Comparative Analysis of the Liver Transcriptome among Cattle Breeds Using RNA-seq

 , , , , , , ,
, , , , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals and Biological Sample Collection
2.2. Laboratory Methods
2.3. Sequence Quality Control and Read Mapping
2.4. Breed Comparisons Analysis of RNA-seq Read Count Data Using DEseq and EdgeR Bioconductor Packages
2.5. Comparative Analysis of GO Terms among Cattle Breeds Using TopGO and ClueGO Packages
2.6. Validation of DE Gene-Transcripts Using RT-PCR/qPCR
2.6.1. RT-PCR/qPCR
2.6.2. qPCR Statistics
3. Results
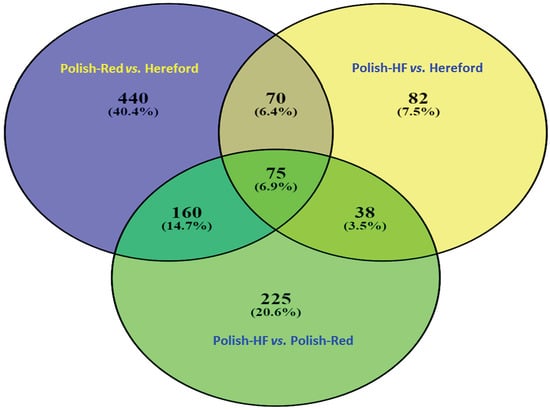
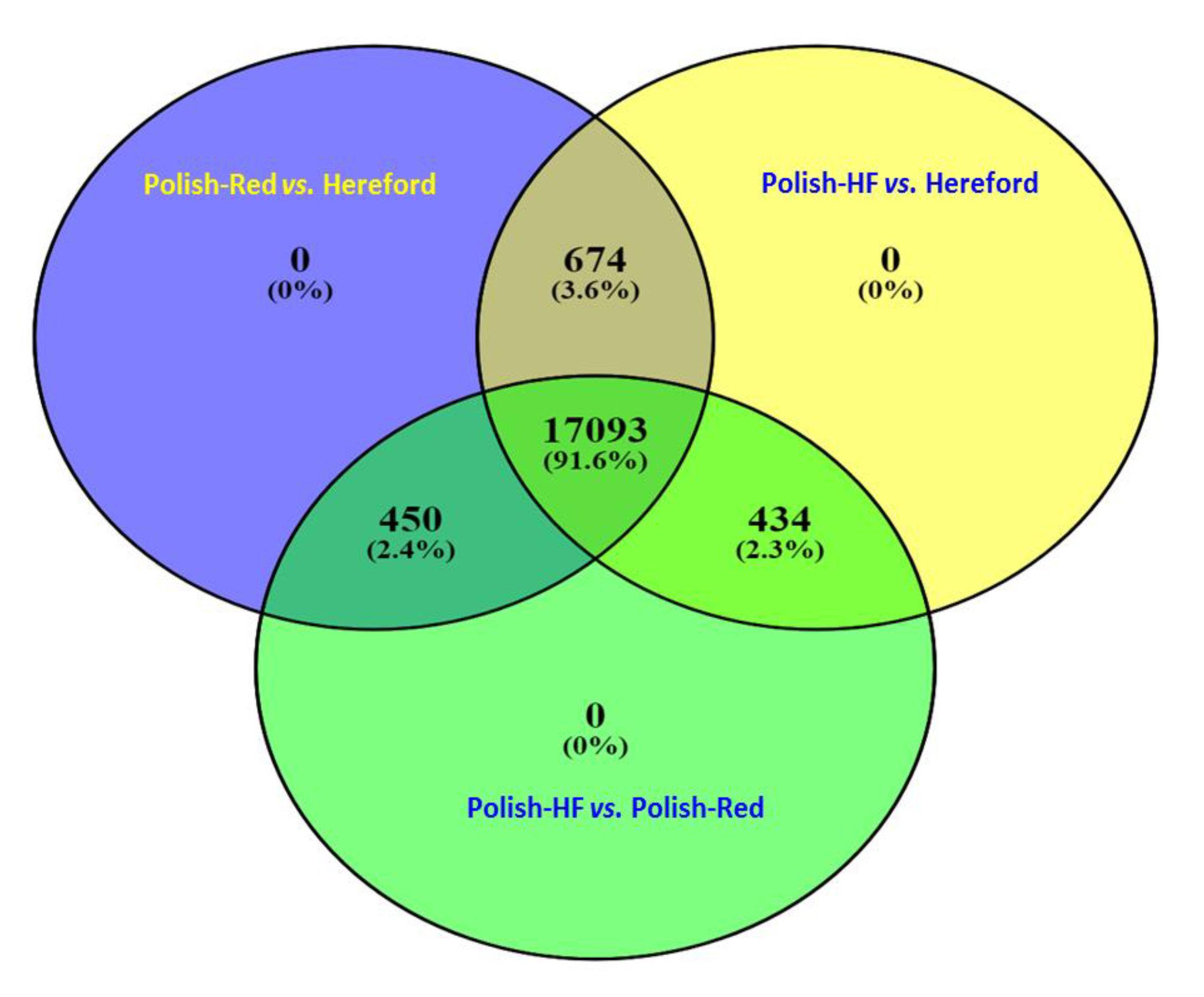
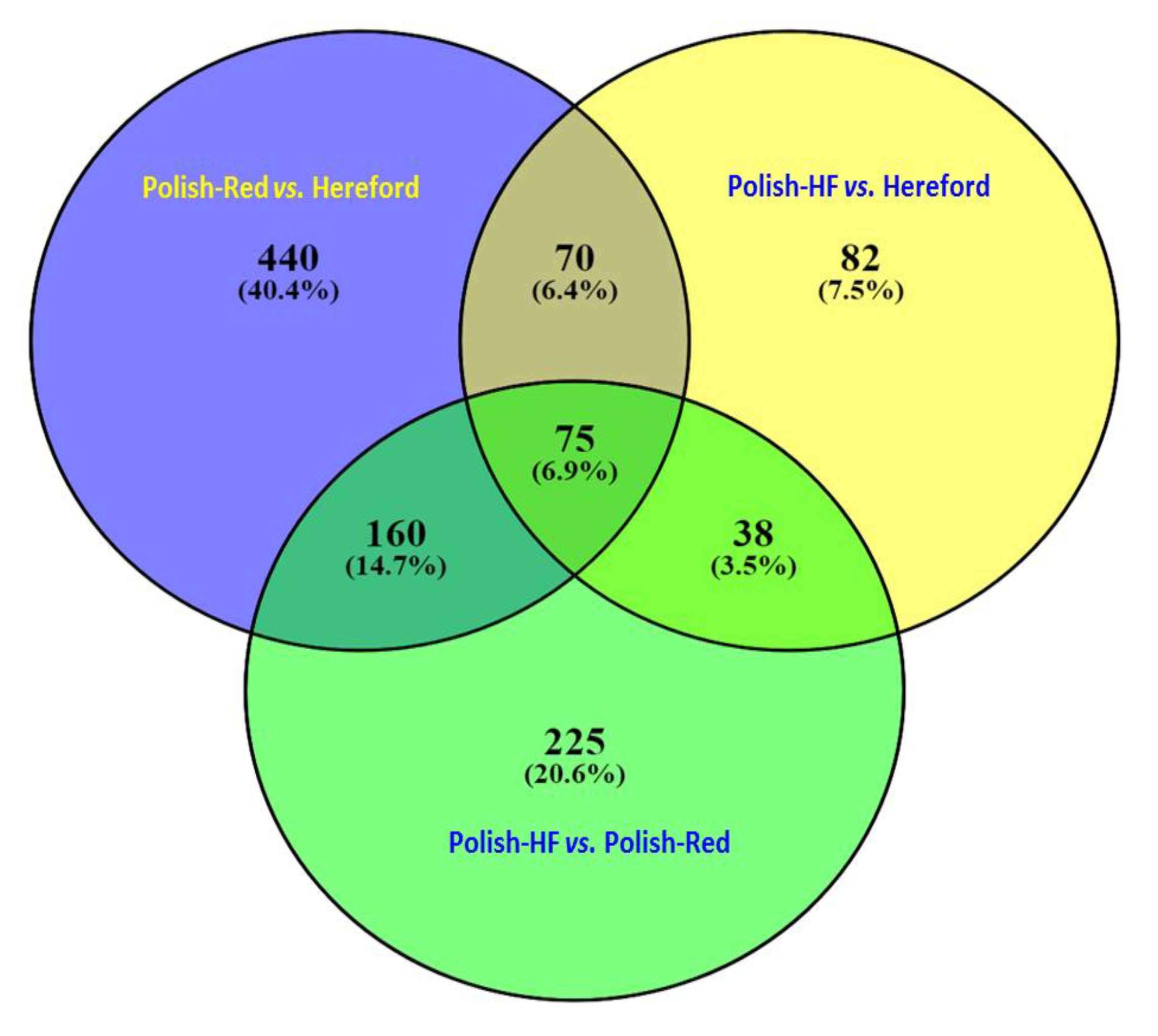
3.1. Comparison of Liver Transcriptome in Cattle Breeds
3.2. Comparison of Gene Ontology Terms among Cattle Breeds Using TopGO Enrichment Analysis
3.3. Comparison of Gene Ontology Terms among Cattle Breeds Using Cytoscape-ClueGO
3.3.1. Assignment of Genes to Functional Pathways
3.3.2. Interpretation and Visualization of Functionally Group Terms in the Form of Gene Networks and Pathways Charts
3.4. Validation of DE Gene-Transcripts Using Quantitative Real Time PCR (qPCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Nadeem, A.; Zhang, B.; Babar, M.; Soller, M.; Khatib, H. Characterization and comparison of the leukocyte transcriptomes of three cattle breeds. PLoS ONE 2012, 7, e30244. [Google Scholar] [CrossRef]
- Litwińczuk, Z.; Żółkiewski, P.; Florek, M.; Chabuz, W.; Domaradzki, P. Semi-intensive fattening suitability and slaughter value of young bulls of three Polish native breeds in comparison with Polish Holstein-Friesian and Simmental. Ann. Anim. Sci. 2014, 14, 453–460. [Google Scholar] [CrossRef]
- Sadkowski, T.; Jank, M.; Zwierzchowski, L.; Oprzadek, J.; Motyl, T. Transcriptomic index of skeletal muscle of beef breeds bulls. J. Physiol. Pharmacol. 2009, 60, 15–28. [Google Scholar] [PubMed]
- Domaradzki, P.; Florek, M.; Staszowska, A.; Litwińczuk, Z. Evaluation of the Mineral Concentration in Beef from Polish Native Cattle. Biol. Trace Elem. Res. 2016, 171, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Berton, M.P.; Fonseca, L.F.; Gimenez, D.F.; Utembergue, B.L.; Cesar, A.S.; Coutinho, L.L.; de Lemos, M.V.; Aboujaoude, C.; Pereira, A.S.; Rafael, M.D.; et al. Gene expression profile of intramuscular muscle in Nellore cattle with extreme values of fatty acid. BMC Genom. 2016, 17, 972. [Google Scholar] [CrossRef]
