Optimized Hepatitis C Virus (HCV) E2 Glycoproteins and their Immunogenicity in Combination with MVA-HCV

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Plasmids Containing HCV E2 Proteins
2.2. E2 Protein Expression
2.3. E2 Protein Purification
2.4. Neutralization Assays
2.5. ELISA Assays
2.6. Determination of Cross-Reactivity against HCV Genotypes
2.7. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
2.8. Blue Native-PAGE (BN-PAGE)
2.9. Dynamic Light Scattering (DLS)
2.10. Mouse Immunizations and Procedures
2.11. Peptides
2.12. Intracellular Cytokine Staining (ICS)
2.13. Statistical Analysis
3. Results
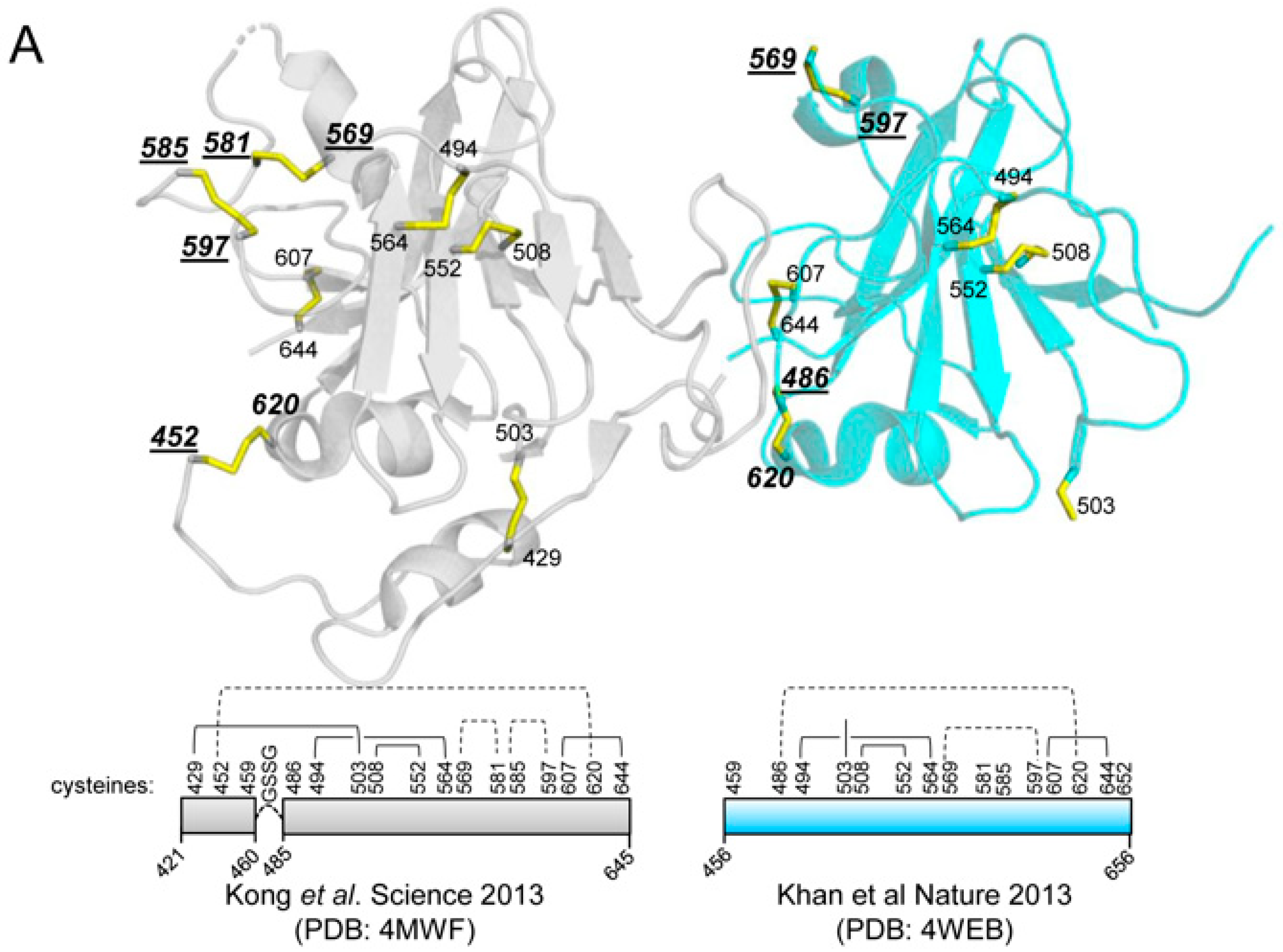
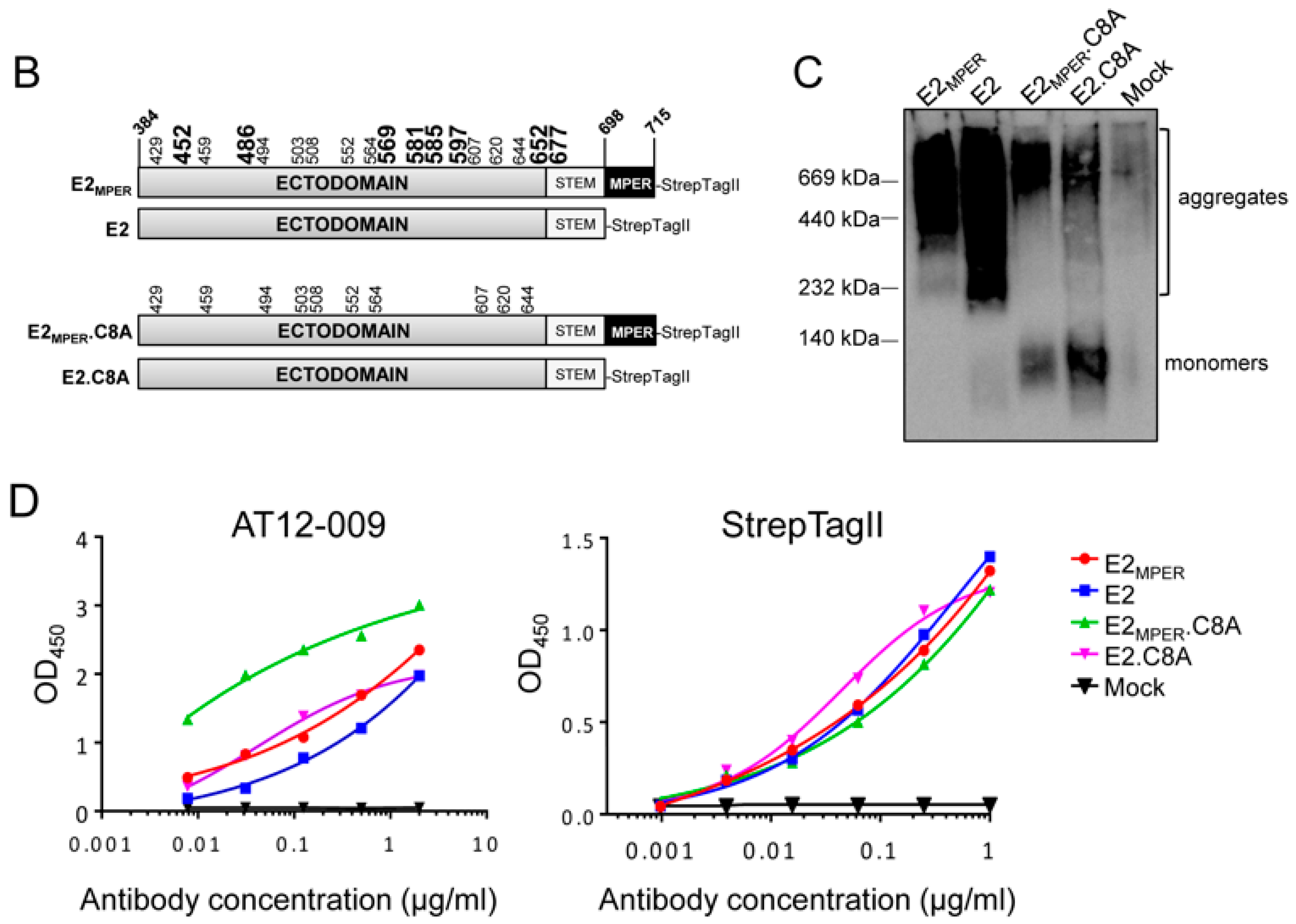
3.1. Design and Generation of a Recombinant HCV E2 Protein Lacking Eight Cysteine Residues
