1. Introduction
A key goal for a wastewater utility is efficient and cost-effective sanitation. In the tropics, wastewater stabilization ponds (WSPs) are favored because they remove enteric pathogens using a simple hydraulic design without chemical intervention [
1]. These systems rely on natural processes like sunlight disinfection, coupled with long hydraulic retention times to treat raw influent [
2]. In essence, WSPs have few financial overheads, are low maintenance and do not require specialist staff for operation [
3]. However, before the construction of a WSP, utilities need to consider whether the site can accommodate the system’s large spatial footprint and mitigate undesirable cyanobacterial blooms and sludge build-up in the ponds [
1,
4]. Sludge-filled ponds are inefficient because they are prone to ‘dead zones’ (pockets of stagnant/anoxic water) and treatment short-circuiting when exposed to wind shear [
5].
The presence of Cyanobacteria in these systems also contributes to increased suspended solids in treated effluent and additional expenditure for the removal of these unwanted bacteria [
4].
Fecal indicator bacteria (FIB) such as
Escherichia coli,
Enterococci and total coliforms are common surrogates for human pathogens, and are used to assess water quality and fecal disinfection in WSPs [
6]. With our expanding knowledge of bacterial communities in diverse environments like WSPs, it is now possible to refocus our attention from a purely fecal-bacterial disinfection perspective, to include how the ponds function in other roles like nutrient cycling in the assessment of the performance of WSPs.
Applying a whole community approach (WCA) to identify complementary bacteria can ultimately lead to a diverse set of monitoring tools. Using the WCA, we can examine the entire wastewater community, which will improve our understanding of wastewater bacterial communities, their dynamics and how they interact with biotic and abiotic factors [
7]. Expanding our bacterial inventory for sewage using the WCA would inform utilities of which bacteria were significantly changing throughout the pond system. Also, this method could show how climatic conditions influence sewage bacteria, and what that means for sanitation, nutrient removal and monitoring frequency [
8]. For example, utilities could answer whether or not the number of ponds is sufficient to cope with storm water [
9]. Furthermore, information from a WCA can identify alternative indicators for WSPs, improve the understanding of the pond function (e.g., nitrogen removal) and determine whether including key WCA-informed pathogenic and non-pathogenic bacteria with routine
E. coli die-off can strengthen WSP monitoring. Moreover, there is growing evidence which suggests that suitable indicator species could also be included from multiple non-fecal origins, since these ‘environmental’ bacteria significantly contribute to sewage microbiomes [
8,
10,
11]. Thus, while
E. coli bacteria are the current monitoring tool, there is a need to find complementary bacteria that reflect the pond function.
Cyanobacteria in WSPs can reduce effluent water quality in tropical regions [
4] and should be considered as part of a robust monitoring plan. Cyanobacterial blooms can release toxins that create public health concerns and kill aquatic animals, therefore utilities need to identify the correct wastewater retention time for safe sanitation before ponds become a reservoir for Cyanobacteria [
12,
13]. Warm, calm waters coupled with high solar radiation and nitrogen and phosphorus nutrient concentrations present ideal conditions for Cyanobacteria [
14]. Since Cyanobacteria are abundant in calm waters, it is likely these bacteria will be more common in ponds that are downstream of other ponds receiving raw influent [
4]. Studies in temperate regions show significant seasonal influences on cyanobacterial numbers, but in the tropics the seasonal effect may be subtle due to year-round warm temperatures and high solar radiation [
9,
14].
Pond systems in the wet-dry tropics including north Australia may have accelerated sludge build-up, water stagnation, short-circuiting and high cyanobacterial populations [
1,
4,
15]. These WSPs experience high seasonal rainfall in the wet season, high rates of evaporation in the dry season along with high solar radiation (UV) and warm air temperatures year-round [
4]. In particular, the dry season conditions of warm air temperatures and constant UV exposure promote Cyanobacteria, the presence of suspended particles and sludge build-up [
15]. Therefore, monitoring tools need to account for the array of year-round climatic and biological challenges that can affect bacterial and chemical levels.
In this study we focused on a suburban WSP (Sanderson WSP), a multi-pond system in the wet-dry tropics. Routine microbiological monitoring of this Sanderson WSP is FIB enumeration (
Escherichia coli,
Enterococci and total coliforms) [
4], which means that other resident WSP bacteria represent a ‘black box’ [
3,
16]. Previous studies on this system indicate that the pond-water chemistry and bacteria fluctuate both spatially and seasonally [
4,
17,
18].
E. coli decay data from chamber studies in the WSP indicate log removal [
17], but this does not shed light on the other bacteria that are performing nutrient removal services or the overall performance of the system [
16].
Because the bacterial ecology of the Sanderson wastewater treatment plant is virtually unknown, this study will address two aims: to describe the bacterial composition throughout the WSP and identify new indicators to complement E. coli to improve monitoring.
In addition, we will test if routinely measured physicochemical parameters are reflective of bacterial community change. As indicated above, space and time are likely drivers of bacterial change in this wet-dry tropical WSP system. Therefore, we expect the bacterial community to significantly change between the wet and dry seasons and between the ponds.