- Driver, A.M.; Peñagaricano, F.; Huang, W.; Ahmad, K.R.; Hackbart, K.S.; Wiltbank, M.C.; Khatib, H. RNA-seq analysis uncovers transcriptomic variations between morphologically similar in vivo- and in vitro-derived bovine blastocysts. BMC Genom. 2012, 13, 118. [Google Scholar] [CrossRef]
- Chitwood, J.L.; Rincon, G.; Kaiser, G.G.; Medrano, J.F.; Ross, P.J. RNA-seq analysis of single bovine blastocysts. BMC Genom. 2013, 14, 350. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Kogelman, L.J.; Santana, M.H.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.; et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 2016, 17, 348. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Coutinho, L.L.; Decker, J.E.; Schnabel, R.D.; Rosa, K.O.; Oliveira, P.S.; Souza, M.M.; Mourão, G.B.; Tullio, R.R.; Chaves, A.S.; et al. Global liver gene expression differences in Nellore steers with divergent residual feed intake phenotypes. BMC Genom. 2015, 16, 242. [Google Scholar] [CrossRef]
- Keogh, K.; Kenny, D.A.; Cormican, P.; Kelly, A.K.; Waters, S.M. Effect of dietary restriction and subsequent re-alimentation on the transcriptional profile of hepatic tissue in cattle. BMC Genom. 2016, 17, 244. [Google Scholar] [CrossRef]
- Mukiibi, R.; Vinsky, M.; Keogh, K.A.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Transcriptome analyses reveal reduced hepatic lipid synthesis and accumulation in more feed efficient beef cattle. Sci. Rep. 2018, 8, 7303. [Google Scholar] [CrossRef] [PubMed]
- Montanholi, Y.R.; Haas, L.S.; Swanson, K.C.; Coomber, B.L.; Yamashiro, S.; Miller, S.P. Liver morphometrics and metabolic blood profile across divergent phenotypes for feed efficiency in the bovine. Acta Vet. Scand. 2017, 59, 24. [Google Scholar] [CrossRef] [PubMed]
- Zarek, C.M.; Lindholm-Perry, A.K.; Kuehn, L.A.; Freetly, H.C. Differential expression of genes related to gain and intake in the liver of beef cattle. BMC Res. Notes 2017, 10, 1. [Google Scholar] [CrossRef]