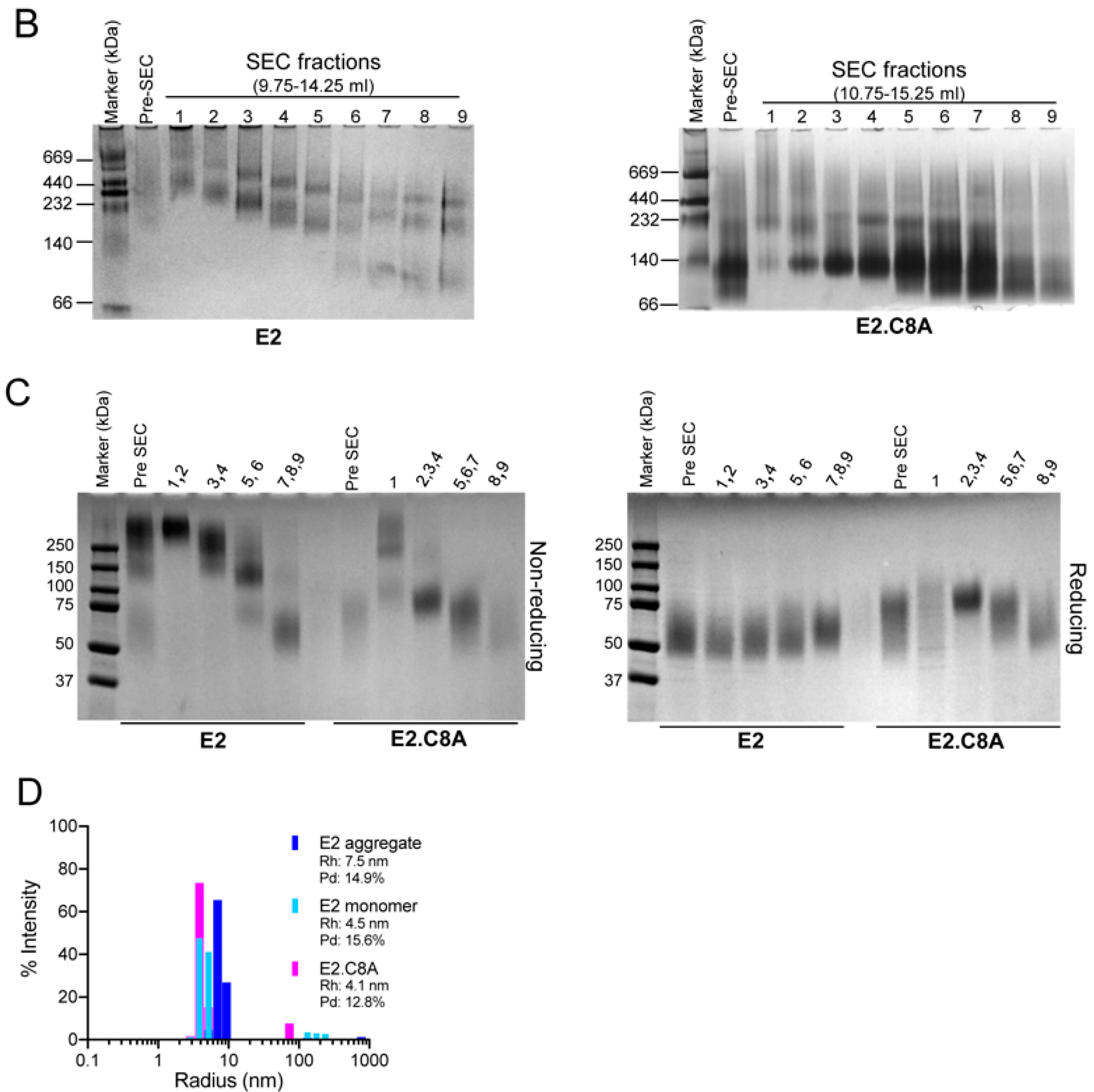
3.2. E2 Proteins Lacking Eight Cysteines are Expressed Less as Aggregates
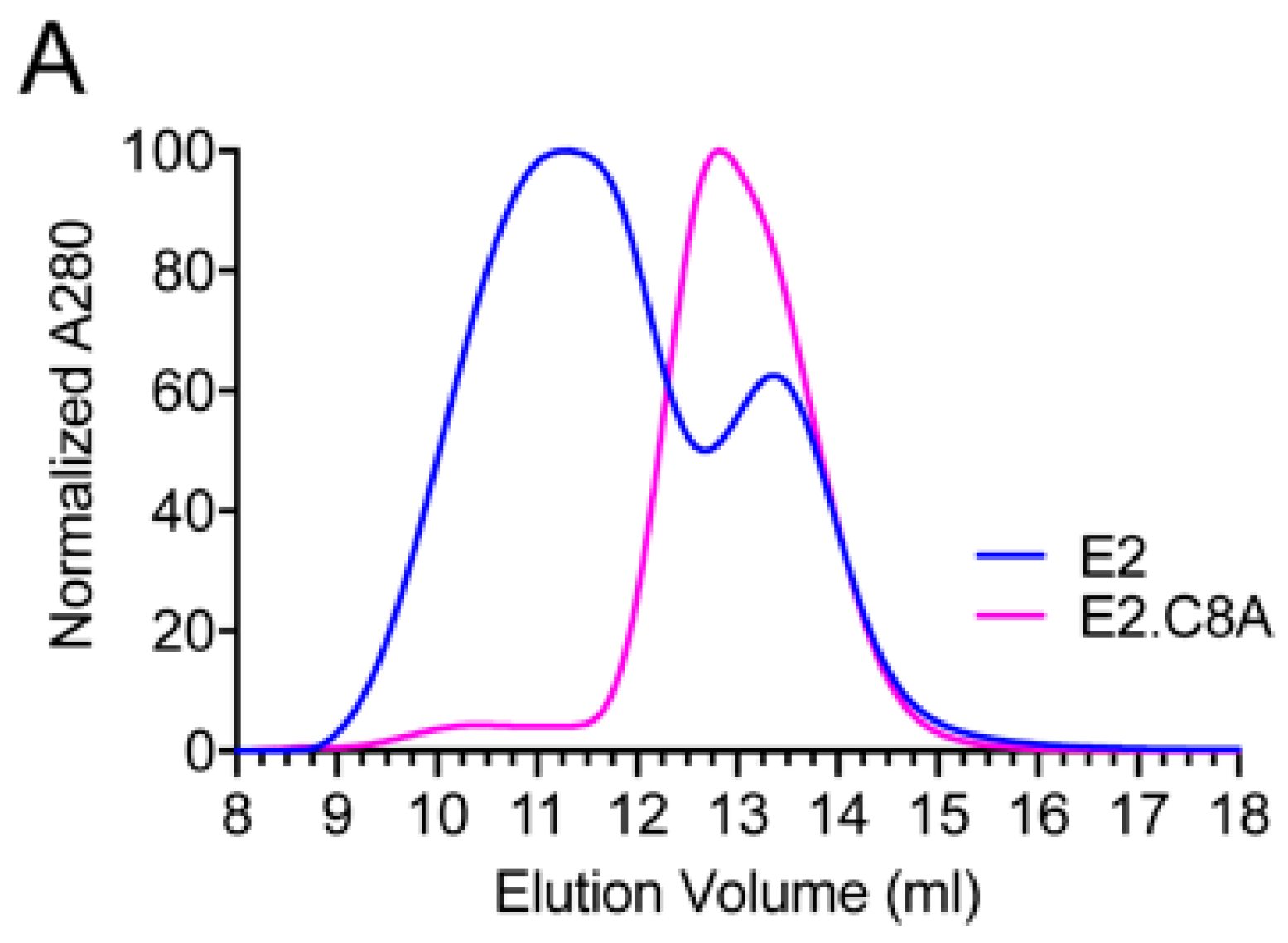
3.3. E2.C8A Is Expressed Mainly as Monomers
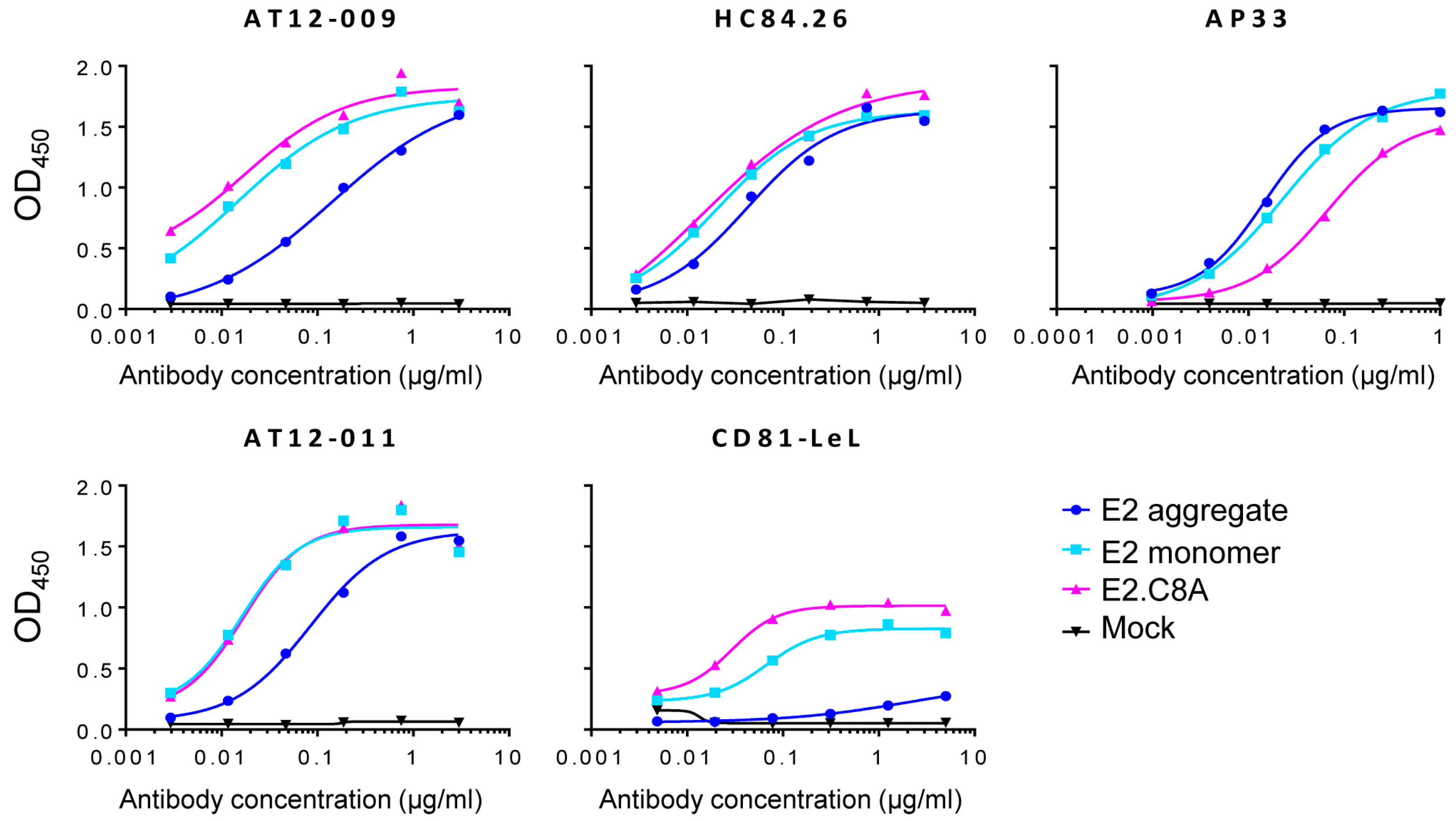
3.4. E2.C8A Monomers Are Efficiently Recognized by Broadly Neutralizing Antibodies (bNAbs) and CD81
