12(~91%), 13(~2%), and 61(~7%).
The mutations at codon 12 are reported to
energize AKT/protein kinase B pathway providing resistance to apoptosis [26]. KRAS mutations
in pancreatic cancer
are due to neoplastic transformation. Most reports about its mutation have been on
relatively small tumors, which lack the statistical justification to determine the appropriate association
with the disease outcome [27]. KRAS oncogene
has a mutational frequency of 20 to 100% and can be used for diagnostic purposes. A subset of tumors contains
multiple mutations in KRAS with some displaying evidence of biallelic mutations.
2.2 TP53 Mutation
TP53
also called antigen NY-CO-13 or p53 provides instructions for producing
a protein called
p53 which acts as a tumor suppressor. One of the functions of TP53
is the activation of target genes during DNA damage or oxidative stress and inducing
apoptosis [28]. It enhances the
expression level of CDKN1A due to which the cell
cycle is arrested [29]. It is regarded as
the Guardian of the Genome as it helps
in cell division and DNA repair. In 70% of pancreatic cancer cases, TP53 is
most frequently mutated resulting in
its binding ability [24,30]. In a study of pancreatic adenocarcinoma patients, less mRNA expression of TP53 was associated with a poor disease prognosis. Clinical evidence suggests
it can be a prognostic marker for diagnosis and therapy.
2.3 CDKN2A Mutation
The
complex of the two cyclins CDK-4 and CDK-6 is involved in the cell cycle's phase transition from G1
to S. The tumor suppressor gene CDKN2A regulates the cell cycle progression by suppressing the CDK-4 and CDK-6 complex.
The CDKN2A gene is located
on chromosome 9p21 in the region that shows high-frequency loss of heterozygosity in various neoplasia. The tumor suppressor region of the CDKN2A
gene encodes two distinct proteins,
P16 and P14. P16 consists of three exons that arrest the cell cycle at the G1
phase thereby stopping cell growth
[31]. The
phosphorylation of retinoblastoma protein is obstructed. The retinoblastoma protein
affects the E2F transcription factor
and participates in the negative regulation of the cell cycle. Another
protein, p14ARF, has a negative
effect on cell growth as it stabilizes p53 activation and targets some CDKs at
G1 and G2 phases thereby inducing apoptosis [32]. Mutations like promoter silencing, heterozygosity, or homozygous
deletion disrupt the operation of CDKN2A. Some
clinical studies on CDKN2A mutation
reported these mutations
as a prognostic and prophetical biomarker.
2.4 SMAD4 Mutation
SMAD4 acts
as tumor suppressor gene. It is known to be deactivated in more than 50% of pancreatic adenocarcinomas, inactivation occurs due to homozygous deletion or intragenic
mutation. SMAD-4 translocate itself in trimeric form into the nucleus activates gene expression and causes cell
growth inhibition [33,34]. SMAD-4 proteins
can transduce signals
from the cell surface to the nucleus.
SMAD-4 mediates TGF-β transduction and gene regulation. Transforming growth factors
regulate proliferation,
differentiation, motility, and
necrobiosis [34].
2.5 BRCA1/2Mutation
BRCA1/2
gene is a tumor suppressor that plays a significant role in the recognition, transcription,
regulation, and double-strand break repair of DNA to forestall cell types
from developing mutations [35].
Somatic mutations in BRCA1 and BRCA2 are reported in about 9% of PDAC patients.
Somatic mutations of BRCA2 appear to be uncommon in tumors of the pancreas. The
mechanism by which mutant BRCA2 contributes to pancreatic cancer development is
unknown. Inactivation of several independent functions of BRCA2, such as remodeling
of chromatin, transcriptional gene control, DNA damage repair, and cell
development and also appears to provide a pathophysiological basis for the
interrelation between BRCA2 mutations and pancreatic cancer [36].
3. Tools for
mutational analysis
Several
tools are available for detecting and analyzing somatic mutations (Table 2),
users can choose the tool depending on the data type and user
interface.
Table 2. List of tools for somatic mutation detection and analysis.
|
S.no |
Name
of tools |
Web-based/
language-based |
Freely
available |
Feature |
Input
data |
Links/source
|
|
1. |
Broad GDAC firehouse [52] |
Web-based |
Yes |
Performs various automated
analyses. Mutational analyses, Correlation analyses, Differential expression
analyses, and Pathway analyses across all types of cancers. |
TCGA |
http//gdac.broadinstitute. org/
|
|
2. |
cBioportal [73] |
Web-based |
Yes |
Allows correlation analyses for
copy number alterations or methylation of genes. The portal also facilitates
users to study gene(s) of interest with access of OncoPrinter and Mutation
Mapper |
TCGA CCLE |
(Ceramiet al., 2012) |
|
3. |
TCGA Clinical explorer [52] |
Web-based |
yes |
Enables users to conclude
relevant clinical information from TCGA data and allows them to translate the
clinical data into the classification of drivers genes, miRNA and proteins |
TCGA |
http//genomeportal.stan ford.edu/pan-tcga/ Weinstein et al., 2013)
|
|
4. |
TCGA4U [74] |
Web-based |
yes |
Genomic alterations that
occurred in the tumor can be understood using this tool to study the
relationship of genomic alterations with clinical data. |
TCGA |
http//www.tcga4u.org, 8888 |
|
5. |
UCSC Xena [75] |
Web-based |
yes |
This tool performs the
comparative analysis of tumor samples to normal samples to explore a gene
expression whether it is up or down-regulated in one or more cancer types. |
TCGA GDC ICGC GTEx TARGET TOIL |
http//xena.ucsc.edu/get ting-started/ |
|
6. |
Vanno [76] |
Web-based |
|
Performs in-depth analysis of
cancer-causing genome sequence alterations. Functional
predictions and mutation landscapes of TCGA data can be derived. |
TCGA |
http//cgts.cgu.edu.tw/ vanno |
|
7. |
MutEnricher [77] |
Python-based software |
yes |
Investigate both coding and
non-coding region for somatic mutation enrichment of the genome. |
TCGA and other cancer
databases. |
https//github.com/asoltis/MutEnricher |
|
8. |
MutaLisk [37] |
Web-based |
yes |
Perform the comparative
analysis of somatic mutations along with physical mapping of the genome. |
Data uploaded in file format
(vcf format) |
http//mutalisk.org/analyze.php |
|
9. |
VarMap [38] |
Web-based |
Yes |
Useful to map the genomic
coordinates to protein. |
The vcf file format is uploaded |
https//www.ebi.ac.uk/thornton-srv/databases/cgi-bin/VarSite/GetPage.pl?varmap=TRUE |
|
10. |
Somatic Sniper [39] |
Software |
Yes implemented in
C |
This tool identifies the single
