
Abstract
Monocyte chemoattractant protein-1 (MCP1) plays a key role in monocyte/macrophage infiltration to the sub-endothelial space of the blood vessel wall, which is a critical initial step in atherosclerosis. In this study, we examined the intracellular signaling pathway of IL-1β-induced MCP1 expression using various chemical inhibitors. The pretreatment of a phosphatidylcholine (PC)-specific PLC (PC-PLC) inhibitor (D609), PKC inhibitors, or an NF-κB inhibitor completely suppressed the IL-1β-induced MCP1 expression through blocking NF-κB translocation to the nucleus. Pretreatment with inhibitors of tyrosine kinase or PLD partially suppressed MCP1 expression and failed to block nuclear NF-κB translocation. These results suggest that IL-1β induces MCP1 expression through activation of NF-κB via the PC-PLC/PKC signaling pathway.
Similar content being viewed by others


Introduction
Inflammation of the blood vessel wall is a principal event in the initiation and maintenance of various vascular diseases, including atherosclerosis and restenosis after angioplasty. Infiltration of monocytes into the vessel wall, a typical phenomenon in inflammation related to vascular diseases, is mediated by chemokines such as monocyte chemoattractant protein-1 (MCP1) (Sasayama et al., 2000). MCP1 is a member of the chemokine family widely expressed in endothelial cells, smooth muscle cells and monocytes in response to several atherogenic stimulants such as CD40 ligand, PDGF, IL-1β and oxidized LDL (Terkeltaub et al., 1998). Several recent in vivo studies have disclosed critical roles of MCP1 in atherosclerosis. Additional depletion of the MCP1 receptor markedly attenuates atherosclerotic lesions by inhibiting macrophage infiltration in apolipoprotein E (ApoE) deficient mice (Boring et al., 1998). Blocking MCP1 function using a dominant negative mutant in rabbit or neutralization of MCP1 with an anti-MCP1 antibody in rat is effective in preventing restenosis after angioplasty (Furukawa et al., 1999; Mori et al., 2002).
Different cell types, including macrophages, lymphocytes, endothelial cells, and smooth muscle cells (SMCs), are involved in atherosclerotic lesion formation (Lusis, 2000). In particular, smooth muscle cells produce cytokines and chemokines that attract and activate leukocytes, induce proliferation of SMCs, and stimulate production of extracellular matrix components.
IL-1β is a multifunctional cytokine responsible for macrophage activation, angiogenesis, and regulation of inflammation (Wu and Ho, 2003). This major proinflammatory cytokine is primarily produced by monocytes, macrophages and polymorphonuclear phagocytes, and acts by inducing numerous genes, including adhesion molecules, proteases, cytokines, and chemokines. Binding of IL-1β to IL-1 receptor I (IL-1RI) activates the NF-κB pathway via activation of the IκB kinase (IKK) complex (Dinarello, 1996; Malinin et al., 1997). Recent studies have demonstrated that the transcription factor NF-κB plays a key role in inflammatory responses against various stimuli (Ghosh and Hayden, 2008; Tu et al., 2008). While it is established that IL-1β induces MCP1 expression via NF-κB and AP-1 activation in endothelial cells, the underlying intracellular signaling pathways are not well understood at present (Martin et al., 1997).
In the present study, we explored the intracellular signaling pathway involved in IL-1β-induced MCP1 expression in primary human aorta smooth muscle cells (HASMCs). Our results show that IL-1β induces MCP1 expression through PC-PLC/PKC pathway-dependent NF-κB activation. Additionally, IL-1β activates PLD and tyrosine kinase, which are also involved in MCP1 expression, but do not require the NF-κB activation.
Results
IL-1β induces MCP1 expression in human aorta smooth muscle cells
To examine the effects of IL-1β on MCP1 expression, primary HASMCs were treated with IL-1β (5 ng/ml) for the indicated time periods. Total RNA was prepared as described in Methods, and the levels of MCP1 mRNA determined by RT-PCR using specific primers. Expression of MCP1 mRNA was increased by IL-1β in a time-dependent manner (Figure 1A). The secreted MCP1 protein level was measured in the supernatant fractions of HASMCs stimulated with IL-1β (5 ng/ml) using the human MCP1 immunoassay kit (R&D systems). IL-1β induced the expression and secretion of MCP1 in a time-dependent manner (Figure 1B).