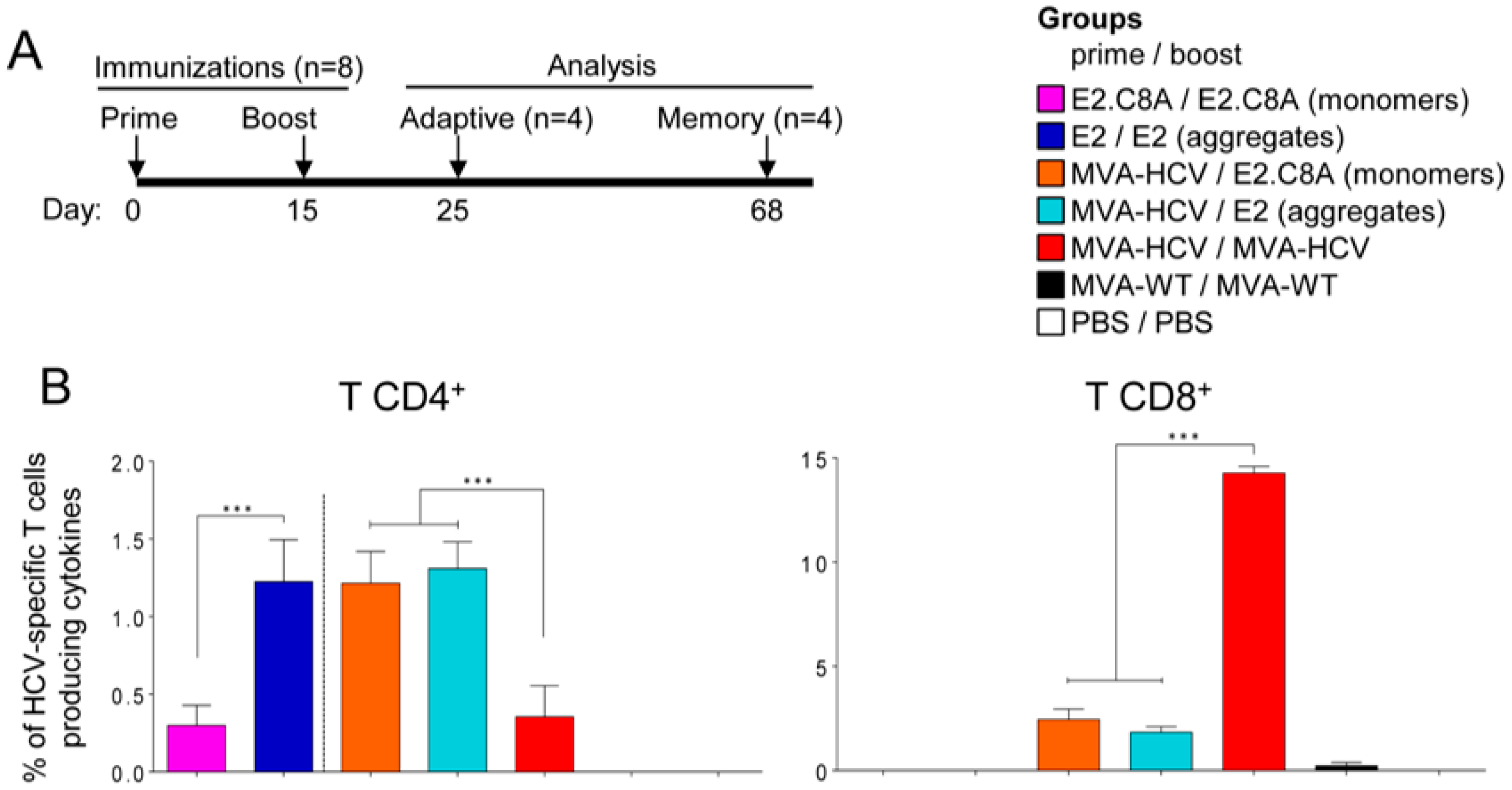
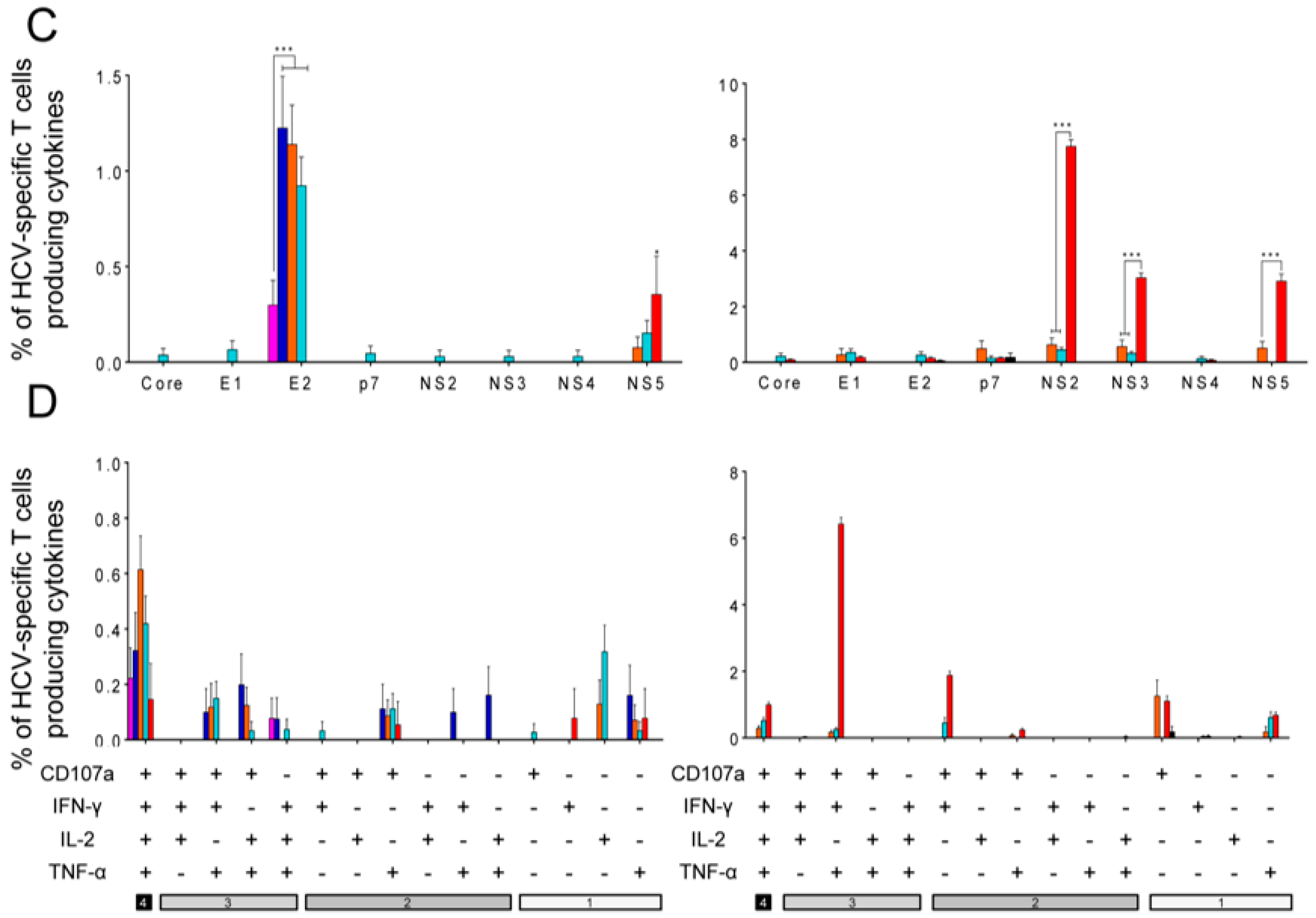
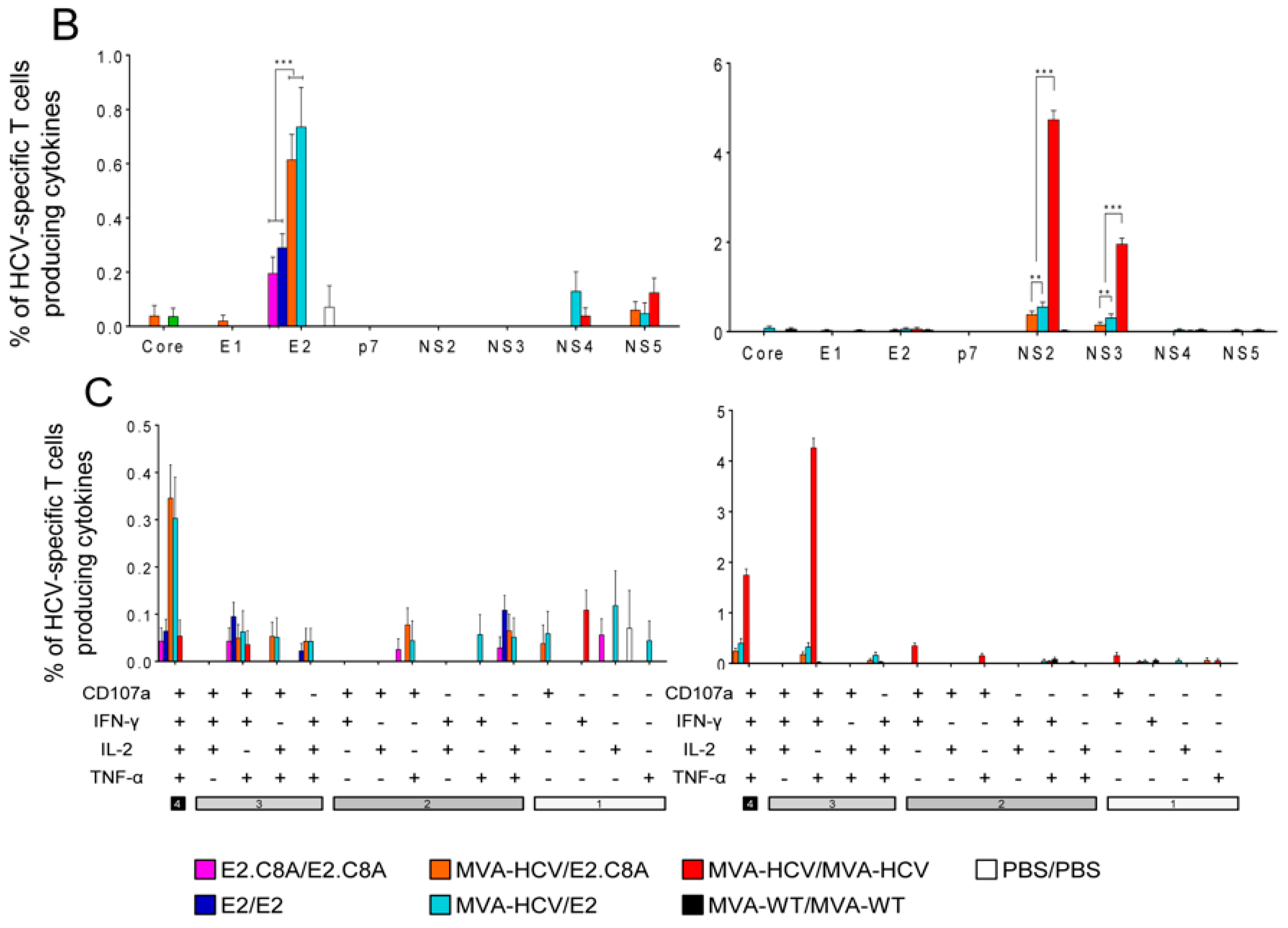
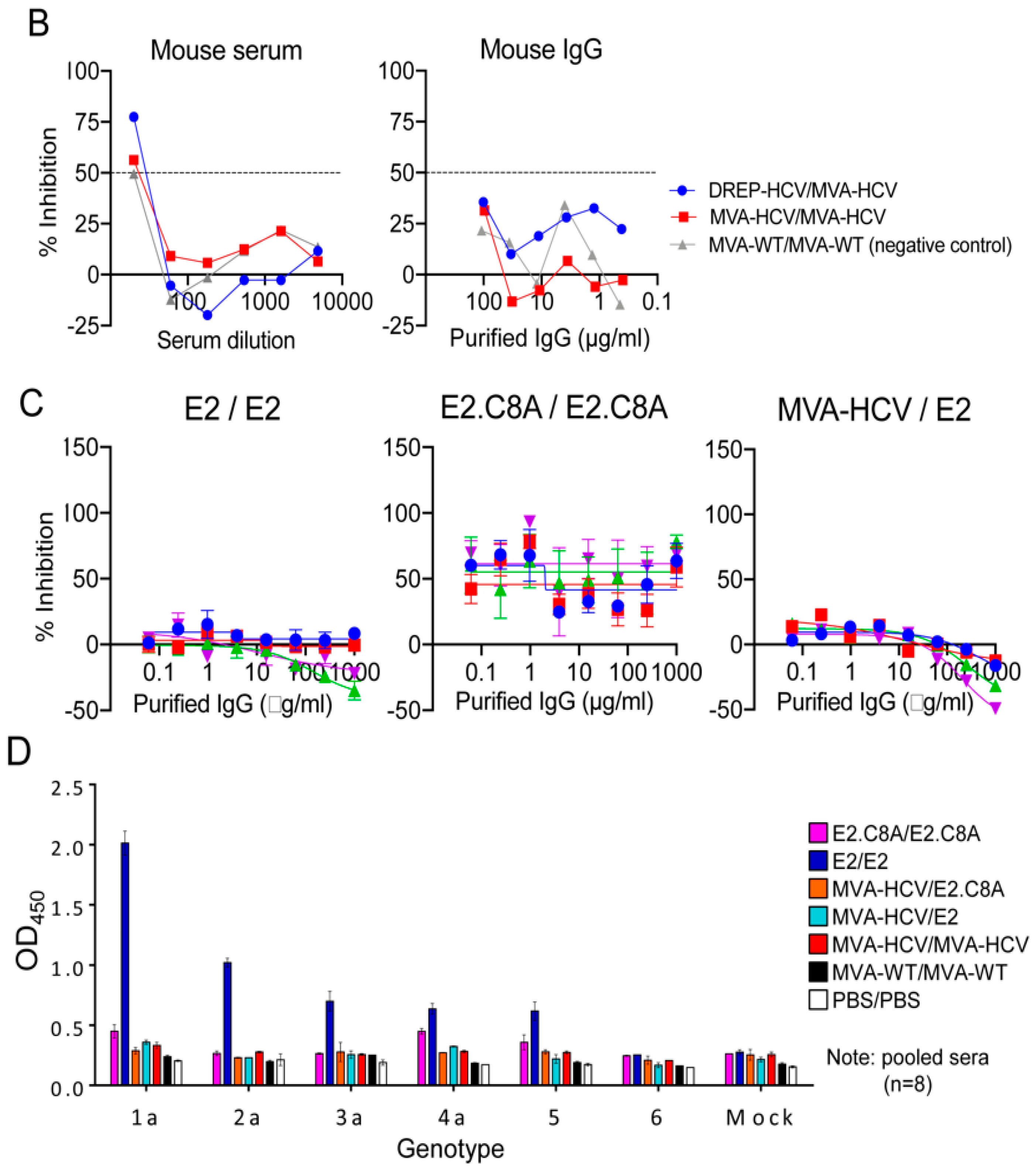
3.5. E2 Aggregates and E2.C8A Monomers Induced Adaptive HCV-Specific CD4+ and CD8+ T Cell Immune Responses in Immunized Mice When Combined with MVA-HCV in Heterologous Prime/Boost Regimens
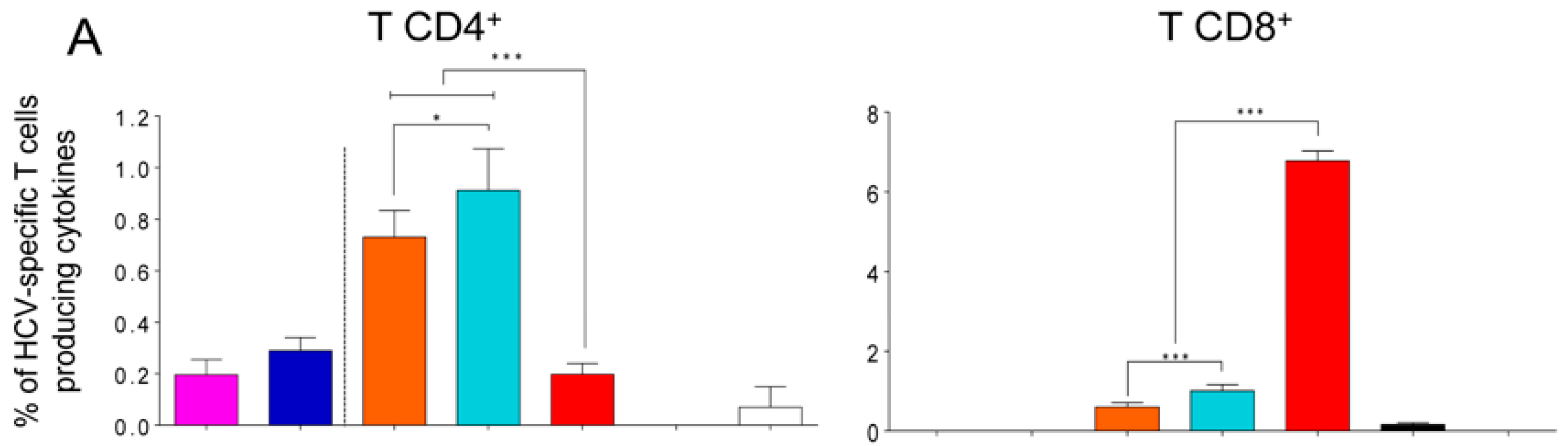