2. Materials and Methods
2.1. Study Site
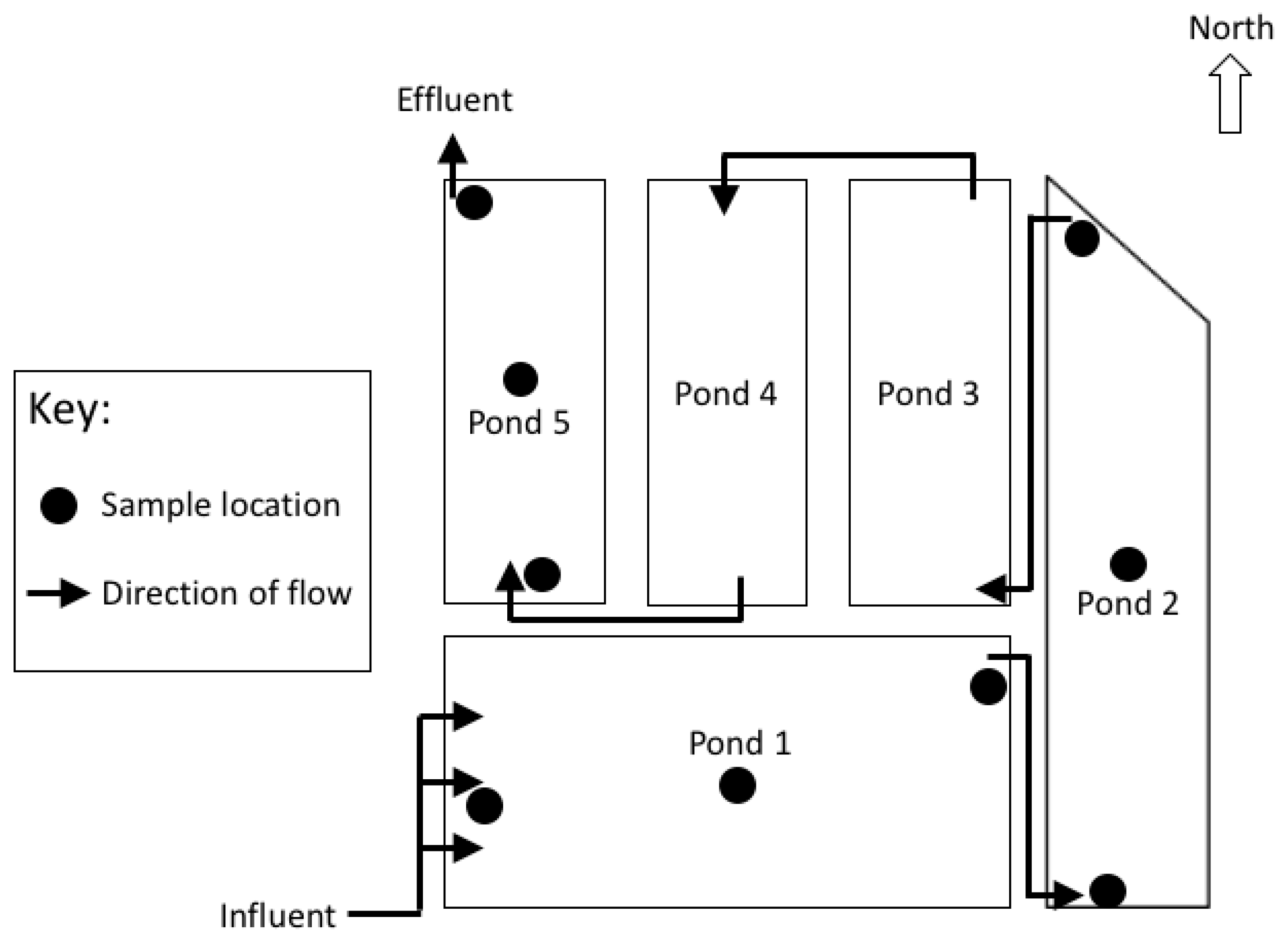
The Sanderson wastewater stabilization ponds (WSP) system comprises five ponds (
Figure 1). Pond 1 is a 2.4 m deep facultative pond which receives raw influent. The remaining four ponds are shallow maturation ponds (
Figure 1). Wastewater entering the Sanderson WSP is retained for an estimated 23 days before the treated effluent is released from the Pond 5 outlet. During the wet season (November to April, Southern Hemisphere), the system is expected to treat approximately 105 ML/day. In peak wet season (Summer, January to March) there is monsoonal rainfall (total rainfall ~1024 mm), high humidity, mean temperatures between 24.7–32 °C, and the highest wind speeds of the year (~134 km/hr) [
19]. The WSPs are managed to cope with flooding and sewage dilution [
4]. The dry season (May to October), is characterized by lower humidity and rainfall (total rainfall ~270 mm), warm, sunny days and cooler nights (mean temperatures between 21.6–31.8 °C). Both the low rainfall and high evaporation (~7.1 mm/d) [
19] concentrate this WSP sewage.
2.2. Wastewater Collection
Wastewater samples (288) were collected in duplicate from ponds 1, 2 and 5 during the early wet (November and December) and dry (Winter, August and July) seasons in 2012 and 2013 (total of four occasions). In a pilot study using total bacterial fingerprinting (denaturing gradient gel electrophoresis), bacterial diversity was at its greatest in ponds 1, 2 and 5 (data not shown), so we focused on these ponds. At each site, surface and benthic water depths were sampled to target aerobes in the oxic top 10 cm of the water column and anaerobic bacteria in the bottom 10 cm from each pond’s inlet, middle and outlet. For each field campaign, samples were collected twice, once in the morning (6–10 am) and again in the afternoon (1–5 pm). In situ measurements of dissolved oxygen (DO), temperature and pH were collected during sampling using a HYDROLAB® Quanta® water quality instrument (Hydrolab Corporation®, Austin, TX, USA).
2.3. Water Physicochemistry and Routine Fecal Indicator Bacteria (FIB) Culture Measurements
To determine if changes in standard water chemistry are associated with changes in microbial communities, we measured the same physicochemical variables that are routinely measured by the WSP operators. Wastewater samples of 1 L, 500 mL, 250 mL, 250 mL and 100 mL were collected for nutrients, biological oxygen demand (BOD), total organic carbon (TOC), total suspended solids (TSS)/total volatile solids (VSS), and alkalinity, respectively. One liter of water was also collected for bacterial community analysis. All samples were kept on ice during sampling. BOD was analyzed using the standard method 5210* [
20] by the Water Chemistry Laboratory, Department of Primary Industry and Fisheries, Northern Territory (NT). TSS and VSS were measured and calculated according to ‘Standard Methods for the examination of water and wastewater’ [
21]. Alkalinity was measured using an in-house Gran method with burette-titration (0.1 N Hydrochloric acid). Alkalinity species were determined using the USGS web-based alkalinity
http://or.water.usgs.gov/alk/calculator. TOC analysis was according to the American Public Health Association method, APHA 5310B Total Organic Carbon [
22] LabMark Pty Ltd. (Melbourne, VIC, Australia). Nutrient water chemistry was also analyzed by LabMark using unfiltered 1 L wastewater samples stored at −20 °C before analysis. Flow injection analysis (FIA) was used to determine ammonia, nitrate, nitrite and orthophosphate [
23]. Prior to analysis, 15 mL of sample were filtered through polyethersulfone (PES), 0.45 µm Minisart
® high flow syringe filters (Sartorius Stedim, Biotech, Göttingen, Germany). For total Kjeldahl nitrogen (TN) and total phosphorus (TP), 10 mL of the remaining unfiltered sample was digested with alkaline potassium persulfate in the autoclave for 1 h at 121 °C, and also analyzed by FIA (Queensland Health Scientific Services, Coopers Plains, QLD, Australia).
E. coli and Enterococci were measured at the Pond 1 inlet and outlet, Pond 2 outlet and Pond 5 outlet by the Water Microbiology Laboratory (Dept. Primary Industry and Fisheries, Darwin, NT, Australia). E. coli were measured using Idexx Colilert AS4276.21-2005 and Enterococci were measured by Idexx Enterolert ASTM D6503-14 (2014) (IDEXX Laboratories Pty Ltd., Rydalmere, NSW, Australia). The detection limit for E. coli and Enterococci was one colony-forming unit (CFU) per 100 mL.