- Cui, X.; Hou, Y.; Yang, S.; Xie, Y.; Zhang, S.; Zhang, Y.; Zhang, Q.; Lu, X.; Liu, G.E.; Sun, D. Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genom. 2014, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Ni, H.; Liu, Y.; Li, J.; Zhang, L.; Guo, Y. RNA-seq analysis of bovine intramuscular, subcutaneous and perirenal adipose tissues. Mol. Biol. Rep. 2014, 41, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Jakhesara, S.J.; Koringa, P.G.; Joshi, C.G. Identification of novel exons and transcripts by comprehensive RNA-seq of horn cancer transcriptome in Bos indicus. J. Biotechnol. 2013, 165, 37–44. [Google Scholar] [CrossRef]
- Yang, J.; Jiang, J.; Liu, X.; Wang, H.; Guo, G.; Zhang, Q.; Jiang, L. Differential expression of genes in milk of dairy cattle during lactation. Anim. Genet. 2016, 47, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, Y.; Du, W.; Zhang, X.; Li, A.; Miao, X. Global transcriptome analysis identifies differentially expressed genes related to lipid metabolism in Wagyu and Holstein cattle. Sci. Rep. 2017, 7, 5278. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.; Kim, K.; Yoon, J.; Jeong, J.Y.; Lee, H.J.; Cho, S.; Kim, H. RNA-seq analysis for detecting quantitative trait-associated genes. Sci. Rep. 2016, 6, 24375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Sahana, G.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Integrating Sequence-based GWAS and RNA-seq Provides Novel Insights into the Genetic Basis of Mastitis and Milk Production in Dairy Cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef]
- Li, C.; Cai, W.; Zhou, C.; Yin, H.; Zhang, Z.; Loor, J.J.; Sun, D.; Zhang, Q.; Liu, J.; Zhang, S. RNA-seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Sci. Rep. 2016, 6, 26813. [Google Scholar] [CrossRef]
- Silva-Vignato, B.; Coutinho, L.L.; Cesar, A.S.M.; Poleti, M.D.; Regitano, L.C.A.; Balieiro, J.C.C. Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genom. 2017, 18, 506. [Google Scholar] [CrossRef] [Green Version]
- Baik, M.; Kang, H.J.; Park, S.J.; Na, S.W.; Piao, M.; Kim, S.Y.; Fassah, D.M.; Moon, Y.S. Triennial growth and development symposium: Molecular mechanisms related to bovine intramuscular fat deposition in the longissimus muscle. J. Anim. Sci. 2017, 95, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Khansefid, M.; Pryce, J.E.; Bolormaa, S.; Chen, Y.; Millen, C.A.; Chamberlain, A.J.; Vander Jagt, C.J.; Goddard, M.E. Comparing allele specific expression and local expression quantitative trait loci and the influence of gene expression on complex trait variation in cattle. BMC Genom. 2018, 19, 793. [Google Scholar] [CrossRef]
- Dekkers, J.C.M. Commercial application of marker- and gene-assisted selection in livestock: Strategies and lessons. J. Anim. Sci. 2004, 82, E313–E328. [Google Scholar] [PubMed]
- Hayes, B.; Goddard, M. Genome-wide association and genomic selection in animal breeding. Genome 2010, 53, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Pareek, C.S.; Smoczyński, R.; Kadarmideen, H.N.; Dziuba, P.; Błaszczyk, P.; Sikora, M.; Walendzik, P.; Grzybowski, T.; Pierzchała, M.; Horbańczuk, J.; et al. Single Nucleotide Polymorphism Discovery in Bovine Pituitary Gland Using RNA-seq Technology. PLoS ONE 2016, 11, e0161370. [Google Scholar] [CrossRef]
- Pareek, C.S.; Błaszczyk, P.; Dziuba, P.; Czarnik, U.; Fraser, L.; Sobiech, P.; Pierzchała, M.; Feng, Y.; Kadarmideen, H.N.; Kumar, D. Single nucleotide polymorphism discovery in bovine liver using RNA-seq technology. PLoS ONE 2017, 12, e0172687. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Brock, G.N.; Rouchka, E.C.; Cooper, N.G.; Wu, D.; O’Toole, T.E.; Gill, R.S.; Eteleeb, A.M.; O’Brien, L.; Rai, S.N. A comparison of per sample global scaling and per gene normalization methods for differential expression analysis of RNA-seq data. PLoS ONE 2017, 12, e0176185. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Wilkinson, L.; Friendly, M. The History of the Cluster Heat Map. Am. Stat. 2009, 63, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Advanced heat map and clustering analysis using heatmap3. Biomed Res. Int. 2014, 2014, 986048. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams (2007–2015). Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 1 March 2007).
- Alexa, A.; Rahnenfuhrer, J. TopGO: Enrichment Analysis for Gene Ontology. R package version 2.34.0 (2018). Available online: http://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 20 October 2018).
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisowski, P.; Pierzchała, M.; Gościk, J.; Pareek, C.S.; Zwierzchowski, L. Evaluation of reference genes for studies of gene expression in the bovine liver, kidney, pituitary, and thyroid. J. Appl. Genet. 2008, 49, 367–372. [Google Scholar] [CrossRef]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; He, T.; Qin, C.; Qiu, K.; Zhang, X.; Luo, Y.; Li, D.; Yin, J. A potential regulatory network underlying distinct fate commitment of myogenic and adipogenic cells in skeletal muscle. Sci. Rep. 2017, 7, 44133. [Google Scholar] [CrossRef] [Green Version]
- Szarek, J.; Adamczyk, K.; Felenczak, A. Polish Red Cattle breeding: Past and present. Anim. Genet. Res. Inf. 2004, 35, 21–35. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Chamberlain, A.J.; Maceachern, S.; Savin, K.; McPartlan, H.; MacLeod, I.; Sethuraman, L.; Goddard, M.E. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim. Genet. 2009, 40, 176–184. [Google Scholar] [CrossRef]
- Hosokawa, D.; Ishii, A.; Yamaji, K.; Sasazaki, S.; Oyama, K.; Mannen, H. Identification of divergently selected regions between Japanese Black and Holstein cattle using bovine 50k SNP array. Anim. Sci. J. 2012, 83, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Pawlina, K.; Frys-Żurek, M.; Bugno-Poniewierska, M. Identification of differential selection traces in two Polish cattle breeds. Anim. Sci. J. 2015, 86, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Salleh, M.S.; Mazzoni, G.; Höglund, J.K.; Olijhoek, D.W.; Lund, P.; Løvendahl, P.; Kadarmideen, H.N. RNA-seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genom. 2017, 18, 258. [Google Scholar] [CrossRef]
- Cesar, A.S.; Regitano, L.C.; Poleti, M.D.; Andrade, S.C.; Tizioto, P.C.; Oliveira, P.S.; Felício, A.M.; do Nascimento, M.L.; Chaves, A.S.; Lanna, D.P.; et al. Differences in the skeletal muscle transcriptome profile associated with extreme values of fatty acids content. BMC Genom. 2016, 17, 961. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Caetano-Anolles, K.; Rodriguez-Zas, S.; Ka, S.; Jeong, J.Y.; Park, S.; Kim, M.J.; Nho, W.G.; Cho, S.; Kim, H.; et al. Comprehensive identification of sexually dimorphic genes in diverse cattle tissues using RNA-seq. BMC Genom. 2016, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.C.O.; Davis, F.J.; Nasser, L.F.; Bowman, P.; Rawlings, N.C. Differences in early patterns of gonadotropin secretion between early and late maturing bulls and changes in semen characteristics at puberty. Theriogenology 1995, 43, 569–578. [Google Scholar] [CrossRef]
- Casas, E.; Lunstra, D.D.; Cundiff, L.V.; Ford, J.J. Growth and pubertal development of F1 bulls from Hereford, Angus, Norwegian Red, Swedish Red and White, Friesian, and Wagyu sires. J. Anim. Sci. 2007, 85, 2904–2909. [Google Scholar] [CrossRef]
- McCabe, M.; Waters, S.; Morris, D.; Kenny, D.; Lynn, D.; Creevey, C. RNA-seq analysis of differential gene expression in liver from lactating dairy cows divergent in negative energy balance. BMC Genom. 2012, 13, 193. [Google Scholar] [CrossRef]
- Pegolo, S.; Cecchinato, A.; Mach, N.; Babbucci, M.; Pauletto, M.; Bargelloni, L.; Schiavon, S.; Bittante, G. Transcriptomic Changes in Liver of Young Bulls Caused by Diets Low in Mineral and Protein Contents and Supplemented with n-3 Fatty Acids and Conjugated Linoleic Acid. PLoS ONE 2016, 11, e0167747. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.S.G.; Liang, G.; Chen, Y.; Stothard, P.; Guan, L.L. Transcriptome profiling of the rumen epithelium of beef cattle differing in residual feed intake. BMC Genom. 2016, 17, 592. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, J.A.; He, Y.; Li, Y.; Liu, J.; Erdman, R.A.; Sonstegard, T.S.; Song, J. Integrated metabolomic and transcriptome analyses reveal finishing forage affects metabolic pathways related to beef quality and animal welfare. Sci. Rep. 2016, 6, 25948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, L.F.; Gimenez, D.F.; dos Santos Silva, D.B.; Barthelson, R.; Baldi, F.; Ferro, J.A.; Albuquerque, L.G. Differences in global gene expression in muscle tissue of Nellore cattle with divergent meat tenderness. BMC Genom. 2017, 18, 945. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breeds | n | Cold Carcass Weight (kg) | Carcass Yield (kg) | Right Half Carcass (kg) | Valuable Cut Meat (kg) | Meat (kg) | Bones (kg) | Fat (kg) |
|---|---|---|---|---|---|---|---|---|
| Polish-HF | 6 | 188.9 ± 5.3 | 53 ± 2.4 | 94.9 ± 1.6 | 59.4 ± 1.9 | 42.3 ± 2.6 | 15.4 ± 3.6 | 9.9 ± 3.8 |
| Polish-Red | 6 | 166.6 ± 6.4 | 56.5 ± 3.6 | 86 ± 2.6 | 54.5 ± 3.4 | 40.7 ± 4.1 | 15.4 ± 5.7 | 10.9 ± 6.0 |
| Hereford | 6 | 193.9 ± 8.9 | 59.6 ± 4.6 | 100.2 ± 2.9 | 62.9 ± 3.1 | 45.4 ± 3.9 | 17.1 ± 5.6 | 13.0 ± 5.8 |
| Gene Full Name | Calpain 11 | Calpain 2 | Insulin Like Growth Factor Binding Protein 1 | Insulin Like Growth Factor Binding Protein 2 | Insulin Like Growth Factor I | Insulin Like Growth Factor Binding Protein, Acid Labile Subunit (IGFALS) | Somatotropin | Family with Sequence Similarity 13 Member A |
|---|---|---|---|---|---|---|---|---|
| Gene symbol | CAPN11 | CAPN2 | IGFBP1 | IGFBP2 | IGF-I | IGFALS | GH | FAM13A |