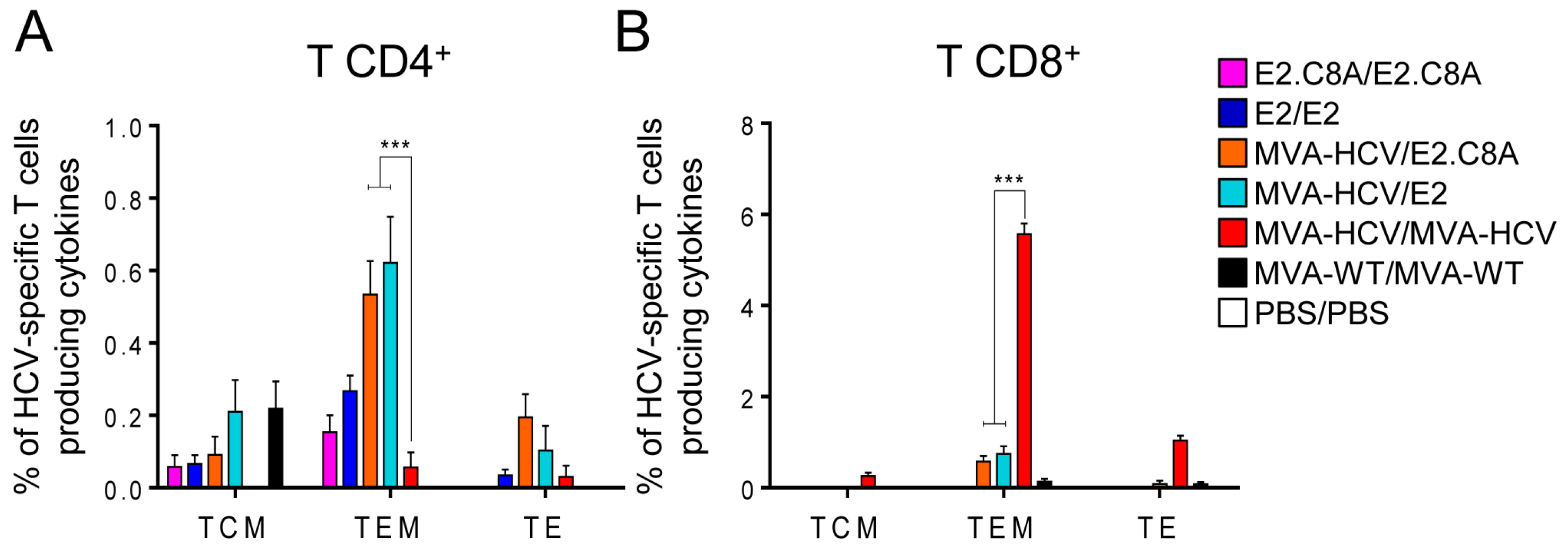
3.6. MVA-HCV Priming Before an E2 Protein Boost Increases the Memory CD4+ T Cell Responses
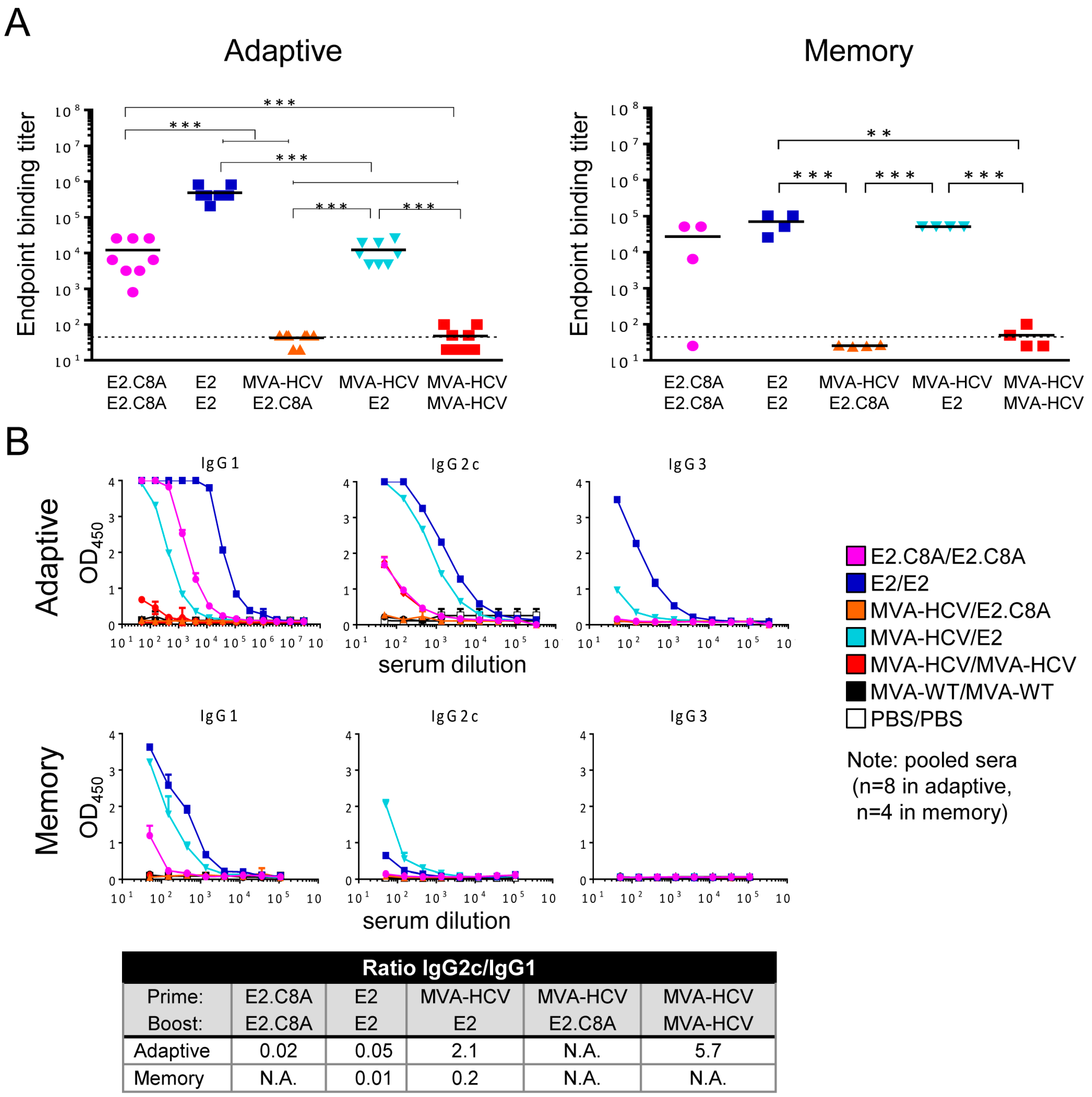
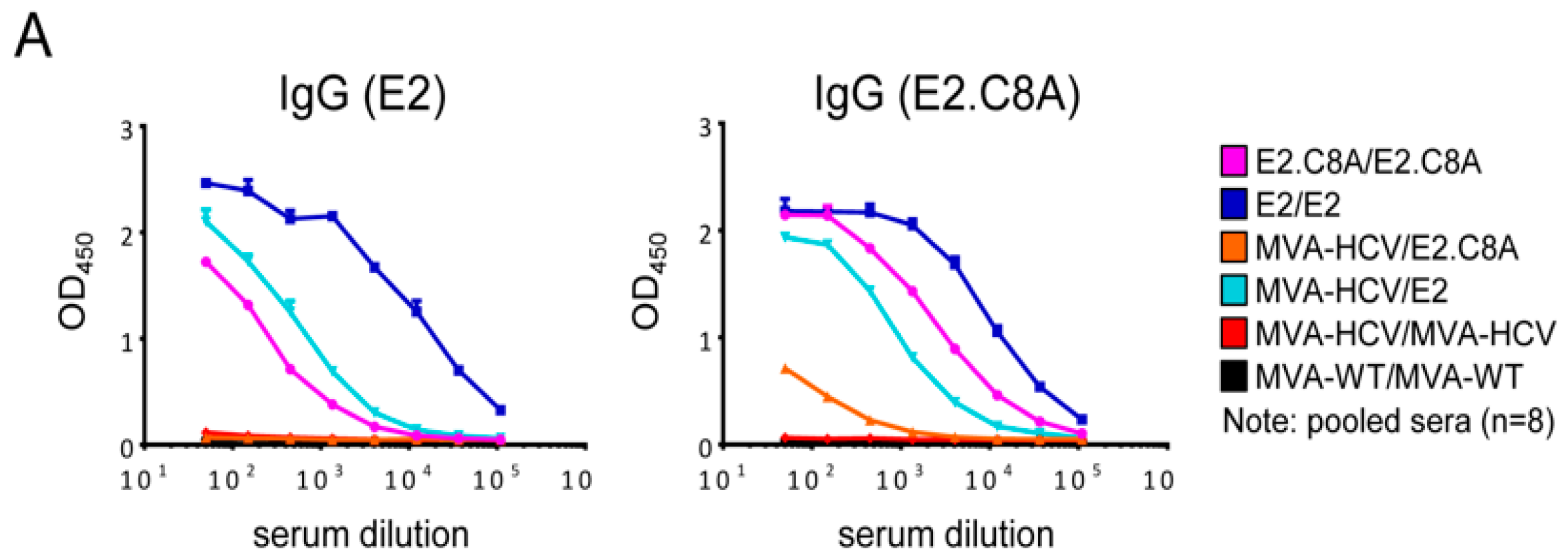
3.7. E2 Aggregates Induced Greater Humoral Immune Response than E2.C8A Either Alone or in Combination with MVA-HCV
3.8. E2/E2 Immunized Mice Elicited Cross Reactive Responses Against Other HCV Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO|Global Hepatitis Report, 2017. Available online: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 13 August 2018).
- WHO|Global Health Sector Strategy on Viral Hepatitis 2016–2021. Available online: http://www.who.int/hepatitis/strategy2016-2021/ghss-hep/en/ (accessed on 10 March 2019).