2.4. Bacterial Community Sequencing
To avoid clogging filter papers with algae, water samples (1 L) were left to settle overnight at 4 °C before filtering 100 mL through 0.45 µm filters (Pall Corporation, New York, NY, USA). DNA was extracted using the PowerWater DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA), following the manufacturer’s protocol.
Extracted DNA was sent to Molecular Research LP (MR DNA, Shallowater, TX, USA) for amplicon sequencing on the Illumina MiSeq platform targeting the V4V5 variable 16S rRNA region. Sequences were edited and classified using the MR DNA proprietary analysis pipeline (
www.mrdnalab.com, Shallowater, TX, USA). Briefly, sequences were depleted of barcodes, and sequences were removed if < 200 bp (base pairs), or they had ambiguous base calls or homopolymer runs exceeding 6 bp. Remaining sequence data was denoized, chimeras and singleton sequences were removed and operational taxonomic units (OTUs) generated using clustering at 3% divergence or 97% similarity [
24,
25,
26,
27,
28,
29]. MR DNA then taxonomically classified the remaining OTUs with BLASTn against the curated GreenGenes database [
30]. OTU sequence data (
Table S1) and OTU metadata (
Table S2) are available at
Supplementaly Materials.
2.5. Statistical Analysis of Physicochemistry and Bacterial Community Sequences
Subsequent filtering of sequences included comparing the sequence number between samples and excluding those with an outlying low sequence number (<10,532). However, initial filtering did not exclude any samples, because all were above the 10,000-sequence number threshold. OTUs found in only one sample were also excluded and data rarefied to 10,000 reads in phyloseq (Bioconductor, Bioconductor, Buffalo, NY). Before rarefying to 10,000 reads, each sample was assessed using rarefaction curves for potential loss of diversity (
Figure A1 and
Figure A2). Sequence data were square root transformed and a resemblance matrix was generated based on Bray-Curtis similarity. Physicochemical data were prepared for analysis by normalizing, removing co-linear variables (VSS, bicarbonate, PO
4+) and generating a resemblance matrix based on Euclidean distance.
Data were analyzed in R (version 1.1.423) using the packages phyloseq in Bioconductor [
31] and IndVal [
32]; in Primer-7 (Primer-E, Plymouth, UK), GenGIS version 2.4.1 [
33], Stata-14 IC (STATA Corp, TX, USA), Cytoscape (version 3.4.0,
www.cytoscape.org) and CoNet [
34].
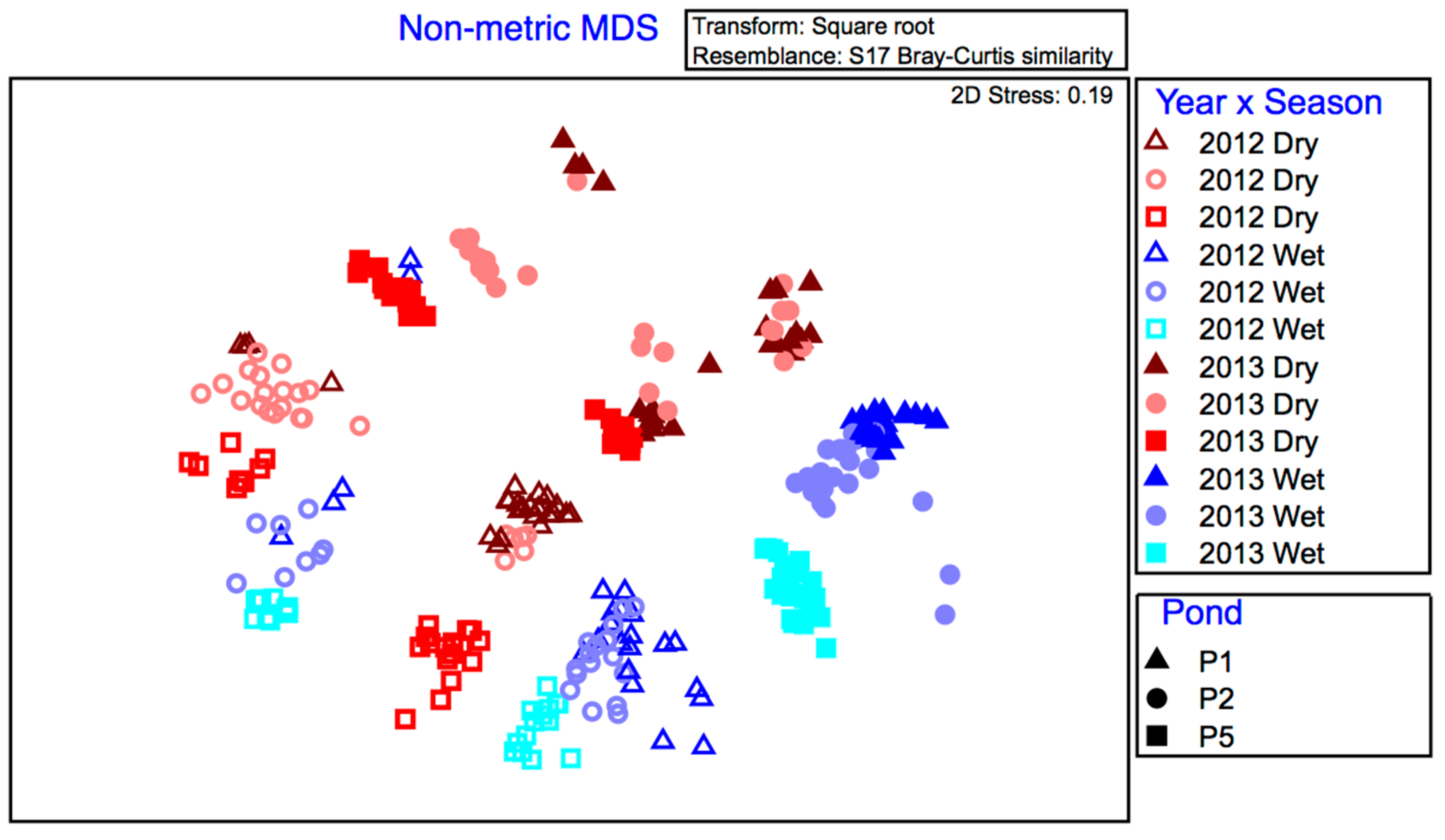
Differences in the bacteria between groups of samples were analyzed by permutational analysis of variance (PERMANOVA) with 9999 permutations. A cross design was used for the PERMANOVAs with six fixed factors: Year (2 levels), Season (2 levels), Pond (3 levels), Location (3 levels), Time (2 levels) and Depth (2 levels). A P value of < 0.05 (two-sided) was considered significant. For multiple comparisons, the Bonferroni correction was applied to P-values to counteract the chance of incorrectly rejecting the null hypothesis. PermDISP (Primer-E Ltd., Plymouth, UK) was used to check for homogeneity of variance between groups. Significant differences between levels of factors were identified using non-parametric pairwise testing.
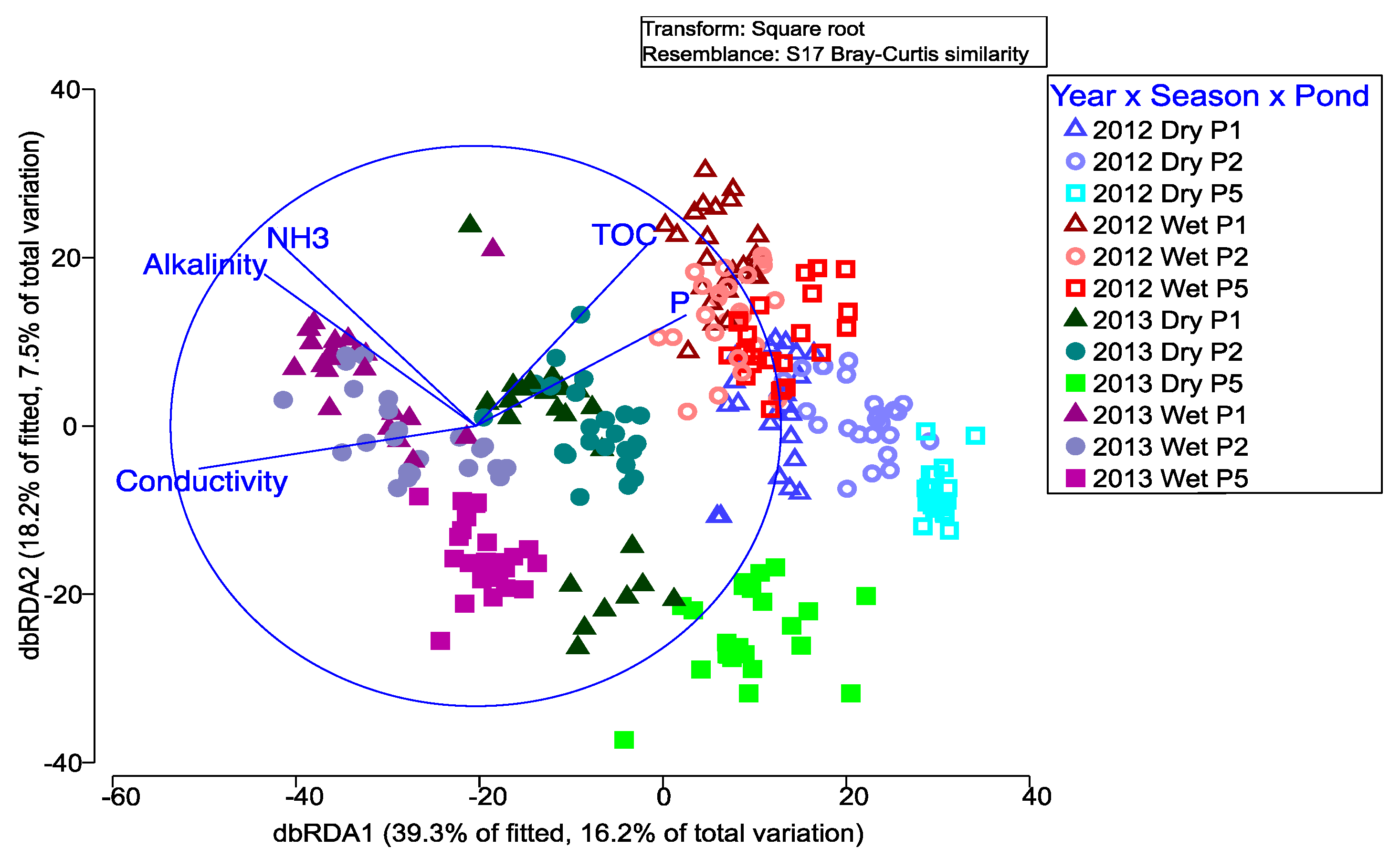
The relationship between the bacteria and physicochemical data was tested using a Distance-based Linear Model (DistLM) and distance-based redundancy analysis (dbRDA). Collinearity between physicochemical variables was checked, and VSS, bicarbonate and orthophosphate excluded from the analysis. Model selection for dbRDA was based on the lowest AIC and BEST elimination. Taxa sampled from each pond location in 2012–2013 were overlaid onto a georeferenced 2013 Google Earth© image using GenGIS. Differences between the phyla were analyzed by PERMANOVA using 9999 permutations and the same six fixed factor cross design described above (
Table A1). Key dominant WSP phyla were examined by calculating their family-level relative abundances with phyloseq and visualizing with ggplot2. Taxa patterns within the Firmicutes, Bacteroidetes and Cyanobacteria phyla were also examined by sub-setting each phyla with phyloseq and calculating the family-level relative abundances. We chose family-level analyses because the 16S region cannot accurately differentiate between closely-related species (Vêtrovský and Baldrian, 2013). For each phyla subset, the family-level change was assessed by PERMANOVA and the percent contribution of families were analyzed by Similarity Percentages (SIMPER) with a 50% contribution cut off (
Table A2,
Table A3 and
Table A4). Indicator bacteria were defined as those taxa that were present in 100% of samples (n = 96) from a particular pond. IndVal [
32] was used to identify indicator taxa, and Cytoscape was used to show their relative abundance across the different ponds. Because these indicator bacteria are not currently used for WSP monitoring, we refer to them as ‘new’ or ‘indicator candidates’, because they are not yet validated. The core microbiome was taken to be taxa present in 90% of all samples [
35] to distinguish between bacteria that were consistently found in wastewater (hereafter referred to as the ‘core microbiome’) with bacteria that are transient and opportunistic [
35,
36,
37,
38].The co-occurrence of core bacteria was determined by CoNet analysis [
34]. To calculate co-occurrence, we set a minimum of 20% occurrences among replicates and transformed (log-2) the data values. An automatic threshold was used to include only the top and bottom 100 edges. Kendall rank correlations (threshold = 0.05) were calculated after generating a Bray Curtis distance matrix (threshold = 0.05), and we tested the strength of the correlations between taxa with Fisher’s Z-test, while accounting for multiple testing using Bonferroni to include only those taxa that significantly (
P < 0.05) co-occurred.
4. Discussion
The bacterial communities in wastewater stabilization ponds (WSPs) at Sanderson were dynamic over space (ponds) and time (season and years). Other wastewater treatment pond studies suggest that the composition of fecal and non-fecal bacteria can change between wastewater systems, and are likely responding to different climatic conditions [
8,
11,
39,
40,
41]. A two-year study of an anaerobic bioreactor also showed that the bacterial composition changed every few months [
42]. Because wastewater systems and ponds can have dynamic bacterial populations, we recommend pond managers collect regular samples over several years to develop a comprehensive baseline of bacterial communities.
We measured standard WSP physicochemical variables and found that conductivity explained 14% of the bacterial change. However, over 76% of the bacterial change was not explained by the physicochemical variables measured, suggesting that other unmeasured variables likely influenced the pond bacteria. There is conflicting information on the physical and chemical drivers of bacterial population change [
16], although it is generally accepted that diverse bacterial communities are likely to consist of a vast array of different ecological niches and nutritional pathways [
11,
41].
Consequently, it is not surprising that the overall influence of a single variable like TOC is not consistent over time or between studies. Previous studies have also concluded that unmeasured variables are likely involved in bacterial community change [
4,
17,
18]. Potentially important factors that have not been included in analyses of the bacterial communities at Sanderson include sewage inflow and specific weather variables. A previous Sanderson diel study showed that the water chemistry changed according to the rate of raw sewage inflow [
18]. Similarly, Shanks et al. [
8] also found inflow affected the bacterial composition because the bacteria that line sewer pipes seed wastewater ponds. Weather variables (e.g., wind direction and speed, cloud cover, irradiance and rainfall) could explain the distinct Sanderson bacterial communities measured in 2013 after the second driest wet season on record [
19]. To fully understand and predict the dynamic bacterial change in wastewater systems, it may be necessary to expand the variables measured to include wind parameters, inflow rates, rainfall and solar radiation.
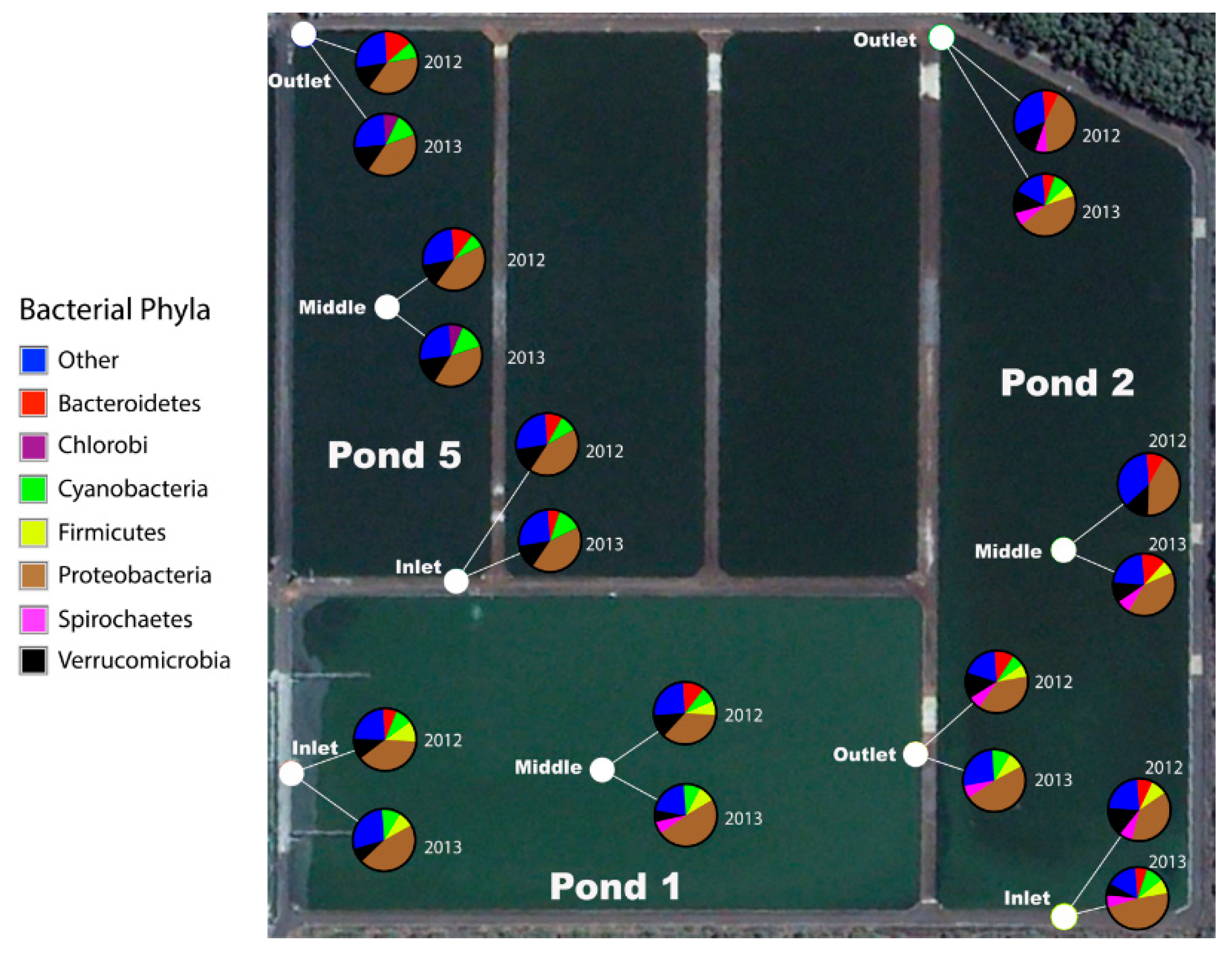
When the bacterial community in the raw influent and ‘treated’ effluent are not significantly different, this is taken as an indication that sanitation in a WSP has failed [
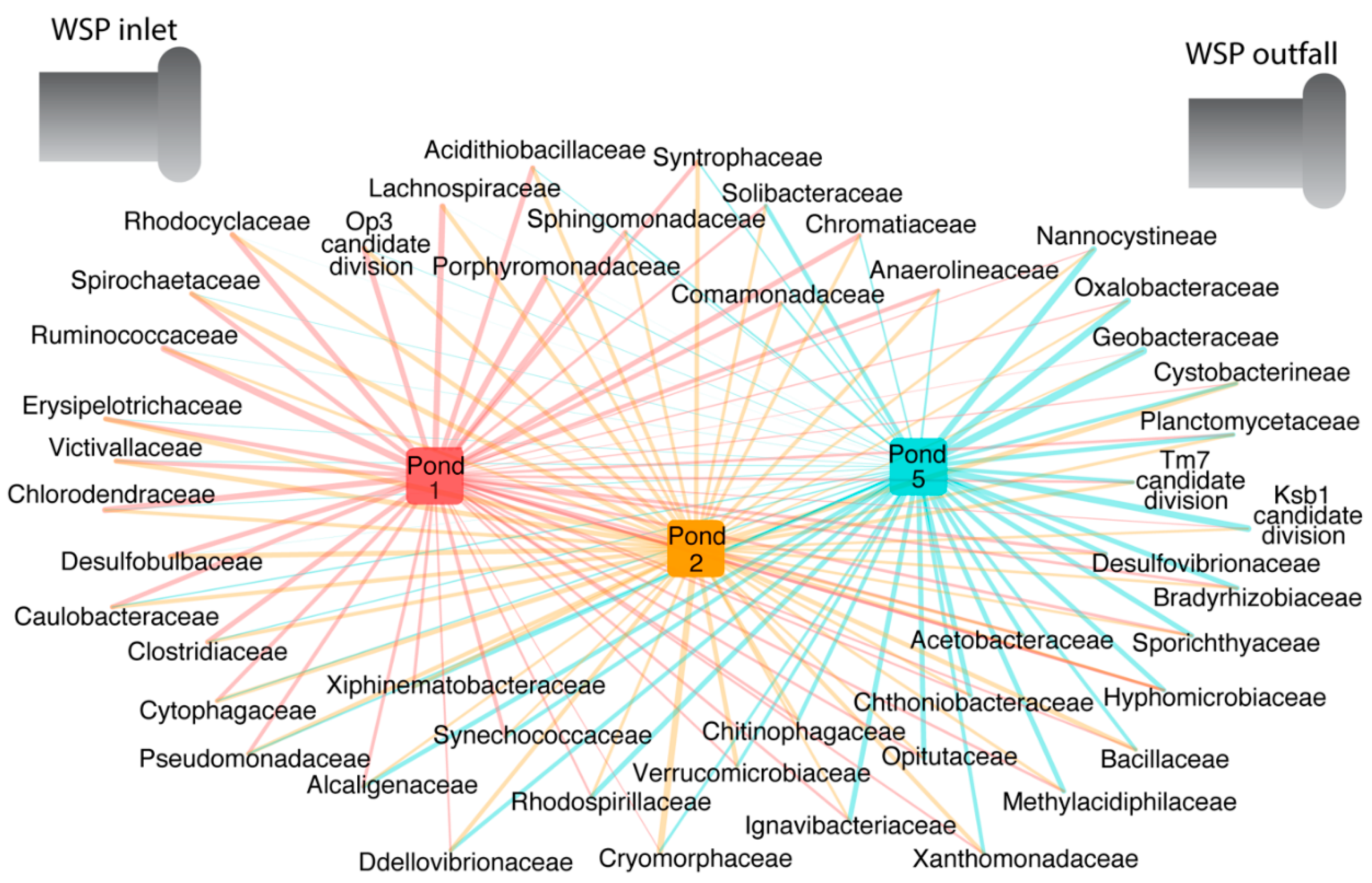
1]. In contrast then, if ponds are functioning and effecting sanitation, it ought to be possible to show this through a measurable difference in the bacterial communities in the influent compared to the treated effluent. Using 16S rRNA gene sequencing, we identified new pond-indicator bacteria that represent each treatment pond and can complement current standard FIB. For the measured facultative and maturation ponds, new pond-indicators were identified using IndVal. Indicators for each pond were defined as those taxa that were present in 100% of the samples from a target pond (
Figure 4). Many of the indicators of the facultative (Pond 1) and first maturation pond (Pond 2) are common in the human gut, such as
Clostridiaceae,
Ruminococcaceae and
Lachnospiraceae [
16,
43]. These bacterial groups were also detected in other sewage studies [
8,
11,
41]. However, non-fecal bacteria were also detected in the first pond and were a conspicuous component of the sewage microbiome (
Figure 5 and
Figure 8). The co-presence of dominant non-fecal bacteria was also reported in other studies, presumably because the influent is a mix of gray water, effluent and pipe biofilms, all of which enter the waste stabilization ponds, mixing both human-associated bacteria with those found in the environment [
8,
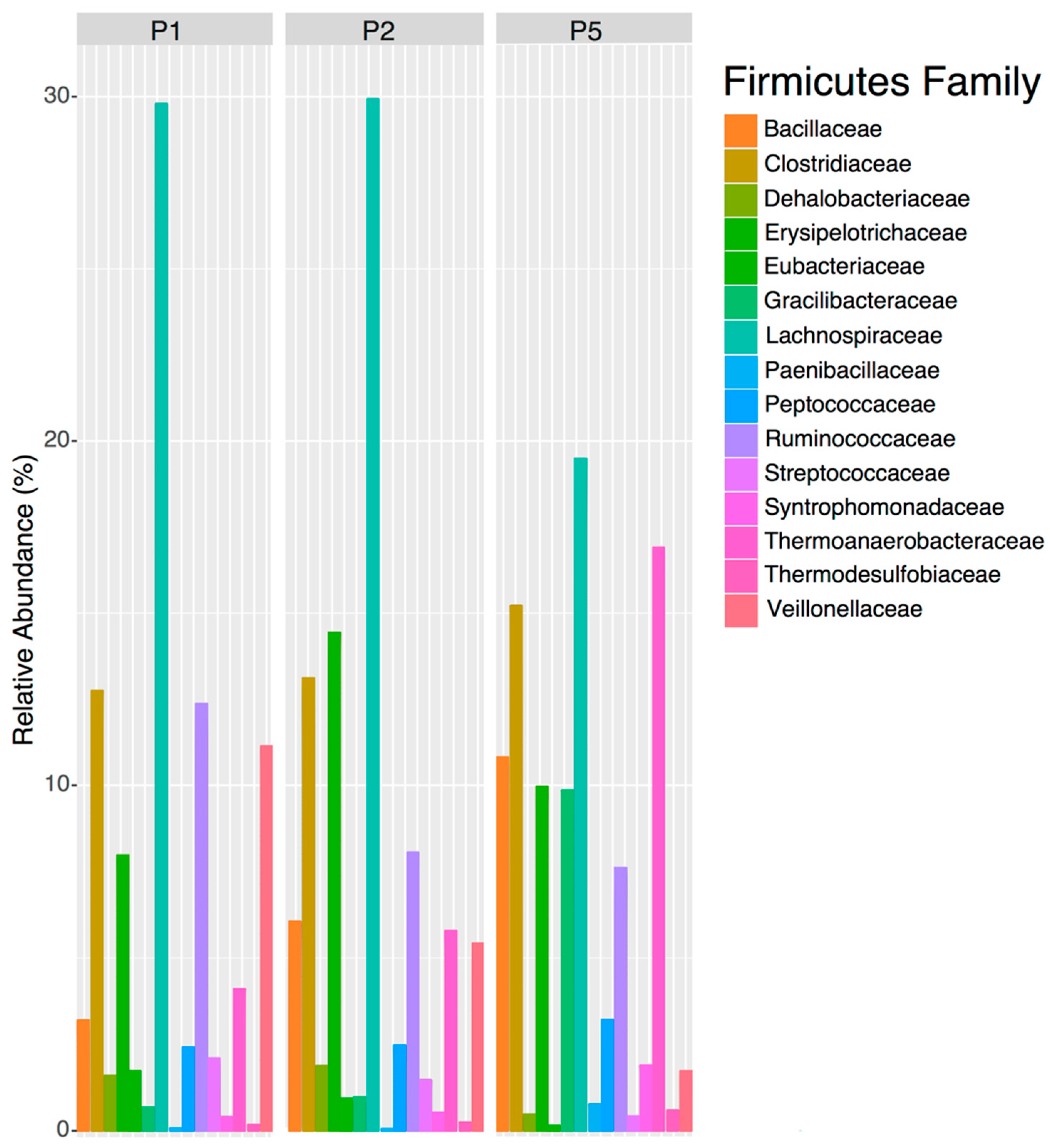
44]. By the final maturation pond (Pond 5) the human-gut bacteria from the first two ponds were largely replaced by bacteria that are typically found in the environment and contribute to ecosystem function, like nitrogen cycling. This pattern suggests that the Sanderson ponds are removing the human-gut bacteria, and that nitrogen removal is highest after Pond 1. Thus, the succession pattern of the new pond-indicators, in which human-gut bacteria in ponds 1 and 2 are supplanted by environmental bacteria in Pond 5, suggests that the Sanderson ponds are performing their expected function. These results suggest that future indicators of human-fecal pollution should target Firmicutes families like
Ruminococcaceae,
Spirochaetaceae and
Clostridiaceae.