nucleotide positions to differentiate between normal and tumours genes in the
form of a somatic scores. |
dbSNP |
http//gmt.genome.wustl.edu/packages/somatic-sniper/ |
|
11. |
MutaNet [40] |
Software Codes in Python |
yes |
Perform the statistical
analysis of mutations in the genome. This tool enables to identify the
impactful mutations. |
UniProt AureoWiki PATRIC Cytoscape NCBI SRA RegulonDB Regprecise |
https//service.bioinformatik.uni-saarland.de/mutanet/ |
|
12. |
VarSim [41] |
Software Code in Java and Python |
yes |
Simulates and
validates the different types of variants such as large structural variants,
SNV, insertions and deletions. |
COSMIC dbSNP, DGV VCF file
format
|
http//bioinform.github.io/varsim/ |
|
13. |
SomVarIUS [42] |
Software written in Python 2.7. |
yes |
Using high-throughput
sequencing can identify a somatic mutation in unpaired tissue samples |
TCGA Takes sorted
alignment files (.bam) as input and Output is in
the variant call format (.vcf) |
https//github.com/kylessmith/SomVarIUS |
|
14. |
MutaGene [43] |
Web-based Python Package |
yes |
Web-based tool for
identification of mutations and mutational processes to analyse genes and
calculate the DNA and protein stability. |
ICGC TCGA PCGP COSMIC(WGS) |
https//www.ncbi.nlm.nih.gov/research/mutagene/ |
|
15. |
VarScan 2 [44] |
Command-line software written in
Java |
yes |
Detects copy number alterations
and other somatic mutations from exome data of normal and tumor pairs.
|
NGS data SOLiD,
Life/PGM, Roche/454 |
http//dkoboldt.github.io/varscan/using-varscan.html
|
|
16. |
CHASM [46] |
Language-based tool |
yes |
This tool discriminates somatic missense mutations as cancer
drivers. |
list of
somatic missense mutations |
http//wiki.chasmsoftware.org |
|
17. |
MutSig CV [47] |
Language-based MATLAB2013a |
yes |
Examines the
mutational changes found in DNA sequencing and identifies mutated genes.
|
MAF file |
https//software.broadinstitute.org/cancer/cga/mutsig |
3.1.1 Mutalisk
[37]
Mutalisk associates somatic mutations with genomic, transcriptional, and epigenomic features
to understand better mutational processes
that contribute to mutation
generation. This web-based technology combines physical genome mapping with somatic mutation
identification. The results are displayed using graphics and charts.
Mutalisk only accepts VCF files
as input.
http//mutalisk.org/analyze.php
3.1.2 VarMap
[38]
VarMap
is a web-based tool for mapping chromosomal coordinates
to canonical UniProt sequences and associated protein 3D structures, including validation checks and structural annotation. It can consider patient
variant information, environmental context, and spatial protein distribution of genetic variants.
https//bio.tools/VarMap
3.1.3 Somatic sniper
[39]
Somatic
sniper detects differences in single-nucleotide location between malignancy and normal samples. It uses the
genotype likelihood model to compute
the somatic score, the likelihood of genotype changes between tumor and normal samples.
http//gmt.genome.wustl.edu/packages/somatic-sniper/
3.1.4 MutaNet [40]
MutaNet
was created to determine the impact of specific mutations on gene regulation and genome performance. MutaNet analyses antibiotic resistance gene alterations and their possible
impact on antibiotic resistance in bacterial strains. MutaNeT analyses mutations
in various genomic
areas statistically. The program also includes mutations
in a given gene regulatory network to assess their global impact.
https//service.bioinformatik.uni-saarland.de/mutanet/
3.1.5 VarSim
[41]
VarSim can simulate and validate various
variants, including single
nucleotide variants, minor indels, and significant structural variants.
It is a comprehensive, automated computing
framework that supports
parallel computing and numerous read simulators. VarSim is the only program
that can mimic SNVs, minor indels, and various types of SVs. VarSim's completeness makes it a near match to real-world sequencing investigations.
https//bioinform.github.io/varsim/
3.1.6 SomVarIUS
[42]
A
computational method for detecting somatic mutations in unpaired tissue samples using high-throughput sequencing data.
SomVarIUS takes sorted alignment
files (.bam) as input and produces predicted somatic mutations in the variant
call format (.vcf) allowing it to be easily integrated into any
conventional genome analysis pipeline.
It also produces an extra output that includes all the information regarding
the status of known cancer disease-associated mutations in samples.
https//github.com/kylessmith/SomVarIUS
3.1.7 MutaGene
[43]
MutaGene can determine the context-dependent mutability of DNA locations and anticipated amino
acid substitutions across
the whole genome. Mutability can be used as a background
model to identify probable driver mutations, relating cancer genetics to phenotype. It aids in decoupling the relative
roles of mutagenesis and selection in
carcinogenesis. Mutations from cancer samples can be submitted in VCF format, Mutagene can recognize them, break them
down into individual mutational signatures, and determine the closely
related cancer kind,
primary location, and cluster of samples with similar mutational profiles.
https//www.ncbi.nlm.nih.gov/research/mutagene/
3.1.8 VarScan2
[44]
VarScan is a platform-independent mutation
caller for targeted
mutations. VarScan 2 detects somatic
mutations and copy number changes
(CNAs) in neoplasia–normal pairs of exome data. It may help discover germline
mutations, multiple sample
variants, somatic mutations, and somatic copy number modifications.
https//dkoboldt.github.io/varscan/using-varscan.html
3.1.9 MuTect
[45]
MuTect was created by the Broad Institute for the accurate
and reliable identification of somatic mutations
in cancer genome
next-generation sequencing data. It
identifies somatic mutations using paired and normal and neoplasia cells as
input. MuTect employs a variant
detection statistic to determine whether a variation is more likely than a sequencing error. MuTect then searches for
and removes six types of known sequencing artefacts
MuTect has been frequently employed in cancer genomes research at the Broad Institute.
https//institute.org/cancer/cga/mutect
3.1.10 CHASM
[46]
CHASM
(Cancer-Specific High Throughput Annotation of
Somatic Mutations) to distinguish and focus on missense mutations most
likely to cause beneficial modifications that increase the normal cell's uncontrolled growth property. CHASM
employs a random classifier forest technique to distinguish between
synthetically manufactured passenger and driver missense mutations.