- Grebely, J.; Conway, B.; Raffa, J.D.; Lai, C.; Krajden, M.; Tyndall, M.W. Hepatitis C virus reinfection in injection drug users. Hepatology 2006, 44, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Fisher, B.E.; Dowd, K.A.; Urban, G.; Liu, L.; Ray, S.C.; Thomas, D.L.; Cox, A.L. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010, 138, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, S.H.; Cox, A.; Hoover, D.R.; Wang, X.-H.; Mao, Q.; Ray, S.; Strathdee, S.A.; Vlahov, D.; Thomas, D.L. Protection against persistence of hepatitis C. Lancet Lond. Engl. 2002, 359, 1478–1483. [Google Scholar] [CrossRef]
- Merat, S.J.; Molenkamp, R.; Wagner, K.; Koekkoek, S.M.; van de Berg, D.; Yasuda, E.; Böhne, M.; Claassen, Y.B.; Grady, B.P.; Prins, M.; et al. Hepatitis C virus Broadly Neutralizing Monoclonal Antibodies Isolated 25 Years after Spontaneous Clearance. PLoS ONE 2016, 11, e0165047. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.J.; Bru, C.; van de Berg, D.; Molenkamp, R.; Tarr, A.W.; Koekkoek, S.; Kootstra, N.A.; Prins, M.; Ball, J.K.; Bakker, A.Q.; et al. Cross-genotype AR3-specific neutralizing antibodies confer long-term protection in injecting drug users after HCV clearance. J. Hepatol. 2019, 71, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Kinchen, V.J.; Zahid, M.N.; Flyak, A.I.; Soliman, M.G.; Learn, G.H.; Wang, S.; Davidson, E.; Doranz, B.J.; Ray, S.C.; Cox, A.L.; et al. Broadly Neutralizing Antibody Mediated Clearance of Human Hepatitis C Virus Infection. Cell Host Microbe 2018, 24, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Thomas, X.V.; Grady, B.P.X.; Van Der Meer, J.T.M.; Ho, C.K.; Vanhommerig, J.W.; Rebers, S.P.; De Jong, M.D.; Van Der Valk, M.; Prins, M.; Molenkamp, R.; et al. Genetic characterization of multiple hepatitis C virus infections following acute infection in HIV-infected men who have sex with men. AIDS Lond. Engl. 2015, 29, 2287–2295. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Shoukry, N.H. Protective immunity against hepatitis C: Many shades of gray. Front. Immunol. 2014, 5, 274. [Google Scholar] [CrossRef]
- Shoukry, N.H. Hepatitis C Vaccines, Antibodies, and T Cells. Front. Immunol. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Swadling, L.; Klenerman, P.; Barnes, E. Ever closer to a prophylactic vaccine for HCV. Expert Opin. Biol. Ther. 2013, 13, 1109–1124. [Google Scholar] [CrossRef] [Green Version]
- Luxenburger, H.; Neumann-Haefelin, C.; Thimme, R.; Boettler, T. HCV-Specific T Cell Responses During and After Chronic HCV Infection. Viruses 2018, 10, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuerst, T.R.; Pierce, B.G.; Keck, Z.-Y.; Foung, S.K.H. Designing a B Cell-Based Vaccine against a Highly Variable Hepatitis C Virus. Front. Microbiol. 2017, 8, 2692. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.-H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV persistence and immune evasion in the absence of memory T cell help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, J.K.; Tarr, A.W.; McKeating, J.A. The past, present and future of neutralizing antibodies for hepatitis C virus. Antivir. Res. 2014, 105, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Osburn, W.O.; Snider, A.E.; Wells, B.L.; Latanich, R.; Bailey, J.R.; Thomas, D.L.; Cox, A.L.; Ray, S.C. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 2014, 59, 2140–2151. [Google Scholar] [CrossRef]
- Pestka, J.M.; Zeisel, M.B.; Bläser, E.; Schürmann, P.; Bartosch, B.; Cosset, F.-L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. USA 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [Green Version]
- Cashman, S.B.; Marsden, B.D.; Dustin, L.B. The Humoral Immune Response to HCV: Understanding is Key to Vaccine Development. Front. Immunol. 2014, 5, 550. [Google Scholar] [CrossRef]
- de Jong, Y.P.; Dorner, M.; Mommersteeg, M.C.; Xiao, J.W.; Balazs, A.B.; Robbins, J.B.; Winer, B.Y.; Gerges, S.; Vega, K.; Labitt, R.N.; et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014, 6, 254ra129. [Google Scholar] [CrossRef] [Green Version]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef]
- Gómez, C.; Perdiguero, B.; Cepeda, M.V.; Mingorance, L.; García-Arriaza, J.; Vandermeeren, A.; Sorzano, C.Ó.S.; Esteban, M. High, broad, polyfunctional, and durable T cell immune responses induced in mice by a novel hepatitis C virus (HCV) vaccine candidate (MVA-HCV) based on modified vaccinia virus Ankara expressing the nearly full-length HCV genome. J. Virol. 2013, 87, 7282–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, M.Q.; Pérez, P.; Gómez, C.E.; Sorzano, C.Ó.S.; Esteban, M.; García-Arriaza, J. Removal of the C6 Vaccinia Virus Interferon-β Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression. Viruses 2018, 10, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, M.Q.; Pérez, P.; Ljungberg, K.; Sorzano, C.Ó.S.; Gómez, C.E.; Liljeström, P.; Esteban, M.; García-Arriaza, J. Potent Anti-Hepatitis C (HCV) T Cell Immune Responses Induced in Mice Vaccinated with DNA-launched RNA Replicons and MVA-HCV. J. Virol. 2019, 93, e00055-19. [Google Scholar] [CrossRef] [Green Version]
- Lavie, M.; Goffard, A.; Dubuisson, J. HCV Glycoproteins: Assembly of a Functional E1–E2 Heterodimer. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.-L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; Chapter 4; ISBN 978-1-904933-20-5. [Google Scholar]
- Falson, P.; Bartosch, B.; Alsaleh, K.; Tews, B.A.; Loquet, A.; Ciczora, Y.; Riva, L.; Montigny, C.; Montpellier, C.; Duverlie, G.; et al. Hepatitis C Virus Envelope Glycoprotein E1 Forms Trimers at the Surface of the Virion. J. Virol. 2015, 89, 10333–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guest, J.D.; Pierce, B.G. Computational Modeling of Hepatitis C Virus Envelope Glycoprotein Structure and Recognition. Front. Immunol. 2018, 9, 1117. [Google Scholar] [CrossRef]
- Luo, K.; Li, S.; Jiang, L.; Zuo, T.; Qing, J.; Shi, X.; Liu, Y.; Wu, H.; Chen, X.; Zhang, L. Combinatorial library-based profiling of the antibody response against hepatitis C virus in humans. J. Gen. Virol. 2015, 96, 52–63. [Google Scholar] [CrossRef]
- Wahid, A.; Dubuisson, J. Virus-neutralizing antibodies to hepatitis C virus. J. Viral Hepat. 2013, 20, 369–376. [Google Scholar] [CrossRef]
- Kong, L.; Jackson, K.N.; Wilson, I.A.; Law, M. Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr. Opin. Virol. 2015, 11, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Colbert, M.D.; Flyak, A.I.; Ogega, C.O.; Kinchen, V.J.; Massaccesi, G.; Hernandez, M.; Davidson, E.; Doranz, B.J.; Cox, A.L.; Crowe, J.E.; et al. Broadly Neutralizing Antibodies Targeting New Sites of Vulnerability in Hepatitis C Virus E1E2. J. Virol. 2019, 93, e02070-18. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, M.; Tello, D.; Yélamos, B.; GómezGutiérrez, J.; Pacheco, B.; Ortega, S.; Serrano, A.G.; Peterson, D.L.; Gavilanes, F.; Rodríguez-Rodríguez, M.; et al. Structural properties of the ectodomain of hepatitis C virus E2 envelope protein. Virus Res. 2009, 139, 91–99. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, K.; Boo, I.; Owczarek, C.M.; Hardy, M.P.; Perugini, M.A.; Fabri, L.; Scotney, P.; Poumbourios, P.; Drummer, H.E. An Optimized Hepatitis C Virus E2 Glycoprotein Core Adopts a Functional Homodimer That Efficiently Blocks Virus Entry. J. Virol. 2017, 91, e01668-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whidby, J.; Mateu, G.; Scarborough, H.; Demeler, B.; Grakoui, A.; Marcotrigiano, J. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J. Virol. 2009, 83, 11078–11089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.; Clementi, N.; Sautto, G.A.; Pfaff, J.; Kahle, K.M.; Barnes, T.; Doranz, B.J.; Dal Peraro, M.; Clementi, M.; Burioni, R.; et al. HCV E2 core structures and mAbs: Something is still missing. Drug Discov. Today 2014, 19, 1964–1970. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C Virus E2 Envelope Glycoprotein Core Structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyak, A.I.; Ruiz, S.; Colbert, M.D.; Luong, T.; Crowe, J.E.; Bailey, J.R.; Bjorkman, P.J. HCV Broadly Neutralizing Antibodies Use a CDRH3 Disulfide Motif to Recognize an E2 Glycoprotein Site that Can Be Targeted for Vaccine Design. Cell Host Microbe 2018, 24, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Tzarum, N.; Giang, E.; Kong, L.; He, L.; Prentoe, J.; Augestad, E.; Hua, Y.; Castillo, S.; Lauer, G.M.; Bukh, J.; et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 2019, 5, eaav1882. [Google Scholar] [CrossRef] [Green Version]