| Forward primer | GAACCTTCAA | CCAACATTGA | TAGCGTAAAT | GTGCAAGAT | GAGTGCAGG | CAGGTAACA | AGCCATCTG | CCTTTCTATTT |
| CTGTCAAGCG | CGAGATTGACA | TGGCAGGGAA | GTCTCTGAACG | AAACAAGAACT | AGCTGGCCTA | TTGTTTGCCC | GAGCAGTGCC | |
| Reverse primer | TTGAGTCGGT | ATTGTCTGCA | ACACTGTGTT | TGCTCGTTGT | TTGGTAGGT | GTGGTCCAG | TATTAGGAAA | CTGAGTCCTCT |
| TCTGGCTTAT | ACTCAAAGGC | CCCATGTTTG | AGAAGAGATGA | CTTCTGGTGTT | GTAGAGTTTCT | GGACAGTGGGAG | GAACTTTGG | |
| Ensembl Gene ID | ENSBTAG000 | ENSBTAG000 | ENSBTAG00 | ENSBTAG00 | ENSBTAG00 | ENSBTAG000 | ENSBTAG00 | ENSBTAG000 |
| 21066 | 12778 | 46768 | 5596 | 11082 | 33299 | 17220 | 11187 | |
| Ensembl Transcript ID | ENSBTAT00 | ENSBTAT00 | ENSBTAT00 | ENSBTAT000 | ENSBTAT00 | ENSBTAT000 | ENSBTAT00 | ENSBTAT00 |
| 4690 | 47532 | 64194 | 7349 | 14713 | 47326 | 22885 | 14855 | |
| BTA chromosome | 23 | 16 | 4 | 2 | 5 | 25 | 19 | 6 |
| Gene Start (bp) | 17,807,298 | 27,781,671 | 76,720,883 | 1.05E + 08 | 66,532,877 | 1,366,647 | 48,768,618 | 37,355,568 |
| Gene End (bp) | 17,820,479 | 27,840,009 | 76,725,301 | 1.05E + 08 | 66,604,734 | 1,368,479 | 48,772,014 | 37,457,493 |
| Transcript Start (bp) | 17,807,298 | 27,781,671 | 76,720,883 | 1.05E + 08 | 66,532,877 | 1,366,647 | 48,768,618 | 37,355,568 |
| Transcript length | 2675 | 3179 | 917 | 1141 | 862 | 1833 | 817 | 5125 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pareek, C.S.; Sachajko, M.; Jaskowski, J.M.; Herudzinska, M.; Skowronski, M.; Domagalski, K.; Szczepanek, J.; Czarnik, U.; Sobiech, P.; Wysocka, D.; et al. Comparative Analysis of the Liver Transcriptome among Cattle Breeds Using RNA-seq. Vet. Sci. 2019, 6, 36. https://doi.org/10.3390/vetsci6020036
Pareek CS, Sachajko M, Jaskowski JM, Herudzinska M, Skowronski M, Domagalski K, Szczepanek J, Czarnik U, Sobiech P, Wysocka D, et al. Comparative Analysis of the Liver Transcriptome among Cattle Breeds Using RNA-seq. Veterinary Sciences. 2019; 6(2):36. https://doi.org/10.3390/vetsci6020036
Chicago/Turabian StylePareek, Chandra Shekhar, Mateusz Sachajko, Jedrzej M. Jaskowski, Magdalena Herudzinska, Mariusz Skowronski, Krzysztof Domagalski, Joanna Szczepanek, Urszula Czarnik, Przymeslaw Sobiech, Dominika Wysocka, and et al. 2019. "Comparative Analysis of the Liver Transcriptome among Cattle Breeds Using RNA-seq" Veterinary Sciences 6, no. 2: 36. https://doi.org/10.3390/vetsci6020036