In addition to identifying new candidates for pond-indicator bacteria, whole-community analysis of the WSP has shed light on the previous microbiological ‘black box’ for these ponds and challenged some of the previous assumptions about non-fecal bacteria and Cyanobacteria. We found that non-fecal bacteria dominated the core wastewater microbiome for Sanderson. This result is similar to other wastewater studies that found 80–90% of bacteria are non-fecal [
8,
11,
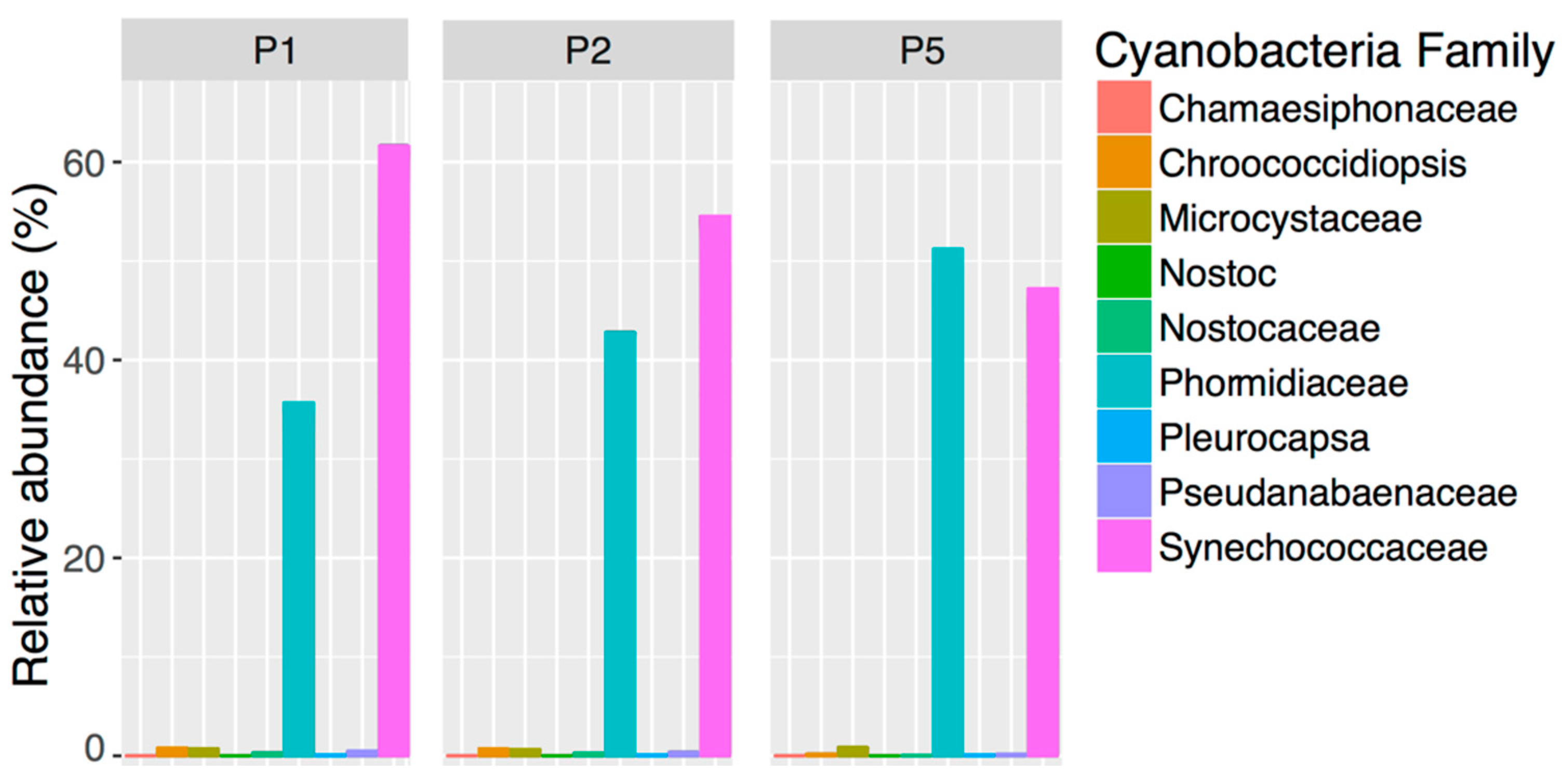
45]. Cyanobacteria represented greater than 6% of the WSP bacterial population regardless of sample timing and location, which was primarily due to the high relative abundance of the families
Synechococcaceae and
Phormidiaceae. Within the wastewater industry, it is assumed that Cyanobacteria become problematic in maturation ponds due to the warm, calm and low organic loading/nutrient conditions [
4,
46]. Consequently, pond managers have considered replacing these ponds with an aerated rock filter to reduce retention time, ammonia levels and Cyanobacteria [
4]. Contrary to expectations of pond managers, Cyanobacteria were abundant at the Pond 1 inlet, suggesting that they may be entering the WSP in the influent. Thus, whole community analysis is a useful tool for identifying the resident bacteria in a WSP and testing assumptions about key taxa before implementing management strategies.