- Kuiken, C.; Combet, C.; Bukh, J.; Shin-I, T.; Deleage, G.; Mizokami, M.; Richardson, R.; Sablon, E.; Yusim, K.; Pawlotsky, J.-M.; et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 2006, 44, 1355–1361. [Google Scholar] [CrossRef]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar] [CrossRef] [Green Version]
- Julien, J.-P.; Lee, J.H.; Cupo, A.; Murin, C.D.; Derking, R.; Hoffenberg, S.; Caulfield, M.J.; King, C.R.; Marozsan, A.J.; Klasse, P.J.; et al. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc. Natl. Acad. Sci. USA 2013, 110, 4351–4356. [Google Scholar] [CrossRef] [Green Version]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.-P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Peña, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, M.; Monrose, V.; Paluch, M.; Techodamrongsin, N.; Rethwilm, A.; Moore, J.P. The production of cleaved, trimeric human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein vaccine antigens and infectious pseudoviruses using linear polyethylenimine as a transfection reagent. Protein Expr. Purif. 2006, 48, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.K.; Crampton, J.C.; Cupo, A.; Ketas, T.; van Gils, M.J.; Sliepen, K.; de Taeye, S.W.; Sok, D.; Ozorowski, G.; Deresa, I.; et al. Murine Antibody Responses to Cleaved Soluble HIV-1 Envelope Trimers Are Highly Restricted in Specificity. J. Virol. 2015, 89, 10383–10398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavillette, D.; Pécheur, E.-I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.-L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, R.W.; Vesanen, M.; Schuelke, N.; Master, A.; Schiffner, L.; Kalyanaraman, R.; Paluch, M.; Berkhout, B.; Maddon, P.J.; Olson, W.C.; et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 2002, 76, 8875–8889. [Google Scholar] [CrossRef] [Green Version]
- Schülke, N.; Vesanen, M.S.; Sanders, R.W.; Zhu, P.; Lu, M.; Anselma, D.J.; Villa, A.R.; Parren, P.W.H.I.; Binley, J.M.; Roux, K.H.; et al. Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 2002, 76, 7760–7776. [Google Scholar] [CrossRef] [Green Version]
- Nájera, J.L.; Gómez, C.E.; García-Arriaza, J.; Sorzano, C.O.; Esteban, M. Insertion of vaccinia virus C7L host range gene into NYVAC-B genome potentiates immune responses against HIV-1 antigens. PLoS ONE 2010, 5, e11406. [Google Scholar] [CrossRef]
- García-Arriaza, J.; Nájera, J.L.; Gómez, C.E.; Sorzano, C.O.S.; Esteban, M. Immunogenic profiling in mice of a HIV/AIDS vaccine candidate (MVA-B) expressing four HIV-1 antigens and potentiation by specific gene deletions. PLoS ONE 2010, 5, e12395. [Google Scholar] [CrossRef]
- Albecka, A.; Montserret, R.; Krey, T.; Tarr, A.W.; Diesis, E.; Ball, J.K.; Descamps, V.; Duverlie, G.; Rey, F.; Penin, F.; et al. Identification of New Functional Regions in Hepatitis C Virus Envelope Glycoprotein E2. J. Virol. 2011, 85, 1777–1792. [Google Scholar] [CrossRef] [Green Version]
- Klasse, P.J.; Depetris, R.S.; Pejchal, R.; Julien, J.-P.; Khayat, R.; Lee, J.H.; Marozsan, A.J.; Cupo, A.; Cocco, N.; Korzun, J.; et al. Influences on Trimerization and Aggregation of Soluble, Cleaved HIV-1 SOSIP Envelope Glycoprotein. J. Virol. 2013, 87, 9873–9885. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, K.; Boo, I.; Tewierek, K.; Edmunds, M.L.; Poumbourios, P.; Drummer, H.E. Role of Conserved Cysteine Residues in Hepatitis C Virus Glycoprotein E2 Folding and Function. J. Virol. 2012, 86, 3961–3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanowicz, R.A.; Wang, R.; Schiel, J.E.; Keck, Z.; Kerzic, M.C.; Lau, P.; Rangarajan, S.; Garagusi, K.J.; Tan, L.; Guest, J.D.; et al. Antigenicity and Immunogenicity of Differentially Glycosylated HCV E2 Envelope Proteins Expressed in Mammalian and Insect Cells. J. Virol. 2019, e01403-18. [Google Scholar] [CrossRef] [Green Version]
- Habel, J.E.; Ohren, J.F.; Borgstahl, G.E. Dynamic light-scattering analysis of full-length human RPA14/32 dimer: Purification, crystallization and self-association. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 254–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owsianka, A.; Tarr, A.W.; Juttla, V.S.; Lavillette, D.; Bartosch, B.; Cosset, F.-L.; Ball, J.K.; Patel, A.H. Monoclonal Antibody AP33 Defines a Broadly Neutralizing Epitope on the Hepatitis C Virus E2 Envelope Glycoprotein. J. Virol. 2005, 79, 11095–11104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, Z.-Y.; Wang, Y.; Lau, P.; Lund, G.; Rangarajan, S.; Fauvelle, C.; Liao, G.C.; Holtsberg, F.W.; Warfield, K.L.; Aman, M.J.; et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute HCV infection. Hepatology 2016, 64, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.; Xia, J.; Wang, Y.; Wang, W.; Krey, T.; Prentoe, J.; Carlsen, T.; Li, A.Y.-J.; Patel, A.H.; Lemon, S.M.; et al. Human Monoclonal Antibodies to a Novel Cluster of Conformational Epitopes on HCV E2 with Resistance to Neutralization Escape in a Genotype 2a Isolate. PLoS Pathog. 2012, 8, e1002653. [Google Scholar] [CrossRef] [Green Version]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Bordo, D.; Galli, G.; Petracca, R.; Falugi, F.; Abrignani, S.; Grandi, G.; Bolognesi, M. CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO J. 2001, 20, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Vietheer, P.T.; Boo, I.; Gu, J.; McCaffrey, K.; Edwards, S.; Owczarek, C.; Hardy, M.P.; Fabri, L.; Center, R.J.; Poumbourios, P.; et al. The core domain of hepatitis C virus glycoprotein E2 generates potent cross-neutralizing antibodies in guinea pigs. Hepatology 2017, 65, 1117–1131. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Suzuki, J.; Sakai, N.; Isome, M.; Nozawa, R.; Tanji, M.; Suzuki, H. Evaluation of T helper-1/-2 balance on the basis of IgG subclasses and serum cytokines in children with glomerulonephritis. Am. J. Kidney Dis. 2004, 44, 42–49. [Google Scholar] [CrossRef]
- Bailey, J.R.; Urbanowicz, R.A.; Ball, J.K.; Law, M.; Foung, S.K.H. Standardized Method for the Study of Antibody Neutralization of HCV Pseudoparticles (HCVpp). Methods Mol. Biol. Clifton NJ 2019, 1911, 441–450. [Google Scholar] [CrossRef]
- Center, R.J.; Boo, I.; Phu, L.; McGregor, J.; Poumbourios, P.; Drummer, H.E. Enhancing the antigenicity and immunogenicity of monomeric forms of hepatitis C virus E2 for use as a preventive vaccine. J. Biol. Chem. 2020, 295, 7179–7192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, J.; Boo, I.; Poumbourios, P.; Drummer, H.E. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J. Biol. Chem. 2011, 286, 31984–31992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzi, A.; Pacheco, B.; Zeng, X.; Kwon, Y.D.; Kwong, P.D.; Sodroski, J. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J. Virol. Methods 2010, 168, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, M.; Ellis, D.; King, N.P. New Vaccine Design and Delivery Technologies. J. Infect. Dis. 2019, 219, S88–S96. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Brinkkemper, M.; Sliepen, K. Nanoparticle Vaccines for Inducing HIV-1 Neutralizing Antibodies. Vaccines 2019, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Kelly, H.G.; Kent, S.J.; Wheatley, A.K. Immunological basis for enhanced immunity of nanoparticle vaccines. Expert Rev. Vaccines 2019, 18, 269–280. [Google Scholar] [CrossRef]
- Denis, J.; Majeau, N.; Acosta-Ramirez, E.; Savard, C.; Bedard, M.-C.; Simard, S.; Lecours, K.; Bolduc, M.; Pare, C.; Willems, B.; et al. