
In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
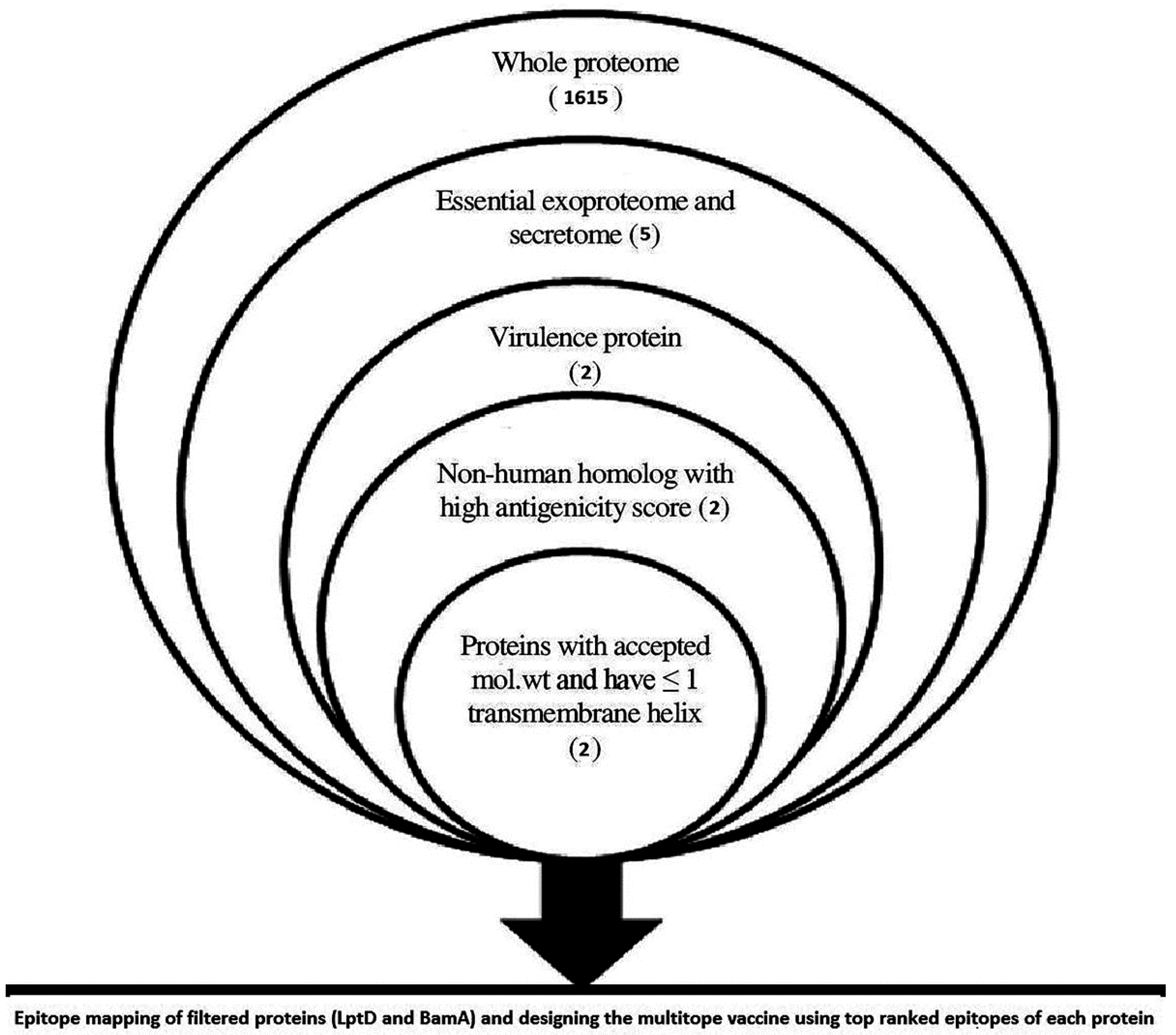
2.1. Data Retrieval and Proteome Analysis
2.2. T and B Cell Epitope Prediction
2.3. Multitope Vaccine Construction
2.4. Physicochemical Characterization, Protein Solubility Assessment and Secondary Structure Prediction
2.5. Tertiary Structure Prediction, Refinement and Validation
2.6. Disulfide Engineering of the Designed Vaccine
2.7. Docking of Designed Vaccine with TLR2
2.8. Molecular Dynamics Simulation
2.9. Reverse Translation and Codon Adaptation
3. Results
3.1. In Silico Proteome Analysis
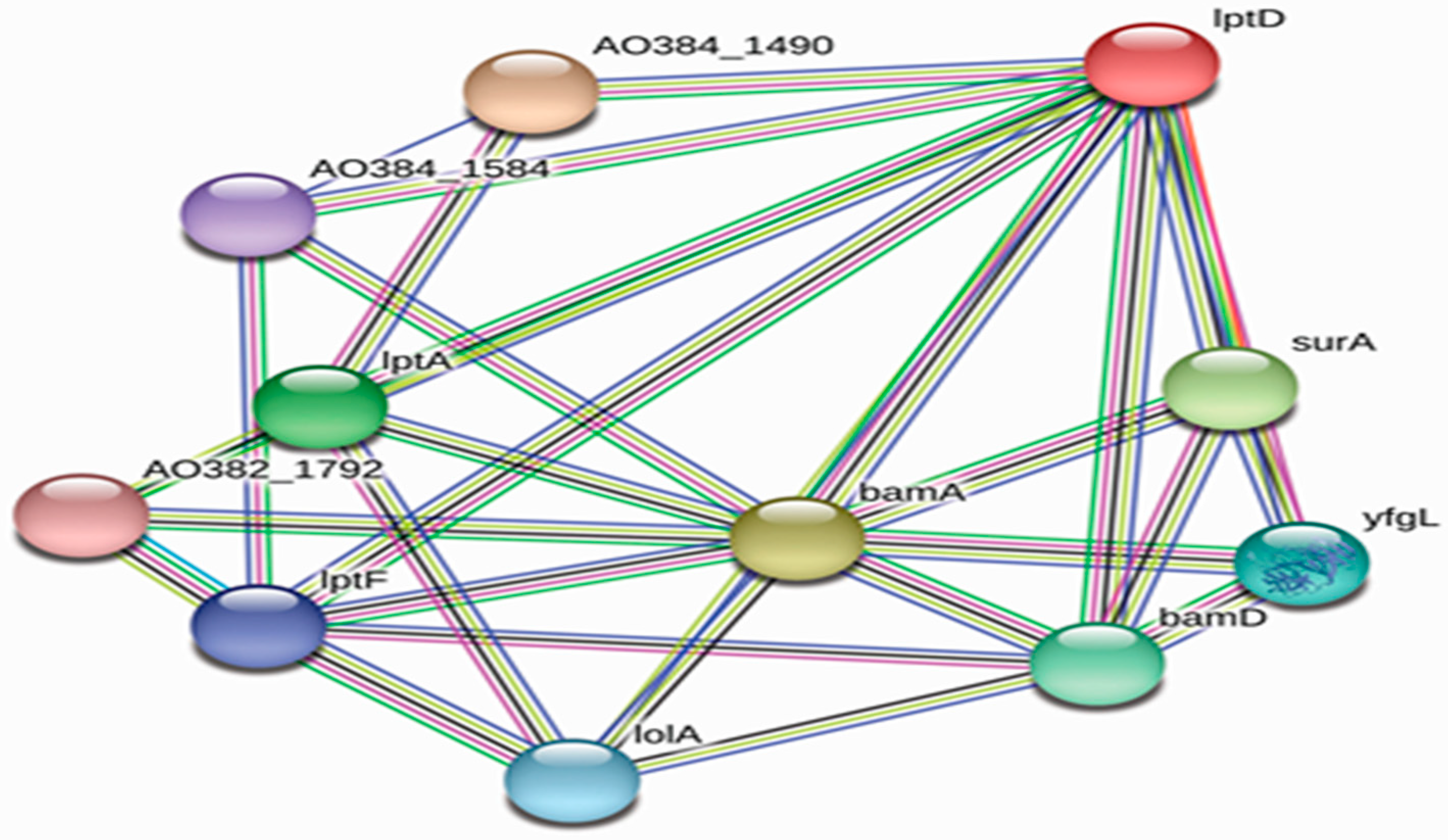
3.2. Protein–Protein Interaction (PPI) Analysis
3.3. T Cell Epitopes
3.4. Construction of Multitope Vaccine with Physiochemical Property Assessment and Secondary Structure Prediction
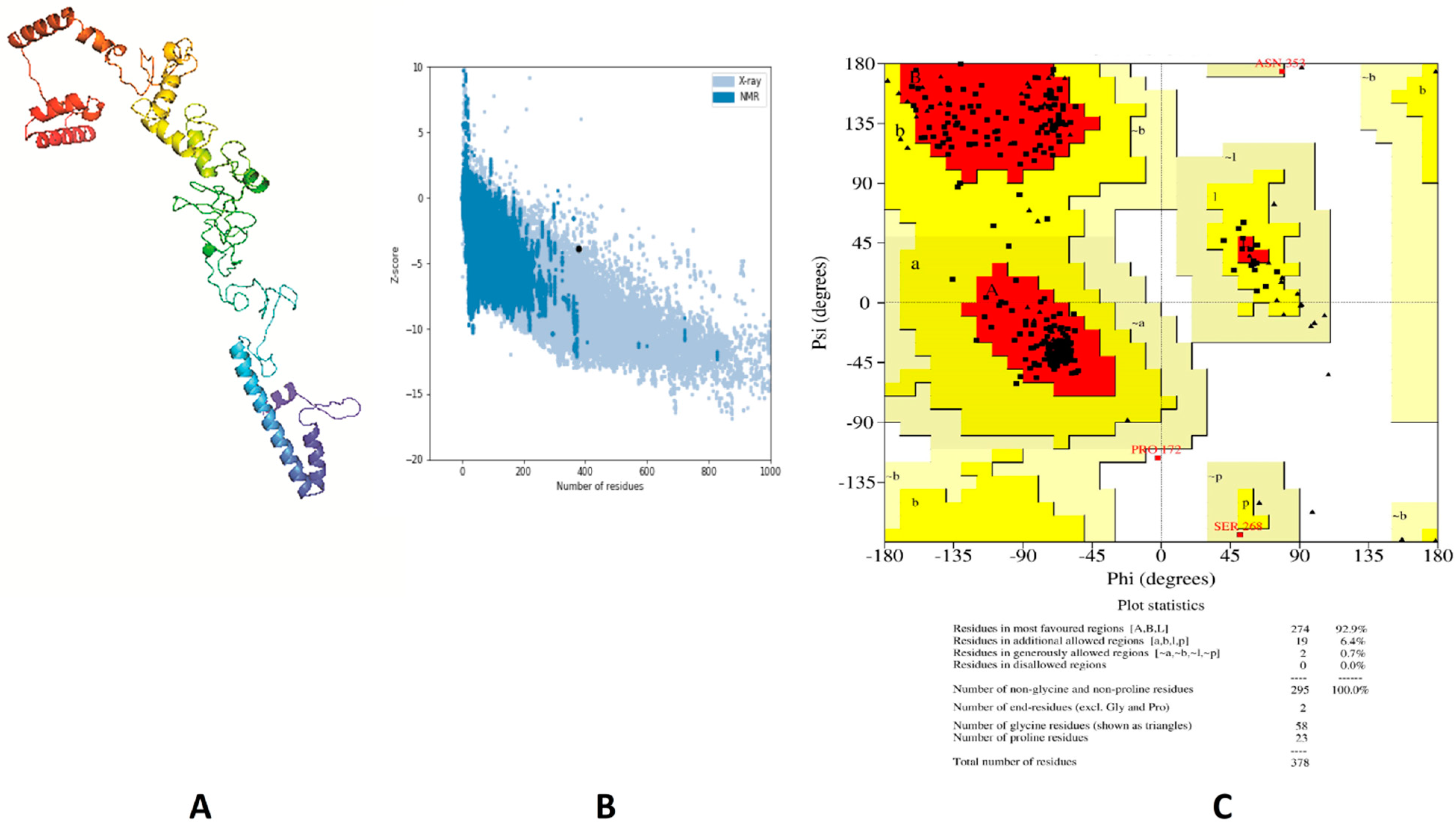
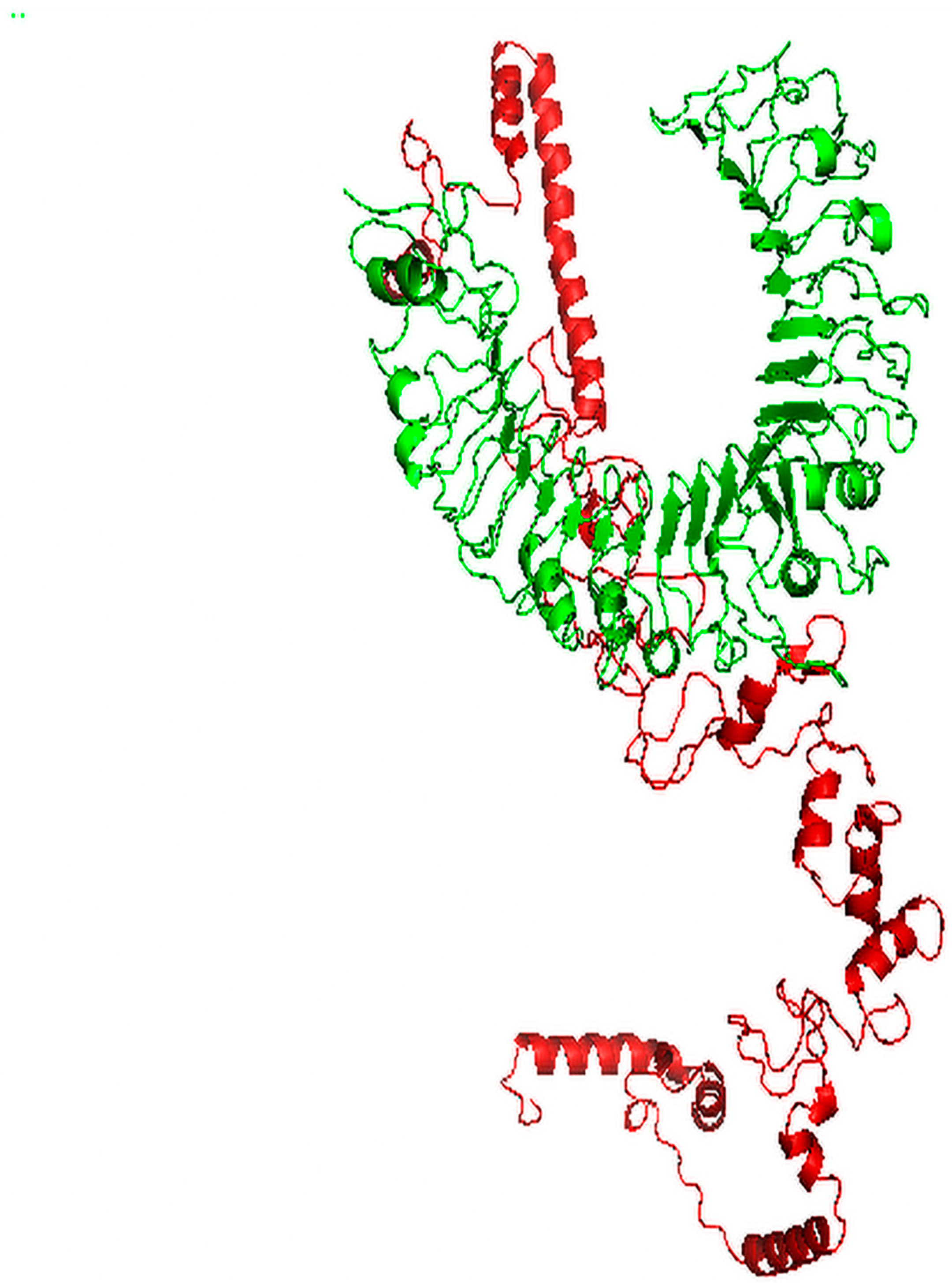
3.5. Tertiary Structure Prediction, Refinement and Validation
3.6. Vaccine Disulfide Engineering
3.7. Molecular Docking of Vaccine with TLR2
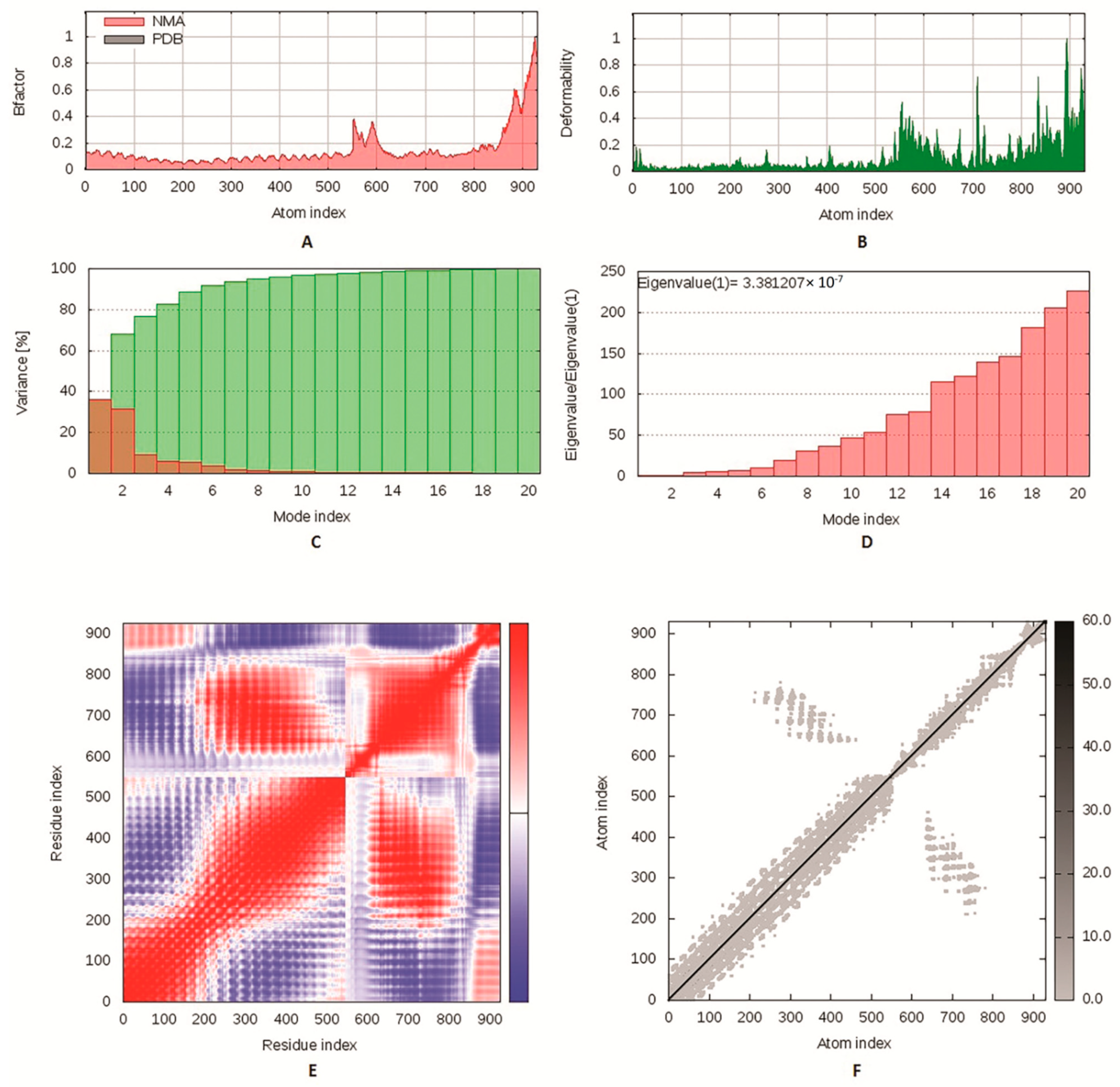
3.8. Molecular Dynamics Simulation
3.9. Vaccine Reverse Translation and Codon Optimization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernhard, S.; Spaniol, V.; Aebi, C. Molecular pathogenesis of infections caused by Moraxella catarrhalis in children. Swiss Med. Wkly. 2012, 142, w13694. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Alvarado-Kristensson, M.; Johansson, M.; Hallgren, O.; Westergren-Thorsson, G.; Mörgelin, M.; Riesbeck, K. The Respiratory Pathogen Moraxella catarrhalis Targets Collagen for Maximal Adherence to Host Tissues. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, A.C.; Pang, B.; King, L.B.; Tan, L.; Murrah, K.A.; Reimche, J.L.; Wren, J.T.; Richardson, S.H.; Ghandi, U.; Swords, W.E. Residence ofStreptococcus pneumoniaeandMoraxella catarrhaliswithin polymicrobial biofilm promotes antibiotic resistance and bacterial persistencein vivo. Pathog. Dis. 2014, 70, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Pichichero, M.E. Vaccine targets againstMoraxella catarrhalis. Expert Opin. Ther. Targets 2016, 20, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertot, G.M.; Becker, P.D.; Guzmán, C.; Grinstein, S. Intranasal Vaccination with Recombinant P6 Protein and Adamantylamide Dipeptide as Mucosal Adjuvant Confers Efficient Protection against Otitis Media and Lung Infection by Nontypeable Haemophilus influenzae. J. Infect. Dis. 2004, 189, 1304–1312. [Google Scholar] [CrossRef] [Green Version]
- Verhaegh, S.J.; de Vogel, C.P.; Riesbeck, K.; Lafontaine, E.R.; Murphy, T.F.; Verbrugh, H.A.; Jaddoe, V.W.; Hofman, A.; Moll, H.A.; van Belkum, A.; et al. Temporal development of the humoral immune response to surface antigens of Moraxella catarrhalis in young infants. Vaccine 2011, 29, 5603–5610. [Google Scholar] [CrossRef] [Green Version]
- Delfani, S.; Fooladi, A.A.I.; Mobarez, A.M.; Emaneini, M.; Amani, J.; Sedighian, H. In silicoanalysis for identifying potential vaccine candidates againstStaphylococcus aureus. Clin. Exp. Vaccine Res. 2015, 4, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Hegde, N.R.; Gauthami, S.; Sampath Kumar, H.M.; Bayry, J. The use of databases, data mining and immunoinformatics in vaccinology: Where are we? Expert Opin. Drug Discov. 2018, 13, 117–130. [Google Scholar] [CrossRef]
- Capelli, R.; Peri, C.; Villa, R.; Nithichanon, A.; Conchillo-Solé, O.; Yero, D.; Gagni, P.; Chiari, M.; Lertmemongkolchai, G.; Cretich, M.; et al. BPSL1626: Reverse and Structural Vaccinology Reveal a Novel Candidate for Vaccine Design Against Burkholderia pseudomallei. Antibodies 2018, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.F.; Haddad, J.P.A.; Paixão, T.A.; Santos, R.L. Meta-Analysis and Advancement of Brucellosis Vaccinology. PLoS ONE 2016, 11, e0166582. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.-F.; Liu, S.; Dong, C.; Guo, H.-X.; Gao, Y.-Z.; Guo, F.-B. Geptop 2.0: An Updated, More Precise, and Faster Geptop Server for Identification of Prokaryotic Essential Genes. Front. Microbiol. 2019, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Wagner, J.R.; Laird, M.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinforma 2006, 4, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskopf, D.; Angelo, M.A.; de Azeredo, E.L.; Sidney, J.; Greenbaum, J.A.; Fernando, A.N.; Broadwater, A.