3.1.11 MutSigCV
[47]
MutSig
is an abbreviation for "mutational significance." MutSig analyses
mutational changes discovered in DNA sequencing to identify genes that were changed
more frequently than expected by chance, given the background mutation process. MutSigCV considers heterogeneity by
employing patient-specific mutation frequencies and spectra and
gene-specific mutation rates, expression, and
replication times.
https//software.broadinstitute.org/cancer/cga/mutsig
3.2 Databases for mutation analyses
Various databases
are available for studying
somatic mutation in different aspects, some of the important databases and case studies
are discussed below (Table 3).
Table 3. List of databases dedicated to mutation analyses.
|
S. no |
Name of
databases |
Description |
Links |
|
1. |
Mutfunc
[48] |
The
mutational analysis includes stability, interaction, modification and TF
binding sites. |
http//www.mutfunc.com/ |
|
2. |
Cancer3D
[49] |
This
database analyses the missense mutation regarding protein structure and helps
the user to analyse the pattern of mutations. |
http//www.cancer3d.org/search
(TCGA CCLE) |
|
3. |
Intogene
[50] |
Intogene
analyses the somatic mutations from tumor genomes for cancer driver genes
identification. It uses different methods for driver genes identification and
compiles the output file for better exploration and analysis. |
https//www.intogen.org/search
(TCGA ICGC) |
|
4. |
TANRIC
[51] |
TANRIC
analysis includes the long non-coding RNAs in the context of clinical and
molecular data. |
https//www.tanric.org (TCGA CCLE) |
|
5 |
TCGA
[52] |
TCGA
has complete data on cancer and is stored in the GDC portal. The generated
information is from a cancer patient and can be used for clinical
significance, mutational analysis and gene expression profiling. |
https
//www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga
|
|
6. |
Cosmic
[53] |
Somatic
mutation database. It has data from expert manual curation and genome-wide
screen. Several browsing tools and datasets are present for comparative
analysis of cancer. |
https
//cancer.sanger.ac.uk/cosmic |
|
7. |
TCPA
[54] |
Portal
is used for the visualization and analysis of functional proteomics |
https//tcpaportal.org/tcpa/
(TCGA) |
|
8. |
GEO
database [55] |
An
NCBI database which has data from various high throughput methods, microarray
experiments, next-generation sequencing etc. This database organised the data
in a very informative form for easily accessible and better understanding. |
https//www.ncbi.nlm.nih.gov/geo/
(NCBI) |
|
9. |
CMPD
[78] |
CMPD
contains more than 2 million genetic alterations, two major components of
CMPD are, a web interface for the database SOLite and another for retrieval
of mutated protein sequences. |
http//cgbc.cgu.edu.tw/cmpd
|
|
10. |
ClinVar
[56] |
ClinVar
provides all the information regarding the relationship between the human
variation and phenotype. |
https//www.ncbi.nlm.nih.gov/clinvar/ |
3.2.1 Mutfunc
[48]
Mutfunc
is a mutational database that includes predictions based on a single nucleotide alteration in three organisms
(Humans, E. coli, Yeast). Protein stability,
interaction interfaces, post-translational changes, and transcription factor
binding sites are among the mechanisms investigated.
3.2.2 Cancer 3D [49]
Cancer
3D is a free and open-source database
that examines missense mutations in the context of protein structure in cancer.
The Cancer3D database contains the
findings of such investigations and data from The Cancer Genome Atlas (TCGA) and the Cancer Cell Line Encyclopedia (CCLE). The database
also assists users in analyzing the distribution patterns
of mutations and their association with changes in pharmacological activity using two algorithms
e-Drug and e-Driver.
https//www.cancer3d.org/search
3.2.3 Intogene [50]
Intogene
collects and analyses somatic mutations in
hundreds of neoplasia genomes to identify cancer-driver genes. Intogene
database employs seven distinct
methods for identifying cancer driver genes and compiles the output data of driver genes and a library
of mutational features
that can be utilized to explain and comprehend the mechanism
of action.
https//www.intogen.org/search
3.2.4 TANRIC
[51]
TANRIC
is an open-source site that analyses long non-coding RNAs (lncRNA). These lncRNAs are crucial in cancer biology.
TANRIC analyses lncRNAs in clinical
and molecular data contexts using the expression patterns of cancer datasets
from TCGA, CCLE, and other
independent datasets. It is a useful tool for determining the function and clinical significance of lncRNAs in
cancer considerably facilitating lncRNA-related biological discoveries
and clinical features.
3.2.5 TCGA [52]
The
NCI and National Human Genome Research Institute collaborated on the Cancer Genome Atlas program. Over 12
years, the TCGA collected comprehensive cancer data from 11000 patients. The full cancer
data set is processed and saved in the GDC portal. The information derived
from the collected
data includes clinical
significance, molecular analysis, and gene expression profiling.
https//www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga
3.2.6 COSMIC
[53]
Cancer somatic
mutations catalogue. Cosmic
is a repository for all
somatic mutations associated with human cancer in a catalogue format with
extensive analysis. There are two
sorts of data in Cosmic expert manual curation and genome-wide screen data. Cosmic is organized
by numerous discrete
projects that present
a variety of datasets and browsing
tools for comparative research.
https//cancer.sanger.ac.uk/cosmic
3.2.7 TCPA
[54]
The
Cancer Proteome Atlas is a comprehensive resource for accessing, visualization and analyzing cancer functional proteomics. This resource provides an idiosyncratic opportunity to verify the findings from TCGA data and identify
model cell lines
for functional investigation.
https//tcpaportal.org/tcpa/
3.2.8 GEO Database [55]
The GEO database is a freely accessible resource
that distributes functional
genomics microarray, next-generation sequencing, and other forms of high throughput data. Platform, sample,
series, datasets, and profiles are several types of geo data. GEO search analysis can be done in numerous ways,
including using GEO datasets to search
for data relevant to their research and GEO profiles. A Gene expression can be
investigated and retrieved at this gene-level base or further analysis.
https//www.ncbi.nlm.nih.gov/geo/
3.2.9 ClinVar
[56]
ClinVar
is a freely accessible database that contains all information about the relationships between human variants and
phenotypes. ClinVar reviews submissions identifying variations
detected in patient samples, claims about their clinical significance, submitter information, and supporting data.
ClinVar enables us to comprehend the
relationship between human variants and observed health states and the history
of that interpretation.
https//www.ncbi.nlm.nih.gov/clinvar/
3.3 Analysis of Mutations using
COSMIC
The
Catalogue of Somatic Mutation (COSMIC) is a comprehensive and systematic
database for studying the role of
somatic mutations in human cancers. It lists various mutations, including gene fusions, copy number
variations, non-coding, drug resistance, and coding mutations. It contains
the library of cancer-causing genes, Cancer Gene Census (CGC) assembled by
specialists from various medical reporting, pharmaceutical development, and laboratory research [53] and tools
for analysis (Fig. 3). The most recent release contains around 6 million coding
mutations from 1.4 million samples
from over 26,000 studies,
this approach uses hidden Markov models to predict protein missense variations'
functional, genetic, and phenotypic
implications. Cosmic uses TCGA gene expression level 3 data and methylation data from the ICGC portal for
TCGA investigations. COSMIC provides for discovering new cancer treatment targets and biomarkers by providing detailed
information on mutation distributions, mutational
signature analyses and effects. Improve the collection of clinical
trial cohorts. Identify
driver mutations and associated genes
to aid in patient diagnosis. For example, researchers in a
study [57] used the COSMIC database, which contains somatic mutations
from The Cancer Genome Atlas (TCGA) and several smaller-scale investigations. Researchers used multi-label classification algorithms and the Disease Ontology hierarchy to find cancer
subtype-specific biomarkers. Saha et al. [21]
used databases such as TCGA and COSMIC to perform mutation annotation and
harmful property prediction analysis. They expected that TP53 would be the most frequently altered gene (41 per cent)
among the 114 reported somatic mutations, followed by KRAS, SMAD4, CTNNB1, and
ERBB3. We uncovered a new TP53 hotspot mutation (p.A138V, in 17 per cent of all
patients).

Figure
3. Overview
of tools and projects available for data analysis and their applications.
3.4 Analysis of TCGA Data for Somatic Mutation
TCGA,
The Cancer Genome Atlas (https//cancergenome.nih.gov/) has genome-wide
data from over 30 cancer types and thousands of somatic mutations that advance the understanding of tumorigenesis. To identify somatic
mutations, exome sequencing data is used that allows the detection of SNVs, Single amino acid substitutions. In
addition to Mutational Analysis, TCGA is used for Survival Analysis,
Correlation Analysis, Methylation Analysis, Exploration
of cancer drivers, Differential Analysis, and Pathway Analysis. In a study,
Baek and Lee [58]
analyzed whole-exome sequencing data of 134 PDAC patients. They discovered five genes, KRAS, CDKN2A, TTN, TP53, and KCNJ18, mutated
in the beginning stages of
tumorigenesis. In another latest study, Hwang et al. [59] used TCGA gene expression data for unsupervised clustering and identified three distinct molecular
subtypes belonging to three different pathways and were also able to validate them in another cohort using each
subtype-specific gene (200 were chosen).
Various powerful yet easy-to-use tools (Fig. 4) are also provided to analyze
and visualize TCGA data, such as The
Broad GDAC portal, TCGA Clinical Explorer, Cancer3D, TCGA4U, and UCSC Xena and Vanno, which allow for performing mutation analysis.

Figure 4. Tools for better
visualization and interpretation of multidimensional data are available from
TCGA.
4. Current treatment and novel opportunities
4.1 Anti-RAS therapy
Surgery
followed by adjuvant chemotherapy is the only possible treatment option for
PDAC, but only 15–20 per cent
of patients are suitable for surgery
[60]. The
only targeted treatment for PDAC is a combination of
gemcitabine and an epidermal growth factor receptor (EGFR) inhibitor, which can improve
life by a statistically significant but clinically unsatisfactory twelve days compared to
gemcitabine alone. Patients with advanced pancreatic cancer are treated
with multiagent combination chemotherapy, such as irinotecan/oxaliplatin/5- fluorouracil or
nab-paclitaxel/gemcitabine, although their median overall survival is > a
year. In PDAC, therapeutic methods
have been mainly ineffective, with no treatment
prolonging life beyond
one year following diagnosis. In 93 per
cent of pancreatic cancers, KRAS mutations are found.
There are additional opportunities for therapeutics targeting individual mutant
KRAS isoforms, particularly with
small molecule inhibitors of KRAS G12C. KRAS testing will be required
to determine the particular KRAS mutation present [61].
4.2
KRAS inhibitors
The development of KRAS inhibitors has proven difficult due to various
reasons. Competitive inhibitors have a very high affinity for
GTP. After binding to the GTP binding site that cannot be overtaken, inhibitors of allosteric groups have been
challenging to create due to the lack of pockets for drug binding
on the KRAS surface [62].
4.3
G12C inhibitors
KRAS
G12C inhibitors are present in only 1% of pancreatic cancer cases, which is
very uncommon [63,64]. In the 12th position,
glycine-to-cysteine mutation triggered the KRAS oncoprotein, increasing
tumor cell cycle progression. The
mutated cysteine is located near a switch II pocket (P2). A small molecule known as Sotorasib
(AMG 510) inhibits
KRAS G12C in a reversible and specific manner
via a unique interaction with the P2 pocket [65].
A study shows that G12C inhibitors
can bind with a recently discovered P2 surface pocket on KRAS and covalently bind to the mutant G12C
protein's reactive cysteine residue, according to a study. Another inhibitor of KRAS G12C, adagrasib (MRTX849), had a confirmed response
in one patient with pancreatic cancer [66].
4.4
G12D inhibitors
RAS-selective inhibitor
RMC-6236 binds to cyclophilin A, a chaperone
protein, and constructs a tri-complex with the specific
RAS protein. Multiple
RAS mutants, notably
KRAS G12V and KRAS G12D, have their signaling inhibited in
their GTP-bound conformations [67]. KRAS
G12D inhibitors are in preclinical development. One direct inhibitor,
which is MRTX1133, is currently undergoing
research trials.
4.5
SOS inhibitors
SOS1 is a Guanine
exchange factor that converts GDP to GTP to activate
KRAS and GTPase-
activating proteins. KRAS signaling is controlled by enzymes that catalyze
the intrinsic hydrolysis of GTP back
to GDP to inactivate KRAS. The guanine exchange factor SOS1 catalyzes the conversion of GDP to GTP to
trigger KRAS and degrade the interaction of the SOS1-KRAS complex, preventing KRAS from storing GTP. Treatment
with a MEK inhibitor reduces SOS1
phosphorylation by ERK and relieves negative response to SOS1, allowing SOS1-mediated feedback loops to
restore RAS-mediated signaling. New small molecule
SOS1 inhibitors impair SOS1-KRAS binding in various
KRAS mutations. The SOS1 inhibitor BI-3406 reduced GTP-bound RAS and reduced
proliferation in practically all KRAS codon 12 and 13
mutants examined. It worked in tandem with MEK inhibitors to prevent
feedback reactivation [65].
4.6
Immunotherapy-based treatment strategies for sporadic PDAC
After surgical resection recurrence
of pancreatic cancer still occurs in a high percentage of patients within the
first two years. Using immunotherapy in conjunction with other treatments like
chemotherapy and/or radiation in both neoadjuvant and adjuvant settings has
improved the survival rate of the patients [69]. In the adjuvant trial, a phase II
multi-institutional study that examined the use of algenpantucel-L
immunotherapy in conjunction with chemotherapy and chemoradiotherapy produced
62% disease-free survival and 86% overall survival after 12 months. Although
the survival of patients did not improve at the time of phase III IMPRESS
clinical trial [70,71]. In a recent trial, 30
patients in Japan received the OCV-C01 multi-peptide vaccine from the KIF20A
protein, which contains peptides from the VEGFR1, VEGFR2, and the vascular
endothelial growth factor receptor (VEGFR)1. Results demonstrated that 58.6% of
patients had cytotoxic lymphocyte responses against KIF20A. In the realm of pancreatic
cancer immunotherapy-based treatment, encouraging outcomes have been seen.
However, the success of the therapy will depend on the prediction of further
combinatorial trials aimed at various mutations [72].
5. Discussion
PDAC is one the most lethal
cancer with a terrible prognosis. Currently, no screening measures can detect cancer in its early
stages which is why its poor overall survival.
Individual characteristics lifestyle diabetes and other diseases are
some risk factors that provide some indication for screening and etiological prevention. Surgical removal of pancreatic
cancer is often difficult due to the organ's location, therefore, studying
mutations and targeting them with
combination drug therapies becomes crucial. The four significant most
significant factors to consider when researching the disease are the four
significant mutant driver genes (KRAS, TP53, CDKN2A, and SMAD4) and their biochemical
pathways pathway, PI3K/AKT
signaling pathway, Janus kinase and activator of transcription (JAK/STAT), and MAPK pathways
are crucial pathways
involved in pancreatic cancer. Current
treatment includes chemo-drugs such as gemcitabine, Folfirinox, and
5-Fluorouracil (5-FU). These drugs are used in combination with other anticancer drugs. The advancement of sequencing
technologies and tumor genetic profiling have reported various genes, pathways, potential prognostic markers, and
mutations involved in pancreatic cancer that have helped in providing detailed insights into the mechanism of onset of the disease. However,
despite these efforts, pancreatic cancer remains unmanageable. Novel
screening and diagnostic methods for detecting resectable PDAC early on,
neoadjuvant therapy to increase the number of patients eligible for curative resection. Somatic mutation detection and
adjuvant therapy to improve postoperative
survival in curative resections and palliative disease patients will overcome
the challenges in PDAC management. Somatic mutations play a significant role in the development
progression of cancer disease; therefore, mutational profiling is a crucial
step in therapeutic decision
making.
6. Conclusion
In this review, a detailed account
of somatic mutation
and its different types, along with top mutations in PDAC and the characterization of driver genes
has been studied
in the present study.
Numerous tools, variant analysis pipelines, and databases for analyzing mutation treatment options and new
possibilities for PDAC are also discussed. Studying somatic mutations in pancreatic cancer can not only help strengthen the disease mechanism
but will also help in dictating the treatment possibilities.
Authors’
contributions
Conceptualized
and drafted the review, S.S. and S.S.; Data mined and analyzed the information,
S.S.; S.T. and M.G.
Conflicts of interest
The
authors declare no conflict of interest
6. References
1.
Sung, H.; Ferlay, J.; Siegel, R.L.;
Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics
2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in
185 Countries. CA Cancer J Clin. 2021, 71 (3), 209–249.
2.
Siegel, R.L.; Miller, K.D.; Fuchs, H.E.;
Jemal, A. Cancer Statistics, 2022. CA Cancer J Clin. 2022, 72 (1), 7–33.
3.
Gaidhani, R.H.; Balasubramaniam, G. An epidemiological
review of pancreatic cancer with special reference to India. Indian J Med Sci.
2021, 73, 99.
4.
Ryan, D.P.; Hong, T.S.; Bardeesy, N.
Pancreatic Adenocarcinoma. New England Journal of Medicine. 2014, 371 (11),
1039–1049.
5.
Maitra, A.; Hruban, R.H. Pancreatic cancer.
Annual Review of Pathology: Mechanisms of Disease. 2008, 3 (1), 157–188.
6.
Alexandrov, L.B.; Nik-Zainal, S.; Wedge,
D.C.; Aparicio, S. A. J. R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.;
Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in
human cancer. Nature. 2013, 500 (7463), 415–421.
7.
Omichessan, H.; Severi, G.; Perduca, V.
Computational tools to detect signatures of mutational processes in DNA from tumours:
A review and empirical comparison of performance. PLoS One. 2019, 14 (9),
e0221235.
8.
Martincorena, I.; Campbell, P.J. Somatic
mutation in cancer and normal cells. Science. (1979). 2015, 349 (6255),
1483–1489.
9.
Cibulskis, K.; Lawrence, M.S.; Carter,
S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.;
Lander, E. S.; Getz, G. Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31 (3), 213–219.
10.
Park, S.; Kim, S.J.; Yu, D.; Peña-Llopis,
S.; Gao, J.; Park, J.S.; Chen, B.; Norris, J.; Wang, X.; Chen, M.; et al. An integrative
somatic mutation analysis to identify pathways linked with survival outcomes
across 19 cancer types. Bioinformatics. 2016, 32 (11), 1643–1651.
11.
Esteller, M. Epigenetics provides a new
generation of oncogenes and tumour-suppressor genes. Br. J. Cancer. 2006, 94
(2), 179–183.
12.
Jones, S.; Zhang, X.; Parsons, D.W.;
Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.;
Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed
by global genomic analyses. Science (1979). 2008, 321 (5897), 1801–1806.
13.
Martini, M.; Vecchione, L.; Siena, S.;
Tejpar, S.; Bardelli, A. Targeted therapies: how personal should we go? Nat.
Rev. Clin. Oncol. 2012, 9 (2), 87–97.
14.
Van Cutsem, E.; Köhne, C.H.; Láng, I.;
Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham,
D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin
as first-line treatment for metastatic colorectal cancer: Updated analysis of
overall survival according to tumor KRAS and BRAF mutation status. J. Clin.
Oncol. 2011, 29 (15), 2011–2019.
15.
Dou, Y.; Gold, H.D.; Luquette, L.J.;
Park, P.J. Detecting somatic mutations in normal cells. Trend. Gen. 2018, 34
(7), 545–557.
16.
Waddell, N.; Pajic, M.; Patch, A.M.;
Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek,
K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer.
Nature. 2015, 518 (7540), 495–501.
17.
Kamisawa, T.; Wood, L.D.; Itoi, T.;
Takaori, K. Pancreatic Cancer. The Lancet. 2016, 388 (10039), 73–85.
18.
Collisson, E.A.; Bailey, P.; Chang,
D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev.
Gastroenterol Hepatol. 2019, 16 (4), 207–220.
19.
Casolino, R.; Paiella, S.; Azzolina, D.;
Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.;
Froeling, F. E. M.; et al. Homologous recombination deficiency in pancreatic
cancer: A systematic review and prevalence meta-analysis. J. Clin. Oncol. 2021,
39 (23), 2617–2631.
20.
Hayashi, A.; Hong, J.;
Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev.
Gastroenterol Hepatol. 2021, 18 (7), 469–481.
21.
Saha, G.; Singh, R.; Mandal, A.; Das,
S.; Chattopadhyay, E.; Panja, P.; Roy, P.; DeSarkar, N.; Gulati, S.; Ghatak,
S.; et al. A novel hotspot and rare somatic mutation p.A138V, at TP53 is
associated with poor survival of pancreatic ductal and periampullary
adenocarcinoma patients. Mol. Med. 2020, 26 (1), 59.
22.
Crowley, F.; Park, W.; O’Reilly, E.M.
Targeting DNA damage repair pathways in pancreas cancer. Cancer Met. Rev. 2021,
40 (3), 891–908.
23.
Lowery, M.A.; Jordan, E.J.; Basturk, O.;
Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.;
Maynard, H.; et al. Real-time genomic profiling of pancreatic ductal
adenocarcinoma: potential actionability and correlation with clinical phenotype.
Clin. Cancer Res. 2017, 23 (20), 6094–6100.
24.
Cicenas, J.; Kvederaviciute, K.;
Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS,
TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers
(Basel) 2017, 9 (12), 42. https://doi.org/10.3390/cancers9050042.
25.
Bamford, S.; Dawson, E.; Forbes, S.;
Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.;
Stratton, M.R.; et al. The COSMIC (catalogue of somatic mutations in cancer) database
and website. Br. J. Cancer. 2004, 91 (2), 355–358.
26.
Vizan, P.; Boros, L.G.; Figueras, A.;
Capella, G.; Mangues, R.; Bassilian, S.; Lim, S.; Lee, W.N. P.; Cascante, M.K.
Ras codon-specific mutations produce distinctive metabolic phenotypes in human
fibroblasts. Cancer Res. 2005, 65 (13), 5512–5515.
27.
Rachakonda, P.S.; Bauer, A.S.; Xie, H.;
Campa, D.; Rizzato, C.; Canzian, F.; Beghelli, S.; Greenhalf, W.; Costello, E.;
Schanne, M.; et al. Somatic mutations in exocrine pancreatic tumors:
Association with patient survival. PLoS One. 2013, 8 (4), e60870.
28.
Levy, N.; Yonish-Rouach, E.; Oren, M.;
Kimchi, A. Complementation by wild-type P53 of interleukin-6 effects on M1 cells:
Induction of cell cycle exit and cooperativity with c-Myc suppression. Mol.
Cell Biol. 1993, 13 (12), 7942–7952.
29.
Bates, S.; Ryan, K.M.; Phillips, A.C.;
Vousden, K.H. Cell cycle arrest and DNA endoreduplication following P21Waf1/Cip1
expression. oncogene 1998, 17 (13), 1691–1703.
30.
Kern, S.; Pietenpol, J.; Thiagalingam,
S.; Seymour, A.; Kinzler, K.; Vogelstein, B. Oncogenic forms of P53 inhibit P53-regulated
gene expression. Science (1979). 1992, 256 (5058), 827–830.
31.
McWilliams, R.R.; Wieben, E.D.; Rabe,
K.G.; Pedersen, K.S.; Wu, Y.; Sicotte, H.; Petersen, G.M. Prevalence of CDKN2A mutations
in pancreatic cancer patients: implications for genetic counseling. Eur. J. Hum.Gen.
2011, 19 (4), 472–478.
32.
Cairns, P.; Mao, L.; Merlo, A.; Lee, D.
J.; Schwab, D.; Eby, Y.; Tokino, K.; van der Riet, P.; Blaugrund, J.E.;
Sidransky, D. Rates of P16 (MTS1 ) Mutations in primary tumors with 9p loss.
Science (1979). 1994, 265 (5170), 415–417.
33.
Zhao, M.; Mishra, L.; Deng, C.X. The role
of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14 (2), 111–123.
34.
Liang, C.; Xu, J.; Meng, Q.; Zhang, B.;
Liu, J.; Hua, J.; Zhang, Y.; Shi, S.; Yu, X. TGFB1-induced autophagy affects
the pattern of pancreatic cancer progression in distinct ways depending on SMAD4
status. Autophagy. 2020, 16 (3), 486–500.
35.
Rosen, M.N.; Goodwin, R. A.; Vickers, M.M.
BRCA Mutated pancreatic cancer: A change is coming. World J. Gastroenterol.
2021, 27 (17), 1943–1958.
36.
Vietri, M.T.; D’Elia, G.; Caliendo, G.;
Albanese, L.; Signoriello, G.; Napoli, C.; Molinari, A.M. Pancreatic cancer with
mutation in BRCA1/2, MLH1, and APC Genes: phenotype correlation and detection
of a novel germline BRCA2 mutation. genes (Basel). 2022, 13 (2), 321.
37.
Lee, J.; Lee, A. J.; Lee, J.K.; Park,
J.; Kwon, Y.; Park, S.; Chun, H.; Ju, Y.S.; Hong, D. Mutalisk: A web-based
somatic mutation analysis toolkit for genomic, transcriptional and epigenomic
signatures. Nucleic Acids Res. 2018, 46 (W1), W102–W108.
38.
Stephenson, J.D.; Laskowski, R.A.;
Nightingale, A.; Hurles, M.E.; Thornton, J.M. VarMap: A web tool for mapping
genomic coordinates to protein sequence and structure and retrieving protein
structural annotations. Bioinfor. 2019, 35 (22), 4854–4856.
39.
Larson, D.E.; Harris, C.C.; Chen, K.;
Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.
K.; Ding, L. Somatic sniper: Identification of somatic point mutations in whole
genome sequencing data. Bioinfor. 2012, 28 (3), 311–317.
40.
Hollander, M.; Hamed, M.; Helms, V.;
Neininger, K. MutaNET: A tool for automated analysis of genomic mutations in
gene regulatory networks. Bioinform. 2018, 34 (5), 864–866.
41.
Mu, J.C.; Mohiyuddin, M.; Li, J.; Bani
Asadi, N.; Gerstein, M.B.; Abyzov, A.; Wong, W.H.; Lam, H.Y.K. VarSim: A high-fidelity
simulation and validation framework for high-throughput genome sequencing with
cancer applications. Bioinform. 2015, 31 (9), 1469–1471.
42.
Smith, K.S.; Yadav, V.K.; Pei, S.;
Pollyea, D.A.; Jordan, C.T.; De, S. SomVarIUS: somatic variant identification
from unpaired tissue samples. Bioinform. 2016, 32 (6), 808–813.
43.
Goncearenco, A.; Rager, S.L.; Li, M.;
Sang, Q.X.; Rogozin, I.B.; Panchenko, A.R. Exploring background mutational
processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017, 45
(W1), W514–W522.
44.
Koboldt, D.C.; Zhang, Q.; Larson, D.E.;
Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.;
Wilson, R.K. VarScan 2: somatic mutation and copy number alteration discovery
in cancer by exome sequencing. Genome Res. 2012, 22 (3), 568–576.
45.
Do Valle, Í.F.; Giampieri, E.;
Simonetti, G.; Padella, A.; Manfrini, M.; Ferrari, A.; Papayannidis, C.;
Zironi, I.; Garonzi, M.; Bernardi, S.; et al. Optimized pipeline of MuTect and
GATK tools to improve the detection of somatic single nucleotide polymorphisms
in whole-exome sequencing data. BMC Bioinform. 2016, 17 (S12), 341.
46.
Carter, H.; Samayoa, J.; Hruban, R. H.;
Karchin, R. Prioritization of driver mutations in pancreatic cancer using
cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer
Biol. Ther. 2010, 10 (6), 582–587.
47.
Lawrence, M.S.; Stojanov, P.; Polak, P.;
Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.;
Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 2013, 499 (7457), 214–218.
48.
Wagih, O.; Galardini, M.; Busby, B.P.;
Memon, D.; Typas, A.; Beltrao, P. A resource of variant effect predictions of
single nucleotide variants in model organisms. Mol. Syst. Biol. 2018, 14 (12).
49.
Porta-Pardo, E.; Hrabe, T.; Godzik, A.
Cancer3D: Understanding cancer mutations through protein structures. Nucleic
Acids Res. 2015, 43 (D1), D968–D973.
50.
Gonzalez-Perez, A.; Perez-Llamas, C.;
Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.;
Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types.
Natz. Methods. 2013, 10 (11), 1081–1082.
51.
Li, J.; Han, L.; Roebuck, P.; Diao, L.;
Liu, L.; Yuan, Y.; Weinstein, J. N.; Liang, H. TANRIC: An interactive open
platform to explore the function of LncRNAs in cancer. Cancer Res. 2015, 75
(18), 3728–3737.
52.
Weinstein, J.N.; Collisson, E.A.; Mills,
G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.;
Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet.
2013, 45 (10), 1113–1120.
53.
Tate, J.G.; Bamford, S.; Jubb, H.C.;
Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.;
Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 2019, 47 (D1), D941–D947.
54.
Li, J.; Lu, Y.; Akbani, R.; Ju, Z.;
Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G. W.; Kane, D.W.;
et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods.
2013, 10 (11), 1046–1047.
55.
Barrett, T.; Wilhite, S.E.; Ledoux, P.;
Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.;
Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data
sets—update. Nucleic Acids Res. 2012, 41 (D1), D991–D995.
56.
Landrum, M.J.; Lee, J.M.; Riley, G.R.;
Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive
of relationships among sequence variation and human phenotype. Nucleic Acids
Res. 2014, 42 (D1), D980–D985.
57.
Amar, D.; Izraeli, S.; Shamir, R.
Utilizing somatic mutation data from numerous studies for cancer research: proof
of concept and applications. Oncogene. 2017, 36 (24), 3375–3383.
58.
Baek, B.; Lee, H. Prediction of survival
and recurrence in patients with pancreatic cancer by integrating multi-omics
data. Sci. Rep. 2020, 10 (1), 18951.