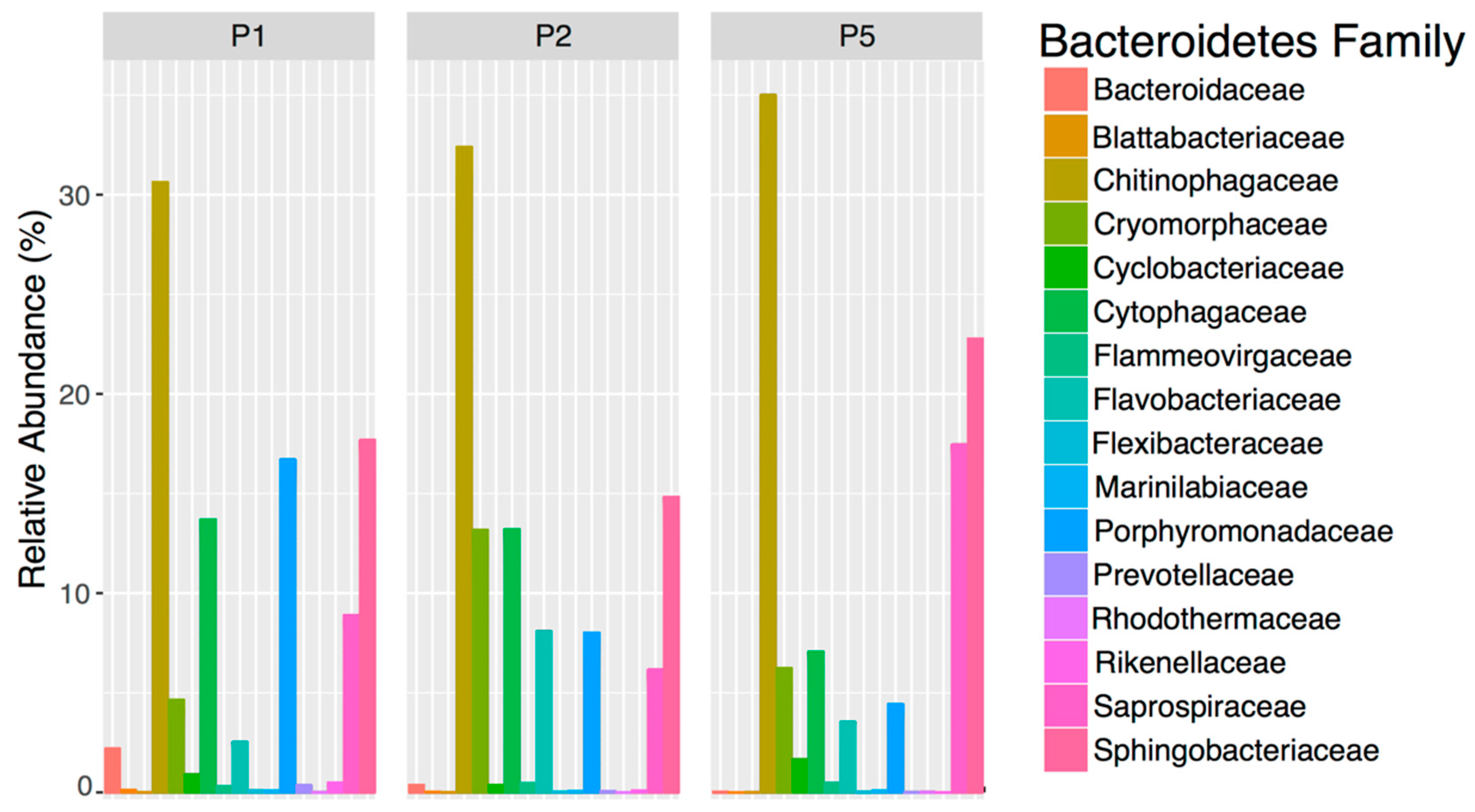
It is imperative to consider the taxonomic classification level when identifying indicators for a bacterial population, because bacteria with the same high-level taxonomic classification can have different ecological roles. For example, we found that Bacteroidetes (phylum) did not decline from ponds 1–5. The continued persistence of Bacteroidetes across the ponds was unexpected because, although a diverse bacterial phylum, they are considered representative of fecal bacteria [
4,
46]. However, a more in-depth investigation of Bacteroidetes at the family level revealed that fecal groups like
Bacteroidaceae did, in fact, decline between ponds 1–5 and that their persistence at the phylum level was likely due to the increase in those families that typically occur in the environment like
Chitinophagaceae [
47]. Currently, there is no consensus for which bacterial taxonomic level to use when classifying indicators, with some studies on wastewater treatment using multiple levels, while others use the family level [
8,
11,
44,
48]. Therefore, to avoid including taxa that may complicate spatial and temporal patterns, we recommend choosing the lowest possible taxonomic level for pond indicators.
In this study,
E. coli sequences were not detected, and
Enterococci sequences were detected in less than 4% of samples, but we note that
E. coli and
Enterococci were cultured from these ponds.
E. coli were not resolved by 16S rRNA sequencing because the short sequence length was not sufficient for taxonomic resolution of this genus [
49,
50]. It is also possible that the DNA extraction method from a highly complex wastewater matrix and diverse microbial population results in different lysis efficiencies for different bacterial groups, and may not have been suitable for gram positive
Enterococci [
51,
52,
53]. There are several other possible explanations: Preferential primer binding during DNA amplification to other bacterial DNA present [
54] or the abundance of these bacteria in samples was rare compared to other taxa and their DNA was not amplified to detectable levels [
53]. Regardless, 16S rRNA sequencing was intended to supplement routine FIB monitoring, and not be utilized as a replacement.
In addition to the current monitoring practices, managers could apply our new pond-indicator candidates, which are a combination of human-gut and environmental bacteria. Because most Sanderson pond-indicators co-occur, it is possible to select a single family in each group as a representative. For example, the pond 1 and 2-indicator family Spirochaetaceae could act as a surrogate for Lachnospiraceae and Desulfobulbaceae because they co-occur in Sanderson wastewater. In future, combining our new pond-indicators with the standard fecal indicator bacteria will lead to a robust monitoring approach that is not only locally relevant, but also provides a bespoke tool-box with indicator candidates for tropical environments worldwide.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