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2046–E2053. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, J.; Sidney, J.; Chung, J.; Brander, C.; Peters, B.; Sette, A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 2011, 63, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.; Ghosh, P.P.; Azim, K.F.; Mukta, S.; Abir, R.A.; Nahar, J.; Khan, M.M.H. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb. Pathog. 2019, 130, 19–37. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, V.; Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 ininfection and immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Pandey, R.K.; Verma, P.; Sharma, D.; Bhatt, T.K.; Sundar, S.; Prajapati, V.K. High-throughput virtual screening and quantum mechanics approach to develop imipramine analogues as leads against trypanothione reductase of leishmania. Biomed. Pharmacother. 2016, 83, 141–152. [Google Scholar] [CrossRef]
- Awan, F.M.; Obaid, A.; Ikram, A.; Janjua, H.A. Mutation-Structure-Function Relationship Based Integrated Strategy Reveals the Potential Impact of Deleterious Missense Mutations in Autophagy Related Proteins on Hepatocellular Carcinoma (HCC): A Comprehensive Informatics Approach. Int. J. Mol. Sci. 2017, 18, 139. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Poland, G.A.; Ovsyannikova, I.G.; Jacobson, R.M. Application of pharmacogenomics to vaccines. Pharmacogenomics 2009, 10, 837–852. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Cai, Y.; Chen, J.; Ye, X.; Mao, S.; Zhu, S.; Xue, X.; Chen, S.; Zhang, L. Evaluation of tandem Chlamydia trachomatis MOMP multi-epitopes vaccine in BALB/c mice model. Vaccine 2017, 35, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Soltan, M.A.; Magdy, D.; Solyman, S.M.; Hanora, A. Design of Staphylococcus aureus New Vaccine Candidates with B and T Cell Epitope Mapping, Reverse Vaccinology, and Immunoinformatics. OMICS A J. Integr. Biol. 2020, 24, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Leow, C.Y.; Kazi, A.; Ismail, C.M.K.H.; Chuah, C.; Lim, B.H.; Singh, K.K.B.; Leow, C.H. Reverse vaccinology approach for the identification and characterization of outer membrane proteins of Shigella flexneri as potential cellular- and antibody-dependent vaccine candidates. Clin. Exp. Vaccine Res. 2020, 9, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Huang, S.; Zhang, Q. Outer membrane proteins: Key players for bacterial adaptation in host niches. Microbes Infect. 2002, 4, 325–331. [Google Scholar] [CrossRef]
- Okuda, S.; Sherman, D.J.; Silhavy, T.J.; Ruiz, N.; Kahne, D. Lipopolysaccharide transport and assembly at the outer mem-brane: The PEZ model. Nat. Rev. Microbiol. 2016, 14, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Majid, M.; Andleeb, S. Designing a multi-epitopic vaccine against the enterotoxigenic Bacteroides fragilis based on immunoinformatics approach. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Yassin, G.M.; Amin, M.A.; Attia, A.S. Immunoinformatics Identifies a Lactoferrin Binding Protein A Peptide as a Promising Vaccine With a Global Protective Prospective AgainstMoraxella catarrhalis. J. Infect. Dis. 2016, 213, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy that Enhances CD4+ T Help in BALB/c Mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | PSORb Result | Essential | Virulent | Non-Human Homolog | Molecular Weight (KDa) | Transmembrane Helices | Antigenicity Score |
|---|---|---|---|---|---|---|---|
| LptD | Outer membrane | √ | √ | √ | 104 | 1 | 0.59 |
| BamA | Outer membrane | √ | √ | √ | 90.9 | 0 | 0.52 |
| BamA | LptD | ||||||
|---|---|---|---|---|---|---|---|
| Epitope | Antigenicity | Allergenicity | Toxicity | Epitope | Antigenicity | Allergenicity | Toxicity |
| YSAGVGATW | 1.16 | None | None | AELSGNVIM | 0.62 | Allergen | None |
| GEVVGGNAL | 1.42 | None | None | FEISTPYYL | 1.29 | Allergen | None |
| LTQDKQLRY | 0.73 | Allergen | None | RPYARLPQL | 0.41 | None | None |
| RYSAGVGATW | 1.31 | Allergen | None | RSRIQFDHTW | 0.49 | None | None |
| SETREVYSL | 0.78 | Allergen | None | SEYRLQHVM | 0.71 | Allergen | None |
| AEINFEGNRL | 0.95 | None | None | YEQLLNNNW | 0.48 | Allergen | None |
| VQFQIGSVF | 0.6 | None | None | SSRSSGLAW | 0.78 | None | None |
| AEEGFSQAM | 0.58 | None | None | SYEQLLNNNW | 0.41 | Allergen | None |
