

Abstract
Increased expression of p202 protein (encoded by the Ifi202 gene) in splenocytes derived from B6.Nba2 mice (congenic for the Nba2 interval derived from the New Zealand Black mice) was correlated with defects in apoptosis of splenic B cells and increased susceptibility to develop systemic lupus erythematosus. We have now investigated the molecular mechanisms by which increased expression of p202 in B6.Nba2 cells contributes to defects in apoptosis. In this study, we report that increased expression of p202 in the B6.Nba2 splenocytes, as compared with cells derived from the parental C57BL/6 (B6) mice, was correlated with increased levels of p53 protein and inhibition of p53-mediated transcription of target genes that encode proapoptotic proteins. Conversely, knockdown of p202 expression in B6.Nba2 cells resulted in stimulation of p53-mediated transcription. We found that p202 bound to p53 in the N-terminal region (aa 44–83) comprising the proline-rich region that is important for p53-mediated apoptosis. Consistent with the binding of p202 to p53, increased expression of p202 in B6.Nba2 mouse embryonic fibroblasts inhibited UV-induced apoptosis. Taken together, our observations support the idea that increased expression of p202 in B6.Nba2 mice increases the susceptibility to develop lupus, in part, by inhibiting p53-mediated apoptosis.
Systemic lupus erythematosus (SLE)6 is an autoimmune disease characterized by production of pathogenic IgG autoantibodies (1, 2, 3, 4, 5, 6, 7). Based on genetic linkage studies, multiple chromosomal loci and genes appear to contribute to the development of the disease in human SLE patients and in mouse models of lupus (1, 2, 7).
IFNs, a family of cytokines, are known to have multiple effects on the immune system, affecting differentiation, proliferation, and survival of B and T cells (8, 9). The family includes two types of IFNs among others: type I (α and β) and type II (γ) (8). Both types of IFNs function through transcriptional activation of IFN-activatable genes encoding the IFN-inducible proteins that mediate the biological activities of IFNs (8). Importantly, increased expression of IFN-activatable genes has been reported in peripheral blood cells from human SLE patients (9, 10, 11). However, the molecular mechanisms by which IFN-inducible proteins contribute to the development of autoimmunity remain unclear.
Generation of B6.Nba2 mice congenic for the Nba2 interval (derived from the New Zealand Black strain), which contains New Zealand Black allele of the IFN-activatable Ifi202 gene on the C57BL/6 (B6) background, has identified Ifi202 as a major candidate gene for lupus susceptibility (12, 13, 14). Studies also revealed that increased expression of p202 protein (encoded by the Ifi202 gene) in B6.Nba2 splenic cells was correlated with defects in apoptosis of B cells (12).
The protein p202 (52 kDa) is an inducible transcriptional modulator (13, 15, 16, 17) that belongs to the p200 protein family (18). The family includes structurally related mouse proteins, such as p202 (both p202a and p202b (13)), p203, and p204 (18, 19). The family also includes human proteins (IFI16, IFIX, MNDA, and AIM2) (19). The ability of p202 to modulate transcription of genes depends on its ability to bind and inhibit the transcriptional activity of factors, such as E2Fs, NF-κB, c-Myc, and AP-1 (13, 20). Additionally, p202 also binds to a segment of 53BP1 protein (21), a p53-binding protein implicated in the DNA damage response in mice (22). Increased expression of p202 in a variety of cell lines inhibits apoptosis (13, 20).
p53 is known to induce apoptosis through various mechanisms (23, 24). For example, binding of p53 to DNA in a sequence-specific manner is known to activate transcription of proapoptotic genes, including PUMA, Gadd45, and Fas (25). Interestingly, recent studies have suggested that p53 can also induce apoptosis through the mitochondrial pathway (26, 27). Furthermore, it is important to note that the N-terminal proline-rich region (aa 63–85) in p53 is important for p53-mediated mitochondrial-dependent and independent apoptosis (24, 28, 29).
Development of SLE in humans is correlated with increases in p53 protein levels in blood lymphocytes (30) and defects in p53-mediated apoptosis (31). Moreover, reduced levels of p53 in peripheral blood cells from patients with rheumatoid arthritis, another autoimmune disease (9), are associated with loss of radiation-induced apoptosis (32). Interestingly, IFN-α or IFN-β treatment of cells is known to result in accumulation of functionally inactive p53 protein (33), which could be activated by posttranslational modifications (33), raising the possibility that increased levels of IFN-inducible proteins in cells contribute to the accumulation of functionally inactive p53 protein.
Importantly, loss of expression of p53 target genes is linked to the development of lupus-like diseases in mice (34, 35, 36). Mice null for the Gadd45a gene, a target of p53 for transcriptional activation, develop lupus-like disease (34). Similarly, loss of expression of p21CIP1 (encoded by the p21 gene), a known transcriptional target of p53, is linked to the development of autoimmunity (35, 36). These observations raise the possibility that defects in p53-mediated transcriptional activation of target genes that encode proapoptotic proteins may contribute to the survival of autoreactive lymphocytes and enhance the development of SLE.