IL-1β induces MCP1 expression in HASMCs. (A) HASMCs were treated with 5 ng/ml IL-1β for the indicated times. Total RNA was isolated and RT-PCR analysis was performed using MCP1 gene-specific primers and the internal control gene, β-actin. Two additional experiments yielded similar results. A representative study is shown. (B) The amount of secreted MCP1 protein was determined in the supernatant after IL-1β treatment for the indicated times using the human MCP1 ELISA kit. Data are presented as mean values obtained from three independent experiments, and the bars represent standard deviations. The significance was determined by Student's t-test (*P < 0.05 vs untreated control).
MCP1 is induced by IL-1β in PC-PLC- and PKC-dependent pathways
To determine whether PLC activity is necessary for IL-1β-induced MCP1 expression, several specific inhibitors were used (Kawakami et al., 2007). Upon pretreatment of cells with 100 µM D609 (a PC-PLC inhibitor) for 30 min, IL-1β-induced MCP1 expression was inhibited at the mRNA and protein levels above 95%, although 50 µM D609 had no effect. While U73122, a phosphatidylinositol-specific PLC (PI-PLC) inhibitor, had no effect, and 100 µM propranolol (a phosphatidate phosphohydrolase inhibitor) suppressed IL-1β-induced MCP1 expression about 33% at secreted protein levels (Figure 2A). To determine whether PKC is necessary for MCP1 induction by IL-1β, cells were pretreated with the PKC inhibitors, staurosporine and bisindolylmaleimide, for 30 min (Pickett et al., 2002). IL-1β-induced MCP1 mRNA and protein expression was completely inhibited by the PKC-specific inhibitors (Figure 3). Especially, the pretreatment of staurosporine completely blocked the basal level of MCP1 expression. This effect of staurosporine on MCP1 expression may due to the broad inhibitory activities against a variety of protein kinases via the prevention of ATP binding to the kinase (Ruegg and Burgess, 1989). These several inhibitors had no effect on cell viability in doses used to these experiments (data not shown). Our results suggest that IL-1β augments MCP1 expression via PC-PLC-dependent PKC activation in HASMCs.

IL-1β-induced MCP1 expression is decreased by the PC-PLC inhibitor, D609 in HASMCs. HASMCs were pretreated with the indicated concentrations of each inhibitor for 30 min prior to IL-1β (5 ng/ml) for 10 h. (A) Total RNA was isolated and RT-PCR analysis was performed using MCP1 gene-specific primers and the internal control gene, β-actin. Two additional experiments yielded similar results. A representative study is shown. (B) The amount of secreted MCP1 protein was determined in the supernatant after IL-1β treatment for 10 h using the human MCP1 ELISA kit. Data are presented as mean values obtained from three independent experiments, and bars represent standard deviations. The significance was determined by Student's t-test (*P < 0.05 vs untreated control).

PKC mediates IL-1β-induced MCP1 expression. HASMCs were pretreated with the indicated concentrations of PKC inhibitors, staurosporine and bisindolylmaleimide, for 30 min prior to IL-1β (5 ng/ml) treatment for 10 h. (A) Total RNA was isolated, and RT-PCR analysis was performed using MCP1 gene-specific primers and the internal control gene, β-actin. Two additional experiments yielded similar results. A representative study is shown. (B) The amount of secreted MCP1 protein was determined in the supernatant after IL-1β treatment for 10 h using the human MCP1 ELISA kit. Data are presented as mean values obtained from three independent experiments, and bars represent standard deviations. The significance was determined by Student's t-test (*P < 0.05 vs untreated control).