| IETELTNQY | 0.93 | Allergen | None | VFYLPYFNF | 2.99 | Allergen | None |
| DSYGGSLSY | 1.34 | None | None | FTASYPLLR | 0.93 | None | None |
| BamA | LptD | ||||||
|---|---|---|---|---|---|---|---|
| Epitope | Antigenicity | Allergenicity | Toxicity | Epitope | Antigenicity | Allergenicity | Toxicity |
| VDVEYYIDPVHPVYV | 0.63 | None | None | DTGRAIAKNTTLRIK | 0.5 | Allergen | None |
| DVEYYIDPVHPVYVR | 0.56 | Allergen | None | GRAIAKNTTLRIKKV | 0.4 | None | None |
| TVDVEYYIDPVHPVY | 0.44 | None | None | TGRAIAKNTTLRIKK | 0.43 | Allergen | None |
| VEYYIDPVHPVYVRR | 0.41 | None | None | STPYYLNLAPNYDAT | 0.91 | Allergen | None |
| TNPYFTVNGVSQSLS | 0.77 | Allergen | None | TPYYLNLAPNYDATI | 0.98 | Allergen | None |
| NPYFTVNGVSQSLSG | 1.02 | Allergen | None | RIKKVPVFYLPYFNF | 1.86 | Allergen | None |
| PYFTVNGVSQSLSGY | 0.92 | Allergen | None | VSYRYIDKMGRTRFE | 0.56 | Allergen | None |
| MTNPYFTVNGVSQSL | 0.75 | None | None | AVSYRYIDKMGRTRF | 0.45 | None | None |
| RPLLTQDKQLRYSAG | 0.6 | Allergen | None | DYNLDYVMDSLMGLN | 0.51 | None | None |
| ALATFGSELILPLPF | 0.99 | None | None | DANYLSDFNALGVES | 0.46 | None | None |
| BamA | LptD | ||||||
|---|---|---|---|---|---|---|---|
| Epitope | Antigenicity | Allergenicity | Toxicity | Epitope | Antigenicity | Allergenicity | Toxicity |
| TGNFKTQDEV | 1.06 | Allergen | None | DGGASDHSAGI | 1.82 | Allergen | None |
| RREMRQLEGALASNQKIQ | 0.6 | None | None | KDQQYHDKD | 0.81 | None | None |
| RKTKYDNKNISNY | 0.7 | None | None | KKSIKDNSEPEKSG | 0.96 | Allergen | None |
| DLTVGFGDKT | 0.85 | None | None | SYDEDSLADQNIAKKNGR | 0.96 | None | None |
| LNKKQNDQT | 1.53 | None | None | APFGMHQDT | 0.42 | Allergen | None |
| HIRVGINDSESYSSRSS | 1.32 | None | None | ||||
| RKENRAFNQSAL | 0.43 | None | None | ||||
| YDYNLDYVMDSLM | 0.62 | None | None | ||||
| YRDAFNPHLSPD | 1.09 | None | None | ||||
| Physicochemical Characteristic | Molecular Weight | Theoretical pI | Extinction Coefficient | GRAVY | Instability Index | Aliphatic Index |
|---|---|---|---|---|---|---|
| Score | 40.2 kDa | 9.71 | 53,290 M−1 cm−1 | −0.625 | 37.3 | 59.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltan, M.A.; Elbassiouny, N.; Gamal, H.; Elkaeed, E.B.; Eid, R.A.; Eldeen, M.A.; Al-Karmalawy, A.A. In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics. Vaccines 2021, 9, 669. https://doi.org/10.3390/vaccines9060669
Soltan MA, Elbassiouny N, Gamal H, Elkaeed EB, Eid RA, Eldeen MA, Al-Karmalawy AA. In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics. Vaccines. 2021; 9(6):669. https://doi.org/10.3390/vaccines9060669
Chicago/Turabian StyleSoltan, Mohamed A., Nada Elbassiouny, Helmy Gamal, Eslam B. Elkaeed, Refaat A. Eid, Muhammad Alaa Eldeen, and Ahmed A. Al-Karmalawy. 2021. "In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics" Vaccines 9, no. 6: 669. https://doi.org/10.3390/vaccines9060669