Forced expression of p202 in cell lines inhibits p53-mediated transcriptional activation of reporter genes (21) and p53-mediated apoptosis (37). Interestingly, p53 represses the transcription of Ifi202 gene (37). Because defects in apoptotic pathways are thought to contribute to the survival of autoreactive lymphocytes and p202 modulates apoptosis (13, 20), we sought to determine whether increased expression of p202 in B6.Nba2 cells has any effect on p53-mediated transcription of genes and apoptosis. In this study, we report that increased expression of p202 in B6.Nba2 splenocytes was associated with inhibition of p53-mediated transcription of target genes that encode proapoptotic proteins. Additionally, we found that p202 bound to p53, and increased expression of p202 in B6.Nba2 cells inhibited p53-mediated apoptosis. In summary, our observations support the idea that increased expression of p202 in B6.Nba2 cells results in inhibition of p53-mediated transcription of genes and defects in p53-mediated apoptosis, thus contributing to increased lupus susceptibility in B6.Nba2 mice.
Materials and Methods
Cells and cell lines
Mouse splenocytes and mouse embryonic fibroblasts (MEFs) were derived from B6.Nba2 and B6 strains of mice (12), maintained in the animal facilities of the University of Colorado Health Sciences Center. Mouse AKR-2B embryonic fibroblasts (16) and the Val5 cell line (expressing a temperature-sensitive mutant of p53) have been described previously (37, 38). B6.Nba2 MEFs were transfected with either the pCMV vector or pCMV-AS-202 plasmid (21) (allowing the expression of an antisense to the Ifi202 mRNA). Transfected cells were selected in G418 (500 μg/ml) for ∼2 wk, and >100 G418-resistant colonies were pooled to analyze reduced expression of p202. MEFs, MEFs transfected with the pCMV vector or pCMV-AS-202 plasmid, mouse AKR-2B fibroblasts, and Val5 cells were maintained at low density in DMEM containing high glucose, supplemented with 10% FBS and antibiotics in an incubator (at 37°C) with 5% CO2. If so indicated, the cultures of Val5 cells were shifted to 32°C for the indicated time. The B6.Nba2 MEFs between passages 2 and 8 were used for experiments.
Plasmids and expression vectors
Plasmids encoding GST-202a (aa 30–255), GST-202a1 (aa 30–158), GST-202a2 (aa 158–255), GST-202b1 (aa 255–372), and GST-202b2 (aa 373–455) have been described previously (39). Plasmids encoding GST-202 (aa 19–445) and GST-202 (aa 255–294) have also been described previously (40). T. Halazonetis (Wistar Institute, University of Pennsylvania, Philadelphia, PA) provided GST-p53 (aa 1–393), GST-p53 (aa 1–83), and GST-p53 (aa 100–300). T. Shenk (Princeton University, Princeton, NJ) provided plasmids encoding the following GST-p53 fusions: 1–160, 160–393, 160–318, and 319–393. Fusions containing residues 1–43 and 44–393 of the human p53 protein were created by PCR amplification of the wild-type pCMV-p53 vector (BD Clontech). S. Fields (University of Washington, Seattle, WA) provided the pCMH6k53 plasmid encoding murine p53 (37).
To generate the plasmid for in vitro transcription and translation of p53, cDNA encoding the wild-type human p53 protein was excised from the pCMV-53 vector using the HindIII and EcoRI restriction enzymes. The cut fragment of 1189 bp was then subcloned into the pcDNA3 vector (Invitrogen Life Technologies) to allow for in vitro transcription using the T7 RNA polymerase.
Immunoblotting
Splenocytes or fibroblasts were collected in PBS, resuspended in a modified radioimmunoprecipitation assay (RIPA)-lysis buffer (50 mM Tris-HCl (pH 8.0), 250 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS), supplemented with protease inhibitors (50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 1 mM PMSF), and incubated at 4°C for 30 min. Cell lysates were sonicated briefly before centrifugation in a microfuge for 10 min at 4°C. The supernatants were collected, and a Bio-Rad protein assay kit measured the protein concentrations. Equal amounts of protein were processed for immunoblotting, as described previously (16). Antiserum to p202 has been described previously (16).
Immunoprecipitations
Total cell lysates were prepared and subjected to immunoprecipitations using preimmune, anti-p202, or anti-p204 sera, as described previously (40). The immunoprecipitates were analyzed by immunoblotting using anti-p53 Abs conjugated to HRP (sc-6243 HRP; Santa Cruz Biotechnology).
In vitro transcription and translation
p53 cDNA in the pcDNA3 vector (Invitrogen Life Technologies) was in vitro transcribed and translated using the TnT T7 Quick Coupled Transcription/Translation System (Promega) supplemented with [35S]methionine. Reaction mixtures were then used for GST pull-down assays.
GST-fusion proteins and GST pull-down assays
Reporter assays
For reporter assays, subconfluent cells were cultured in six-well plates. Cells were transfected with the 202-luc (5 μg) or the indicated other luciferase reporter plasmid and pRL-TK reporter plasmid (0.5 μg), using a calcium phosphate transfection kit (Invitrogen Life Technologies), as suggested by the supplier. Unless otherwise indicated, cells were harvested between 43 and 48 h after transfections. Cells were lysed, and the firefly and Renilla luciferase activities were determined, as described previously (37).
RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen Life Technologies), and was digested with DNase I (to remove any DNA in the preparation). Isolated total RNA (1 μg) was subjected to cDNA synthesis using SuperScript First-Stand Synthesis system (Invitrogen Life Technologies), as suggested by the supplier, followed by PCR, using a pair of primers specific to the Ifi202a (forward primer, 5′-ggtcatctaccaactcagaat-3′; reverse primer, 5′-ctctaggatgccactgctgttg-3′), as described previously (17). Primer sequences for other genes are available upon request. For RT-PCR, we used the Superscript one-step RT-PCR system from Invitrogen Life Technologies.
Flow cytometric analysis
Flow cytometry was performed on single cell suspensions from adherent (after trypsin and EDTA treatment), as well as the floating cells after pooling them. Briefly, to measure accumulation of cells with a sub-G1 DNA content, cells were stained with propidium iodide (50 μg/ml; Sigma-Aldrich) and subjected to flow cytometry, as described previously (37). To detect cells undergoing early stages of apoptosis, the Annexin VFITC apoptosis kit (BioSource International) was used, as suggested by the supplier, and cells were subjected to flow cytometry.
Results
p202 inhibits p53-mediated transcriptional activation of genes
To elucidate the molecular mechanisms by which increased expression of p202 in B6.Nba2 splenocytes inhibited apoptosis, we tested whether p202 has any effect on p53 levels and/or the expression of p53 target genes. As shown in Fig. 1,A, splenocytes derived from B6.Nba2 female mice expressed detectable levels of p202. Consistent with our earlier observations (12), we could not detect the expression of p202 in splenocytes derived from the age-matched B6 female mice. Notably, increased expression of p202 in the B6.Nba2 splenocytes was associated with increased levels of p53 protein (Fig. 1,A), a >70% reduction in levels of Gadd45a and PUMA mRNAs, and a 50–60% decrease in levels of p21 and Mdm2 mRNAs (Fig. 1,B). Of note, increased levels of Ifi202 mRNA in B6.Nba2 splenocytes did not correlate with increases in levels of p53 mRNA (Fig. 1 B). Taken together, these observations indicated that increased expression of p202 protein in B6.Nba2 splenocytes results in increases in p53 protein levels by posttranscriptional mechanisms and decreases in steady state levels of mRNAs for p53 target genes.
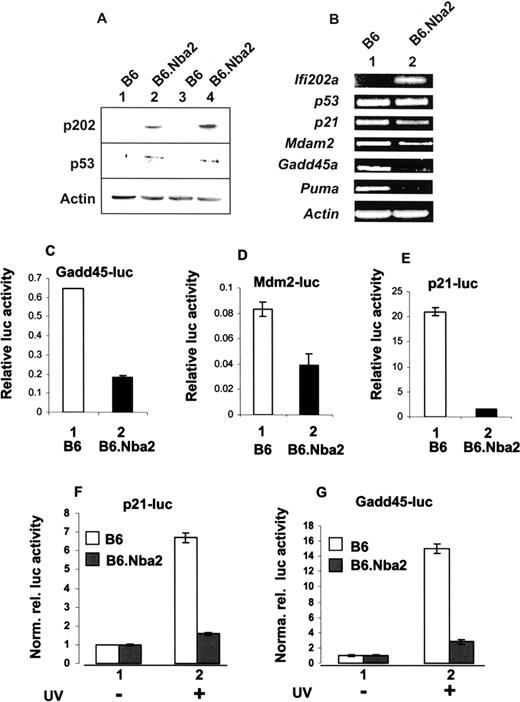
Increased expression of p202 in B6.Nba2 splenocytes correlates with increased levels of p53 protein and inhibition of basal and induced transcription of p53 target genes. A, Extracts prepared from age-matched B6 (lanes 1 and 3) or B6.Nba2 (lanes 2 and 4) female mice were analyzed by immunoblotting using Abs specific to the indicated proteins. Each lane represents one animal. B, Total RNA isolated from age-matched B6 (lane 1) or B6.Nba2 (lane 2) female mice was analyzed by RT-PCR using a pair of primers specific to the indicated genes. RNA isolated from two different B6 or B6.Nba2 female mice gave essentially similar results. C–E, Subconfluent cultures of B6 (column 1) or B6.Nba2 (column 2) MEFs (passage 6) were transfected with Gadd45-luc-reporter (C), Mdm2-luc-reporter (D), or p21-luc-reporter plasmid (E), along with pRL-TK reporter plasmid, as described in Materials and Methods. Forty-two hours after transfections, cell lysates were analyzed for dual luciferase activity. Relative luciferase activity is indicated. The bars indicate the SD. Similar results were obtained in two independent experiments. F and G, Subconfluent cultures of B6 (□) or B6.Nba2 (▪) MEFs (passage 6) were transfected with p21-luc-reporter plasmid (F) or Gadd45-luc-reporter plasmid (G) along with pRL-TK plasmid, as described in C. Twenty-four hours after transfections, one set of cells (column 1) was either left untreated or exposed to UV-B light (5 mJ/m2; column 2), and 42 h after transfections, dual luciferase activity was determined. Normalized relative luciferase activity is indicated. The bars indicate the SD.
Increased expression of p202 in B6.Nba2 splenocytes correlates with increased levels of p53 protein and inhibition of basal and induced transcription of p53 target genes. A, Extracts prepared from age-matched B6 (lanes 1 and 3) or B6.Nba2 (lanes 2 and 4) female mice were analyzed by immunoblotting using Abs specific to the indicated proteins. Each lane represents one animal. B, Total RNA isolated from age-matched B6 (lane 1) or B6.Nba2 (lane 2) female mice was analyzed by RT-PCR using a pair of primers specific to the indicated genes. RNA isolated from two different B6 or B6.Nba2 female mice gave essentially similar results. C–E, Subconfluent cultures of B6 (column 1) or B6.Nba2 (column 2) MEFs (passage 6) were transfected with Gadd45-luc-reporter (C), Mdm2-luc-reporter (D), or p21-luc-reporter plasmid (E), along with pRL-TK reporter plasmid, as described in Materials and Methods. Forty-two hours after transfections, cell lysates were analyzed for dual luciferase activity. Relative luciferase activity is indicated. The bars indicate the SD. Similar results were obtained in two independent experiments. F and G, Subconfluent cultures of B6 (□) or B6.Nba2 (▪) MEFs (passage 6) were transfected with p21-luc-reporter plasmid (F) or Gadd45-luc-reporter plasmid (G) along with pRL-TK plasmid, as described in C. Twenty-four hours after transfections, one set of cells (column 1) was either left untreated or exposed to UV-B light (5 mJ/m2; column 2), and 42 h after transfections, dual luciferase activity was determined. Normalized relative luciferase activity is indicated. The bars indicate the SD.
To determine whether the observed decreases in the basal levels of mRNAs for the p53 target genes in the above experiments were due to an inhibition of transcription of genes, we chose to compare basal and induced transcriptional activity of p53 in B6 and B6.Nba2 MEFs. B6.Nba2 MEFs express increased levels of p202 (41). As shown in Fig. 1, C–E, the basal activity of three well-known p53-responsive reporters (the Gadd45-luc, Mdm2-luc, and p21-luc) was significantly lower in B6.Nba2 compared with B6 MEFs. Because p53 is also known to activate the transcription of the above reporter genes in response to DNA damage caused by exposure of cells to DNA damaging agents, such as UV light (42), we also compared the activity of p21-luc and Gadd45-luc reporters in B6.Nba2 vs B6 MEFs after UV exposure. As shown in Fig. 1, F and G, UV light-induced activity of the two p53-responsive reporters was significantly lower in B6.Nba2 than B6 MEFs. Taken together, these observations indicated that increased expression of p202 in B6.Nba2 cells is associated with inhibition of basal as well as induced transcription of p53 target genes.
To determine whether inhibition of p53-mediated transcription in B6.Nba2 cells is dependent on the expression of p202, we chose to down-regulate the expression of p202. For this purpose, we transfected B6.Nba2 MEFs with either an empty vector (pCMV) or a plasmid (pCMV-AS-202 (21)) that allows the expression of antisense to Ifi202 mRNA. Consistent with our previous observations (21), transfection of the pCMV-AS-202 plasmid in B6.Nba2 MEFs resulted in knockdown of Ifi202 mRNA and protein (Fig. 2, A and B). Additionally, consistent with our previous observations (21), transfection of pCMV-AS-202 plasmid in B6.Nba2 cells did not result in alterations in the expression of p204 protein (data not shown), another member of the p200 family (18). Importantly, the knockdown of p202 expression resulted in stimulation of the activity of p21-luc, Mdm2-luc, and Gadd45-luc reporters albeit to different extents (Fig. 2 C). Our observations provided support for the idea that inhibition of p53-mediated transcription of these reporter genes in B6.Nba2 cells is dependent on the expression of p202 protein.

Knockdown of p202 expression in B6.Nba2 MEFs results in stimulation of the activity of p53-responsive reporters. A, Total RNA isolated from B6.Nba2 MEFs, either transfected with an empty vector (pCMV) or pCMV-AS-202 plasmid, was subjected to RT-PCR using a pair of primers specific to the indicated mRNA. B, Total cell extracts from B6.Nba2 MEFs, either transfected with an empty vector (pCMV) or pCMV-AS-202 plasmid, were subjected to immunoblotting using Abs specific to the indicated proteins. C, B6.Nba2 MEFs transfected with vector (□) or pCMV-AS-202 plasmid (▪) were transfected with either p21-luc-reporter (column 1), Mdm2-luc-reporter (column 2), or Gadd45-luc-reporter (column 3) along with pRL-TK-reporter plasmid. After 40–45 h of transfections, dual luciferase activity was determined, as described in Materials and Methods. The normalized relative luciferase reporter activity is indicated.
Knockdown of p202 expression in B6.Nba2 MEFs results in stimulation of the activity of p53-responsive reporters. A, Total RNA isolated from B6.Nba2 MEFs, either transfected with an empty vector (pCMV) or pCMV-AS-202 plasmid, was subjected to RT-PCR using a pair of primers specific to the indicated mRNA. B, Total cell extracts from B6.Nba2 MEFs, either transfected with an empty vector (pCMV) or pCMV-AS-202 plasmid, were subjected to immunoblotting using Abs specific to the indicated proteins. C, B6.Nba2 MEFs transfected with vector (□) or pCMV-AS-202 plasmid (▪) were transfected with either p21-luc-reporter (column 1), Mdm2-luc-reporter (column 2), or Gadd45-luc-reporter (column 3) along with pRL-TK-reporter plasmid. After 40–45 h of transfections, dual luciferase activity was determined, as described in Materials and Methods. The normalized relative luciferase reporter activity is indicated.
p202 binds to p53 in the proline-rich region
To investigate the molecular mechanisms by which p202 inhibits p53-mediated transcription of genes, we tested whether p202 could bind to p53. As shown in Fig. 3, A and D, the full-length p53 (aa 1–393 fused with GST, but not GST alone) bound to p202 present in the cellular extracts prepared from mouse AKR-2B cells treated with IFN-α (to increase p202 levels) in GST pull-down assays. Interestingly, the N-terminal segment (aa 1–83) of p53, comprising the transcription activation domains 1 and 2, as well as the proline-rich segment (aa 63–85) (28, 29), was sufficient to associate with p202. In contrast, the DNA binding domain (aa 100–300) of p53 or the C-terminal segment, containing the tetramerization domain, did not bind p202 (Fig. 3 D).

Identification of p202 binding domain in p53 (GST-pull down assays). A, Full-length p53 and indicated p53 deletion mutants were expressed fused to GST protein in bacteria, and the fusion proteins were purified. Cellular lysates, prepared from mouse AKR-2B cells treated with IFN (1000 U/ml, 24 h), were incubated with glutathione S-Sepharose beads bound with the indicated purified GST-fusion proteins. The beads were washed, and the bound proteins were analyzed by immunoblotting, using polyclonal Abs specific to p202 (sc-6054; Santa Cruz Biotechnology). Similar results were obtained in three experiments. B, Beads bound with the indicated GST-fusion proteins were incubated with total cell lysates, as described in A. The bound proteins were analyzed by immunoblotting using Abs to p202. Similar results were obtained in two experiments. C, Beads bound with indicated GST protein were incubated with 250 ng (1×; lanes 1 and 3) or 500 ng (2×; lanes 2 and 4) of recombinant purified p53 expressed in the Sf9 insect cells (catalogue 66011T; purchased from BD Pharmingen). Beads were washed, and the bound proteins were analyzed by immunoblotting using specific Abs to p53. Similar results were obtained in two experiments. D, A schematic representation of various structural and functional domains of human p53 and their ability to bind cellular p202. The numbers in parentheses indicate the starting and ending amino acid residues in GST fusion proteins. AD-1, activation domain-1; AD-2, activation domain-2; +, binding; −, no binding.
Identification of p202 binding domain in p53 (GST-pull down assays). A, Full-length p53 and indicated p53 deletion mutants were expressed fused to GST protein in bacteria, and the fusion proteins were purified. Cellular lysates, prepared from mouse AKR-2B cells treated with IFN (1000 U/ml, 24 h), were incubated with glutathione S-Sepharose beads bound with the indicated purified GST-fusion proteins. The beads were washed, and the bound proteins were analyzed by immunoblotting, using polyclonal Abs specific to p202 (sc-6054; Santa Cruz Biotechnology). Similar results were obtained in three experiments. B, Beads bound with the indicated GST-fusion proteins were incubated with total cell lysates, as described in A. The bound proteins were analyzed by immunoblotting using Abs to p202. Similar results were obtained in two experiments. C, Beads bound with indicated GST protein were incubated with 250 ng (1×; lanes 1 and 3) or 500 ng (2×; lanes 2 and 4) of recombinant purified p53 expressed in the Sf9 insect cells (catalogue 66011T; purchased from BD Pharmingen). Beads were washed, and the bound proteins were analyzed by immunoblotting using specific Abs to p53. Similar results were obtained in two experiments. D, A schematic representation of various structural and functional domains of human p53 and their ability to bind cellular p202. The numbers in parentheses indicate the starting and ending amino acid residues in GST fusion proteins. AD-1, activation domain-1; AD-2, activation domain-2; +, binding; −, no binding.
Because the N-terminal segment (aa 1–83) of p53, which bound p202 in cellular extracts, contains two transcription activation domains (28, 29), we tested whether both domains are involved in binding to p202. As shown in Fig. 3 B, a segment containing the activation domain-1 transcription activation domain (aa 1–43) did not bind p202. These observations suggested that the proline-rich region (aa 44–83) in p53 may be responsible for binding to p202.
To examine whether interaction between p202 and p53 proteins is independent of other proteins, we incubated increasing amounts of recombinant full-length wild-type p53 (without GST; purchased from BD Pharmingen), which was expressed in Sf9 insect cells and immunopurified using DO-7 anti-p53 Ab, with beads bound with GST alone or GST-p202. As shown in Fig. 3 C, the recombinant p53 did not bind to GST protein. In contrast, p53 bound to GST-p202, indicating that interaction between p53 and p202 is independent of other cellular proteins.
p53 binds to p202 in the C-terminal 200-aa repeat
The protein p202 contains two partially conserved repeats of 200 aa, which are known to bind several proteins in vitro and in vivo (13, 20). Therefore, to identify the segment(s) in p202 sufficient to bind p53, we used binding of GST-p202 fusion proteins with p53. As shown in Fig. 4, in vitro translated p53 is detected as two prominent p53 protein bands, which could arise due to the initiation of translation at an internal protein translation initiation site. Importantly, neither p53 protein bound to GST protein. However, p53 did bind to almost full-length p202 (aa 9–445). Interestingly, a segment in p202 (aa 255–294), which binds other p202-binding proteins, such as E2F1 (20, 40), also bound to p53, albeit weakly. The a-repeat in p202 or its segments a1 or a2, did not bind to p53 detectably. In contrast, the b-repeat or its segment b1 or b2 bound to p53 relatively strongly. We also found that cellular p53 selectively bound to GST-p202 (aa 19–445), but not GST (data not shown).

Identification of a p53-binding region in p202. Upper panel, The indicated p202 deletion mutants were expressed fused to GST protein in bacteria and the fusion proteins were purified. p53 protein was synthesized in vitro in rabbit reticulocyte lysate using coupled in vitro transcription and translation system. An aliquot (10 μl) of translation reaction was incubated with beads bound with the indicated GST-p202 fusion proteins, as described previously (39 ). The beads were washed, and the bound proteins were analyzed by SDS-PAGE, followed by fluorography. As a control, an aliquot of translation reaction was also subjected to electrophoresis (lanes labeled as input). Similar results were obtained in two experiments. Lower panel, A schematic representation of various segments in p202 and their ability to bind in vitro translated p53. The two repeats (a and b) in p202 and their two segments (a1 and a2 and b1 and b2, respectively) are also indicated. The numbers in the parentheses indicate the starting and the ending amino acids residues in p202 in indicated GST-fusion proteins. ++, Strong binding; +, binding; ±, weak binding.
Identification of a p53-binding region in p202. Upper panel, The indicated p202 deletion mutants were expressed fused to GST protein in bacteria and the fusion proteins were purified. p53 protein was synthesized in vitro in rabbit reticulocyte lysate using coupled in vitro transcription and translation system. An aliquot (10 μl) of translation reaction was incubated with beads bound with the indicated GST-p202 fusion proteins, as described previously (39 ). The beads were washed, and the bound proteins were analyzed by SDS-PAGE, followed by fluorography. As a control, an aliquot of translation reaction was also subjected to electrophoresis (lanes labeled as input). Similar results were obtained in two experiments. Lower panel, A schematic representation of various segments in p202 and their ability to bind in vitro translated p53. The two repeats (a and b) in p202 and their two segments (a1 and a2 and b1 and b2, respectively) are also indicated. The numbers in the parentheses indicate the starting and the ending amino acids residues in p202 in indicated GST-fusion proteins. ++, Strong binding; +, binding; ±, weak binding.
p202 associates with p53 in vivo
Binding of p202 to p53 in the above experiments prompted us to determine whether p202 associates with p53 in vivo. For this purpose, we chose to use the Val5 cell line (37, 38), which overexpresses a temperature-sensitive mutant of p53. Incubation of Val5 cells at 32°C results in accumulation of an active p53 protein, whereas incubation of cells at 37°C results in an inactive p53 protein (37, 38). Because the wild-type, but not the mutant, p53 represses the transcription of the Ifi202 gene (37), expression of the temperature-sensitive mutant of p53 in Val5 cells allowed us to study interactions between p202 and p53 in vivo. As shown in Fig. 5, p53 was immunoprecipitated with specific Abs to p202, but not by preimmune serum or Abs to p204 (also a member of the 200 protein family (43), from total cell extracts prepared from Val5 cells incubated at 32°C. Of note, we could not detect p53 in p202 immunoprecipitates from the Val5 total cell extracts after incubation of cells at 37°C (data not shown), indicating that p53 in mutant conformation does not associate with p202. Taken together, these experiments indicated that a significant fraction of p202 was associated with the wild-type p53 in Val5 cells.

p202 and p53 are detected in a complex in vivo. Val5 cells were cultured at 37°C and treated with IFN to increase p202 levels. Cells were shifted to 32°C for 3 h, and total cell extracts were subjected to immunoprecipitations using antiserum to p202 (lane 3) or p204 (lane 4). As a negative control, we also incubated lysates with preimmune serum (lane 1). Immunoprecipitates were analyzed by immunoblotting using specific Abs to p53 conjugated to HRP (sc-6243; Santa Cruz Biotechnology). As a control, we also analyzed a fraction of the input lysate for levels of p53 (lane 1). Similar results were obtained in two experiments.
p202 and p53 are detected in a complex in vivo. Val5 cells were cultured at 37°C and treated with IFN to increase p202 levels. Cells were shifted to 32°C for 3 h, and total cell extracts were subjected to immunoprecipitations using antiserum to p202 (lane 3) or p204 (lane 4). As a negative control, we also incubated lysates with preimmune serum (lane 1). Immunoprecipitates were analyzed by immunoblotting using specific Abs to p53 conjugated to HRP (sc-6243; Santa Cruz Biotechnology). As a control, we also analyzed a fraction of the input lysate for levels of p53 (lane 1). Similar results were obtained in two experiments.
We also attempted to detect in vivo interactions between p53 and p202 using Abs directed against different regions of p53 in cell extracts derived from the Val5 cells incubated at 32°C. These attempts were unsuccessful because p202 (52 kDa) comigrated with the H chain of the IgG protein, thus making it difficult to detect p202 by immunoprecipitation-Western assays.
Increased expression of p202 inhibits UV-induced apoptosis in B6.Nba2 MEFs
Because p53 levels increase in cells upon exposure to UV light (42) and MEFs are known to undergo apoptosis after exposure to UV light that is mediated by p53 (44), we compared apoptosis between B6 and B6.Nba2 MEFs after UV treatment. As shown in Fig. 6,A, UV treatment of B6 and B6.Nba2 MEFs resulted in up-regulation of p53. Consistent with our observation using splenocytes (Fig. 1), the induced levels of p53 were measurably higher in B6.Nba2 than B6 MEFs (compare lane 4 with lane 2). As expected from previous studies (45), basal levels of p53 were not detectable in low passage (passages 2–8) B6 and B6.Nba2 MEFs. Interestingly, in contrast to our previous observation that UV-induced p53 protein levels in cells of mouse AKR-2B cell line down-regulated p202 mRNA and protein levels (37), UV-induced p53 levels in B6.Nba2 MEFs did not result in down-regulation of p202 protein levels (Fig. 6 A). Instead, we noted a measurable (∼2-fold) increase in p202 protein levels (compare lane 4 with lane 3).

Increased expression of p202 in B6.Nba2 MEFs decreases apoptosis in response to UV light. A, Subconfluent cultures of MEFs (passage 5) from B6 (lanes 1 and 2) or B6.Nba2 (lanes 3 and 4) mice were either left untreated (lanes 1 and 3) or treated with UV-B light (5 mJ/m2; lanes 2 and 4). Total cell lysates were prepared after 14 h of the treatment of MEFs and analyzed by immunoblotting using Abs specific to the indicated protein. The extent of p53 induction (middle panel, lanes 2 and 4) is indicated at the bottom of the figure. Similar results were obtained in three independent experiments. B, Phase-contrast photographs of B6 and B6.Nba2 MEFs without any treatment or after UV treatment. C, Subconfluent cultures of B6 or B6.Nba2 MEFs were either left untreated (top two panels) or treated with UV-B light (5 mJ/m2; bottom two panels). Cells were incubated for 14 h, and at the end of the incubations, floating as well as attached cells were subjected to flow cytometry after propidium iodide staining, as described in Materials and Methods. The percentage of the accumulation of sub-G1 cells is indicated. Similar results were obtained in four experiments.
Increased expression of p202 in B6.Nba2 MEFs decreases apoptosis in response to UV light. A, Subconfluent cultures of MEFs (passage 5) from B6 (lanes 1 and 2) or B6.Nba2 (lanes 3 and 4) mice were either left untreated (lanes 1 and 3) or treated with UV-B light (5 mJ/m2; lanes 2 and 4). Total cell lysates were prepared after 14 h of the treatment of MEFs and analyzed by immunoblotting using Abs specific to the indicated protein. The extent of p53 induction (middle panel, lanes 2 and 4) is indicated at the bottom of the figure. Similar results were obtained in three independent experiments. B, Phase-contrast photographs of B6 and B6.Nba2 MEFs without any treatment or after UV treatment. C, Subconfluent cultures of B6 or B6.Nba2 MEFs were either left untreated (top two panels) or treated with UV-B light (5 mJ/m2; bottom two panels). Cells were incubated for 14 h, and at the end of the incubations, floating as well as attached cells were subjected to flow cytometry after propidium iodide staining, as described in Materials and Methods. The percentage of the accumulation of sub-G1 cells is indicated. Similar results were obtained in four experiments.
Importantly, cultures of B6 MEFs (as compared with B6.Nba2 MEFs) accumulated many more cells that morphologically appeared apoptotic after exposure to UV light (Fig. 6,B). Because apoptosis in primary MEFs is not synchronized, to compare the extent of apoptosis between B6 and B6.Nba2 MEFs, we collected both adherent (after trypsin and EDTA treatment) and floating cells and analyzed them by flow cytometry after propidium iodide staining. As shown in Fig. 6,C, we detected a ∼5-fold increase in apoptosis of B6 MEFs after UV treatment. In contrast, much small (∼30%) increases in apoptosis of B6.Nba2 MEFs were detected after UV treatment. Because propidium iodide staining allows detection of cells in the late phase of apoptosis, we also analyzed MEFs after annexin V staining, which allows the detection of cells in early stages of apoptosis. Using this approach, we noted that UV treatment of B6 MEFs (after ∼14 h) resulted in increases (from 5.09 to 13.24% in one experiment and from 5.75 to 17.6% in the second experiment) in annexin V-positive MEFs. In contrast, no measurable increases in annexin V-positive cells were detected in B6.Nba2 MEFs after UV treatment in two experiments. Taken together, our observations show that increased expression of p202 in B6.Nba2 MEFs inhibits UV-induced transcription of p53-responsive reporter genes (Fig. 1) and inhibits UV-induced apoptosis (Fig. 6).
Discussion
Our observations are as follows: 1) increased expression of p202 in B6.Nba2 cells was correlated with increased levels of p53 protein (Figs. 1 and 6); 2) increased expression of p202 in B6.Nba2 cells was associated with inhibition of p53-mediated transcription of target genes (Fig. 1); 3) knockdown of p202 expression stimulated transcription of p53-responsive reporter genes (Fig. 2); 4) p202 bound to p53 protein both in vitro and in vivo (Figs. 3–5); and 5) increased expression of p202 in B6.Nba2 MEFs inhibited UV-induced apoptosis that is mediated by p53 (Fig. 6). These observations are consistent with the idea that increased expression levels of p202 protein in B6.Nba2 cells result in increased levels of p53 protein and defects in p53-mediated apoptosis, thus contributing to increased lupus susceptibility in B6.Nba2 mice. Consistent with this idea, the development of SLE in human patients has been associated with increases in p53 protein levels in blood lymphocytes (30) and defects in p53-mediated apoptosis (31). Furthermore, loss of the expression of p53 transcriptional target genes, such as the Gadd45a (34) and p21 (35, 36) in mice, is associated with the development of autoimmunity.
We have reported previously that increased expression of p202 in cell lines up-regulates p53 protein levels and inhibits p53-mediated transcription of reporter genes (21). Our current observations that p202 binds to p53 and that increased expression of p202 in B6.Nba2 splenocytes was correlated with up-regulation of p53 protein levels and inhibition of p53-mediated transcription of genes indicate that increased levels of p202 in B6.Nba2 cells result in the accumulation of functionally inactive p53 protein. Because IFN-αβ treatment of cells is known to increase p53 protein levels in an inactive form (33), our observations are consistent with the possibility that IFN-induced levels of p202 protein in cells contribute to functional inactivation of p53 and its accumulation.
Increases in cellular levels of p53 protein have largely been attributed to increases in the t1/2 (42). However, a recent study has provided evidence that increased translation of p53 mRNA also contributes to increases in p53 protein levels after DNA damage (46). Although it remains unclear which mechanism contributes to the increased levels of p53 protein in B6.Nba2 splenocytes, our previous (21) and current observations (Fig. 1) that increased expression of p202 inhibits p53-mediated transcription of the mdm2 gene provide support for the idea that p202 increases the p53 protein levels by down-regulating the expression of MDM2 protein, a known negative regulator of the p53 protein levels (29).
p53 induces apoptosis by nuclear-dependent and independent mechanisms (23, 24). In the cytoplasm, p53 is known to induce apoptosis by the mitochondrial pathway (26, 27). Interestingly, p202 is detected both in cytoplasm and nucleus of mouse fibroblasts (13, 16, 41). Moreover, in the cytoplasm, p202 associates with mitochondrial membranes (13, 16, 41). Therefore, our observations that p202 binds to p53 and inhibits p53-mediated transcription of genes raise an interesting possibility that binding of p202 to p53, which results in accumulation of functionally inactive p53 (Fig. 1), inhibits p53-mediated functions by inhibiting both nuclear-dependent and independent functions. It may be relevant to note that binding of SV40 large T Ag to p53 results in accumulation of functionally inactive p53 and protein complexes containing the SV40 large T Ag and p53 are known to contribute to autoimmunity by inducing high titers of anti-p53 Abs (47, 48). Therefore, our current observations also raise the possibility that accumulation of functionally inactive p53 in complexes with p202 contributes to the development of autoimmunity.
Differences in posttranslational modification of p53 in splenocytes derived from C57BL/6 and DBA/2 mice are linked to increased sensitivity to radiation-induced apoptosis in C57BL/6 mice (49). Because expression of p202 is not detectable in C57BL/6 splenocytes, but detectable in DBA/2 splenocytes (12), our observations raise the possibility that the lack of p202 expression in C57BL/6 mice contributes to increased susceptibility to radiation-induced apoptosis in splenocytes. Further work is in progress to determine how mouse strain-specific posttranslational modifications in p53 affect its functional interactions with p202 protein.
Inhibition of NF-κB activity is known to abrogate p53-mediated apoptosis (50). Therefore, inhibition of NF-κB-mediated transcription by p202 in certain cells (20) is predicted to decrease p53-mediated apoptosis. Consistent with this prediction, decreased p65-Rel A protein expression in T lymphocytes from SLE patients is associated with defects in T cell activation and apoptosis (51). Therefore, it will be interesting to determine whether the p202-mediated inhibition of NF-κB activity in B6.Nba2 cells also contributes to the inhibition of p53-mediated apoptosis.
In summary, observations described in this work provide support for our model (Fig. 7). The model predicts that: 1) a variety of factors may contribute to increased expression of p202 in cells of the immune system (13, 20); and 2) increased levels of p202 in B6.Nba2 lymphocytes and MEFs decrease susceptibility to apoptosis, in part, by inhibiting p53-mediated transcription. Our observations contribute to an understanding of the molecular mechanisms by which increased levels of IFN-inducible p202 protein in B6.Nba2 mice increase susceptibility to develop lupus. Importantly, these observations will serve as the molecular basis to understand the role of structurally and functionally related IFN-inducible proteins in human SLE.

Regulation and role of p202 in the development of lupus susceptibility through inhibition of p53-mediated transcription in the B6.Nba2 mouse model of SLE.
Regulation and role of p202 in the development of lupus susceptibility through inhibition of p53-mediated transcription in the B6.Nba2 mouse model of SLE.
Acknowledgments
We thank Drs. T. Halazonetis, T. Shenk, M. Murphy, S. Fields, G. Lozano, C. Prives, and A. Levine for generously providing reagents.
Disclosures
The authors have no financial conflict of interest.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported, in part, by National Institutes of Health Grant AI066261 (to D.C.).
Abbreviations used in this paper: SLE, systemic lupus erythematosus; MEF, mouse embryonic fibroblast.