59.
Hwang, J.W.; Jang, S.K.; Lee, D.J.
Genomic analysis of pancreatic cancer reveals 3 molecular subtypes with
different clinical outcomes. Medicine. 2021, 100 (14), e24969.
60.
Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.;
Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; Liu, C. Molecular alterations
and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol.
2020, 13 (1), 130.
61.
Lambert, A.; Schwarz, L.; Borbath, I.;
Henry, A.; Van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An update on
treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019,
11, 175883591987556.
62.
Lee, M.S.; Pant, S. Personalizing medicine
with germline and somatic sequencing in advanced pancreatic cancer: current
treatments and novel opportunities. American Society of Clinical Oncology
Educational Book. 2021, No. 41, e153–e165.
63.
Spencer-Smith, R.; O’Bryan, J.P. direct
inhibition of RAS: quest for the holy Grail? Semin. Cancer Biol. 2019, 54,
138–148.
64.
Bailey, P.; Chang, D.K.; Nones, K.;
Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner,
T. J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of
pancreatic cancer. Nature. 2016, 531 (7592), 47–52.
65.
Simanshu, D.K.; Nissley, D.V.;
McCormick, F. RAS Proteins and their regulators in human disease. Cell. 2017,
170 (1), 17–33.
66.
Hayashi, A.; Hong, J.; Iacobuzio-Donahue,
C.A. The Pancreatic Cancer Genome Revisited. Nat Rev Gastroenterol Hepatol.
2021, 18 (7), 469–481.
67.
Sakamoto, K.; Masutani, T.; Hirokawa, T.
Generation of KS-58 as the First K-Ras(G12D)-inhibitory peptide presenting
anti-cancer activity in vivo. Sci. Rep. 2020, 10 (1), 21671.
68.
Ye, Y.; Zheng, S. Successful immunotherapy
for pancreatic cancer in a patient with TSC2 and SMAD4 mutations: a case report.
Front. Immunol. 2021, 12.
69.
Kole, C.; Charalampakis, N.; Tsakatikas,
S.; Frountzas, M.; Apostolou, K.; Schizas, D. Immunotherapy in combination with
well-established treatment strategies in pancreatic cancer: current insights.
Cancer Manag Res. 2022, 14, 1043–1061.
70.
Hardacre, J.M.; Mulcahy, M.; Small, W.;
Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz,
H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard
adjuvant therapy for pancreatic cancer: a phase 2 study. J. Gastro. Surg. 2013,
17 (1), 94–101.
71.
Hewitt, D.B.; Nissen, N.; Hatoum, H.; Musher,
B.; Seng, J.; Coveler, A.L.; Al-Rajabi, R.; Yeo, C.J.; Leiby, B.; Banks, J.; et
al. A phase 3 randomized clinical trial of chemotherapy with or without
algenpantucel-L (hyperacute-pancreas) immunotherapy in subjects with borderline
resectable or locally advanced unresectable pancreatic cancer. Ann. Surg. 2022,
275 (1), 45–53.
72.
Torphy, R.J.; Zhu, Y.; Schulick, R.D.
Immunotherapy for pancreatic cancer: barriers and breakthroughs. Ann. Gastro. Surg.
2018, 2 (4), 274–281.
73.
Cerami, E.; Gao, J.; Dogrusoz, U.;
Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.;
Larsson, E.; et al. The CBio cancer genomics portal: an open platform for
exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2 (5),
401–404.
74.
Huang, Z.; Duan, H.; Li, H.
Identification of gene expression pattern related to breast cancer survival
using integrated TCGA datasets and genomic tools. Biomed Res. Int. 2015, 2015,
1–10.
75.
Goldman, M.J.; Craft, B.; Hastie, M.;
Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks,
A.N.; et al. Visualizing and interpreting cancer genomics data via the xena
platform. Nat. Biotechnol. 2020, 38 (6), 675–678.
76.
Huang, P.J.; Lee, C.C.; Tan, B. C.M.;
Yeh, Y.M.; Huang, K.Y.; Gan, R.C.; Chen, T.W.; Lee, C.Y.; Yang, S.T.; Liao, C.S.;
et al. Vanno: a visualization-aided variant annotation tool. Hum Mutat. 2015,
36 (2), 167–174.
77.
Soltis, A.R.; Dalgard, C.L.; Pollard,
H.B.; Wilkerson, M.D. MutEnricher: A flexible toolset for somatic mutation
enrichment analysis of tumor whole genomes. BMC Bioinform. 2020, 21 (1), 338.
78.
Huang, P.J.; Lee, C.C.; Tan, B. C.M.;
Yeh, Y.M.; Julie Chu, L.; Chen, T.W.; Chang, K.P.; Lee, C.Y.; Gan, R.C.;
Liu, H.; et al. CMPD: Cancer mutant proteome database. Nucleic Acids Res. 2015,
43 (D1), D849–D855.
This work is licensed under the
Creative Commons Attribution
4.0
License (CC BY-NC 4.0).
Abstract
Pancreatic
ductal adenocarcinoma (PDAC) is one of the fatal cancers among all critical cancers. The progression of the disease is
primarily due to the oncogene activation and
inactivation of tumor suppressor genes causing genome instability and
contributing to this malignancy in human cells.
Somatic mutations drive
cancer progression, and thus identification of such molecular alterations has the potential to deliver a
deeper understanding of the nature of
that tumor. Even though next-generation sequencing has discovered several
functional mutations in KRAS,
TP53, CDNK2A, SMAD4, and BRCA1/2, their clinical effects remain unclear.
Pancreatic cancer remains unmanageable, with a 5-year survival rate of 5-10%.
The biological significance of core
driver genes, the importance of studying somatic mutations leading
to the disease diagnosis their use in clinical
practice and an account
of computational tools and databases that assist in a
detailed mutational analysis have been discussed in this review.
Abstract Keywords
Pancreatic
ductal adenocarcinoma, somatic mutation, KRAS, tumor suppressor, driver
genes, mutational analyses,
diagnosis, computational tools,
databases
This work is licensed under the
Creative Commons Attribution
4.0
License (CC BY-NC 4.0).


This work is licensed under the
Creative Commons Attribution 4.0
License.(CC BY-NC 4.0).