Inhibitors of tyrosine kinase and PLD decrease IL-1β-induced MCP1 expression
In previous studies, a phosphatidate phosphohydrolase inhibitor, propranolol, suppressed IL-1β-induced MCP1 expression. Since phosphatidate phosphohydrolase converts phosphatidic acid (the primary glycerol-based product of PLD) to DAG, which is involved to PKC activation, we examined the involvement of PLD in IL-1β-induced MCP1 expression. Pretreatment with 1% butanol (a PLD inhibitor) for 30 min decreased the MCP1 induction by IL-1β about 69% but treatment with propanol, which is three carbon alcohol not to inhibit the PLD activity, did not (Figures 4A and 4B). To determine whether tyrosine kinase is required for MCP1 expression induced by IL-1β, cells were pretreated with two tyrosine kinase inhibitors, genistein and AG18 (tyrphostin 23) for 30 min (Frasor et al., 2001; Delva et al., 2008). These tyrosine kinase inhibitors, genistein and AG18 decreased IL-1β-induced MCP1 mRNA and protein expression about 56% and 78% at highest dose, respectively (Figure 4A and 4B). The results clearly suggest that PLD and the tyrosine kinase pathway are involved in IL-1β-induced MCP1 expression.

Inhibitors of tyrosine kinase and PLD attenuate IL-1β-induced MCP1 expression. HASMCs were pretreated with the indicated concentrations of tyrosine kinase inhibitors, genistein and AG18, and the PLD inhibitor, BuOH, for 30 min prior to IL-1β (5 ng/ml) for 10 h. (A, C) Total RNA was isolated, and RT-PCR analysis was performed using MCP1 gene-specific primers and the internal control gene, β-actin. Two additional experiments yielded similar results. A representative study is shown. (B, D) The amount of secreted MCP1 protein was determined in the supernatant after IL-1β treatment for 10 h using the human MCP1 ELISA kit. Data are presented as mean values obtained from three independent experiments and bars represent standard deviations. The significance was determined by Student's t-test (*P < 0.05 vs untreated control).
NF-κB translocates to the nucleus and activates MCP1 expression
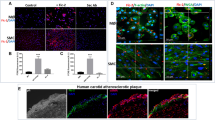
Several reports show that NF-κB is involved in TNF-κ-induced MCP1 expression (Iseki et al., 2000; Chen et al., 2004). To establish whether NF-κB activation is required for IL-1β-induced MCP1, cells were pretreated with the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), prior to IL-1β treatment. PDTC suppressed IL-1β-induced MCP1 expression in a dose-dependent manner (Figures 5A and 5B). We assayed NF-κB DNA-binding activity in IL-1β-treated HASMCs using EMSA. IL-1β induced an increase in the NF-κB-specific DNA-protein complex, while PDTC led to a decrease in complex formation (Figure 5C). To examine the upstream signaling pathway of IL-1β-induced NF-κB activation, we pretreated cells with several inhibitors, and examined NF-κB translocation promoted by IL-1β to the nucleus. D609 (a PC-PLC inhibitor), staurosporine (a PKC inhibitor) and PDTC (a NF-κB inhibitor) blocked IL-1β-induced NF-κB translocation to the nucleus, but not butanol (a PLD inhibitor) or AG18 (a tyrosine kinase inhibitor) (Figure 5D). These results clearly indicat that IL-1β induces MCP1 expression via NF-κB activation in PC-PLC- and PKC-mediated pathways, while PLD and tyrosine kinase are not required to activate NF-κB. Accordingly, we propose that IL-1β induces MCP1 expression via both NF-κB-dependent and -independent pathways (Figure 6).

IL-1β induces MCP1 expression via both NF-κB-dependent and -independent pathways. (A) HASMCs were pretreated with the indicated concentrations of the NF-κB inhibitor, PDTC, for 30 min prior to IL-1β (5 ng/ml) treatment for 10 h. Total RNA was isolated, and RT-PCR analysis was performed using MCP1 gene-specific primers and the internal control gene, β-actin. Two additional experiments yielded similar results. A representative study is shown. (B) The amount of secreted MCP1 protein was determined in the supernatant after IL-1β treatment for 10 h using the human MCP1 ELISA kit. Data are presented as mean values obtained from three independent experiments, and bars represent standard deviations. The significance was determined by Student's t-test (*P < 0.05 vs untreated control). (C) HASMCs were pretreated with PDTC at the indicated concentrations prior to IL-1β for 6 h. Nuclear extracts were prepared, and EMSA was conducted using [32P]-labeled NF-κB-specific oligonucleotide. (D) HASMCs were seeded onto 8-well chamber slides and pretreated with several inhibitors prior to IL-1β treatment for 6 h. After fixation and permeabilization, cells were stained with FITC-labeled anti-p65 antibody. FITC fluorescence was visualized under a confocal microscope.

Schematic representation of the signaling pathway of IL-1β-induced MCP1 expression in HASMCs. IL-1β activates PC-PLC to induce PKC activation and translocation of NF-κB to the nucleus. This activation cascade results in increased expression of MCP1. IL-1β additionally activates PLD and tyrosine kinase, which are involved in MCP1 expression independently of NF-κB translocation.
Discussion
Here, we describe the intracellular signaling pathways involved in IL-1β-induced MCP1 expression. IL-1β stimulated MCP1 expression in a time-dependent manner (Figure 1), which was completely suppressed by the PC-PLC specific inhibitor, D609, and PKC inhibitors, staurosporine and bisindolylmaleimide (Figure 2 and 3). PKC is activated by DAG, which is generated directly by the action of PLC. Our results show that PKC activation via PC-PLC-dependent DAG is involved in IL-1β-induced MCP1 expression (Figure 2 and 3). DAG is additionally produced indirectly via a pathway involving the production of phosphatidic acid by PLD, followed by a dephosphorylation reaction catalyzed by phosphatidate phosphohydrolase. This finding suggests that PLD is involved in IL-1β-induced MCP1 expression via PKC activation. However, the PKC inhibitor, staurosporine, interfered with NF-κB nuclear localization, but not the PLD inhibitor, butanol (Figure 5), indicating that PLD is not required for NF-κB activation responsible for MCP1 induction. Therefore, it appears that there are several signaling pathways underlying IL-1β-induced MCP1 expression. A variety of cis-elements are present in the promoter region of the MCP1 gene, including NF-κB, STAT, AP-1, and SP-1 sites (Ueda et al., 1994; Lim and Garzino-Demo, 2000; Lee et al., 2003). Recent reports show that PLD induces the production of cytokines via JNK activity, followed by activation of the transcription factor, AP-1 (Zhao et al., 2007). It is possible that PLD is involved in IL-1β-induced MCP1 expression through activation of the JNK/AP-1 pathway. IL-1β-stimulated MCP1 was completely suppressed by the NF-κB inhibitor, PDTC, but only partially blocked by the PLD inhibitor, butanol. PLD is possibly involved in IL-1β-induced MCP1 expression via activation of AP-1 (Figures 4A, 4B and 5). The tyrosine kinase inhibitors, genistein and AG18, blocked IL-1β-induced MCP1 expression, but not NF-κB translocation to the nucleus (Figures 4C, 4D and 5D). Thus, tyrosine kinase may be involved in MCP1 induction through another signaling pathway that does not require NF-κB translocation, such as PLD.
In the present study, we demonstrate that IL-1β mainly induces MCP1 expression via the PC-PLC/PKC/NF-κB signaling cascade, while another mechanism to induce MCP1 expression is mediated by PLD or tyrosine kinase. These results contribute to elucidating the signaling pathway promoting MCP1 expression in the preliminary step of atherosclerosis.
Methods
Cells and materials
Primary human aorta smooth muscle cells were kindly provided by Dr. In-Kyu Lee (Kyungpook National University, Taegu, Korea). Primary culture of human aorta smooth muscle cells (Cryo NHMC) and its corresponding growth medium (CC-3146 MsGM) were purchased from Clonetics (San Diego, CA). DMEM, containing 10% FCS, 20 mM HEPES buffer and 100 mg/ml gentamicin was used throughout the experiments. Human recombinant IL-1β and human MCP1 immunoassay kit were purchased from R&D systems (Minneapolis, MN). D609, U73122, genistein, AG18, staurosporine, bisindolylmaleimide, butanol, propanol, propranolol, and PDTC were acquired from Calbiochem (San Diego, CA), dissolved in DMSO and freshly diluted in culture medium for all in vitro experiments. The anti-p65 antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Other reagents were purchased from Sigma- Aldrich (St. Louis, MO).
RNA Isolation and RT-PCR
Total cellular RNA was extracted from cells using the TRIzol reagent (Life Technologies, Carlsbad, CA). cDNA was synthesized from 2 µg of total RNA using M-MLV reverse transcriptase (Gibco-BRL, Gaithersburg, MD). MCP1 cDNA was amplified by PCR with the following specific primers: (sense) 5'-CTCGCTCAGCCAGATGCAATCAAT-3' and (anti-sense) 5'-CCCAGGGGTAGAACTGTGGTTCAA-3'. PCR products were analyzed by agarose gel electrophoresis, and visualized using ethidium bromide.
Quantification of MCP1 expression
Following treatment of HASMCs with IL-1β (5 ng/ml) for 12 h, supernatant fractions were collected. MCP1 concentrations in culture medium were measured using a commercially available immunoassay kit (R&D systems), according to the manufacturer's instructions.
Immunofluorescence staining
HASMCs were grown on a chamber slide and treated with each inhibitor in the presence of 5 ng/ml IL-1β for 6 h. Cells were fixed with 4% paraformaldehyde, and washed twice with PBS. After washing, cells were permeabilized in 0.25% Triton X-100, and blocked with 1% BSA in PBS for 30 min before successive incubation with anti-p65 antibody (1:100) for 2 h and FITC-labeled anti-mouse antibody (1:200) for 1 h. Next, cells were washed twice with PBS. After additional washes, slides were mounted using anti-Fade mounting solution (Molecular Probes). Immuno-stained cells were visualized and photographed under a confocal microscope. Control cells were subjected to similar manipulations, but not treated with IL-1β.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from control or drug-treated cells as described previously (Beak et al., 2002). The double-stranded oligonucleotide used to detect the DNA-binding activities of NF-κB was 5'-AGTTGAGGGGACTTTCCCAGGC-3'. The reaction mixture for EMSA contained 20 mM Tris-HCl, pH 7.6, 1 mM DTT, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 1% NP-40, 1 µg poly (dI-dC) and 5 µg nuclear proteins. Unlabeled wild-type oligonucleotide was added to the reaction mixture and incubated for 10 min at room temperature. [32P]-labeled probe DNA (300,000 rpm) was added, and the binding reaction allowed to proceed for another 20 min. Mixtures were resolved on 8% polyacrylamide gels at 150 V for 4 h. Gels were dried, and subjected to autoradiography.
Statistical analysis
All data are presented as mean ± SD. Significant differences between the groups were determined using the unpaired Student's t-test. A value of *P < 0.05 was accepted as indication of statistical significance. All the figures shown in this article were obtained from at least three independent experiments with a similar pattern.
Abbreviations
- HASMCs:
-
human aorta smooth muscle cells
- MCP1:
-
monocyte chemoattractant protein-1
References
Baek WK, Park JW, Lim JH, Suh SI, Suh MH, Gabrielson E, Kwon TK . Molecular cloning and characterization of the human budding uninhibited by benomyl (BUB3) promoter . Gene 2002 ; 295 : 117 - 123
Boring L, Gosling J, Cleary M, Charo IF . Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis . Nature 1998 ; 394 : 894 - 897
Chen CC, Chen JJ, Chou CY . Protein kinase C alpha but not p44/42 mitogen-activated protein kinase, p38, or c-Jun NH(2)-terminal kinase is required for intercellular adhesion molecule-1 expression mediated by interleukin-1beta: involvement of sequential activation of tyrosine kinase, nuclear factor-kappaB-inducing kinase, and IkappaB kinase 2 . Mol Pharmacol 2000 ; 58 : 1479 - 1489
Chen YM, Chiang WC, Lin SL, Wu KD, Tsai TJ, Hsieh BS . Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: modulation by type III phosphodiesterase inhibition . J Pharmacol Exp Ther 2004 ; 309 : 978 - 986
Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP . Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism . J Biol Chem 2008 ; 283 : 18303 - 18313
Dinarello CA . Biologic basis for interleukin-1 in disease . Blood 1996 ; 87 : 2095 - 2147
Frasor J, Barkai U, Zhong L, Fazleabas AT, Gibori G . PRL-induced ERalpha gene expression is mediated by Janus kinase 2 (Jak2) while signal transducer and activator of transcription 5b (Stat5b) phosphorylation involves Jak2 and a second tyrosine kinase . Mol Endocrinol 2001 ; 15 : 1941 - 1952
Furukawa Y, Matsumori A, Ohashi N, Shioi T, Ono K, Harada A, Matsushima K, Sasayama S . Anti-monocyte chemoattractant protein-1/monocyte chemotactic and activating factor antibody inhibits neointimal hyperplasia in injured rat carotid arteries . Circ Res 1999 ; 84 : 306 - 314
Ghosh S, Hayden MS . New regulators of NF-kappaB in inflammation . Nat Rev Immunol 2008 ; 8 : 837 - 848
Iseki A, Kambe F, Okumura K, Niwata S, Yamamoto R, Hayakawa T, Seo H . Pyrrolidine dithiocarbamate inhibits TNF-alpha-dependent activation of NF-kappaB by increasing intracellular copper level in human aortic smooth muscle cells . Biochem Biophys Res Commun 2000 ; 276 : 88 - 92
Kawakami A, Aikawa M, Nitta N, Yoshida M, Libby P, Sacks FM . Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation . Arterioscler Thromb Vasc Biol 2007 ; 27 : 219 - 225
Lee YW, Hennig B, Toborek M . Redox-regulated mechanisms of IL-4-induced MCP1 expression in human vascular endothelial cells . Am J Physiol Heart Circ Physiol 2003 ; 284 : 185 - 192
Lim SP, Garzino-Demo A . The human immunodeficiency virus type 1 Tat protein up-regulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of SP-1, AP-1, and NF-kappaB consensus sites . J Virol 2000 ; 74 : 1632 - 1640
Lusis AJ . Atherosclerosis . Nature 2000 ; 407 : 233 - 241
Malinin NL, Boldin MP, Kovalenko AV, Wallach D . MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1 . Nature 1997 ; 385 : 540 - 544
Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR . Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1 . Eur J Immunol 1997 ; 27 : 1091 - 1097
Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, Usui M, Egashira K, Sugimachi K . Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits . Circulation 2002 ; 105 : 2905 - 2910
Pickett CA, Manning N, Akita Y, Gutierrez-Hartmann A . Role of specific protein kinase C isozymes in mediating epidermal growth factor, thyrotropin-releasing hormone, and phorbol ester regulation of the rat prolactin promoter in GH4/GH4C1 pituitary cells . Mol Endocrinol 2002 ; 16 : 2840 - 2852
Ruegg UT, Burgess GM . Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases . Trends Pharmacol Sci 1989 ; 10 : 218 - 220
Sasayama S, Okada M, Matsumori A . Chemokines and cardiovascular diseases . Cardiovasc Res 2000 ; 45 : 267 - 269
Terkeltaub R, Boisvert WA, Curtiss LK . Chemokines and atherosclerosis . Curr Opin Lipidol 1998 ; 9 : 397 - 405
Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC . Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice . Cancer Cell 2008 ; 14 : 408 - 419
Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T . NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene . J Immunol 1994 ; 153 : 2052 - 2063
Wu MY, Ho HN . The role of cytokines in endometriosis . Am J Reprod Immunol 2003 ; 49 : 285 - 296
Zhao Y, He D, Zhao J, Wang L, Leff AR, Spannhake EW, Georas S, Natarajan V . Lysophosphatidic acid induces interleukin-13 (IL-13) receptor alpha2 expression and inhibits IL-13 signaling in primary human bronchial epithelial cells . J Biol Chem 2007 ; 282 : 10172 - 10179
Acknowledgements
This work was supported by MRC at Keimyung University (R13-2002-028-03001-0).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lim, J., Um, H., Park, JW. et al. Interleukin-1β promotes the expression of monocyte chemoattractant protein-1 in human aorta smooth muscle cells via multiple signaling pathways. Exp Mol Med 41, 757–764 (2009). https://doi.org/10.3858/emm.2009.41.10.082
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2009.41.10.082
Keywords
This article is cited by
-
Glutathione, polyamine, and lysophosphatidylcholine synthesis pathways are associated with circulating pro-inflammatory cytokines
Metabolomics (2022)
-
5-LO-derived LTB4 plays a key role in MCP-1 expression in HMGB1-exposed VSMCs via a BLTR1 signaling axis
Scientific Reports (2021)
-
Cytokines as therapeutic targets for cardio- and cerebrovascular diseases
Basic Research in Cardiology (2021)
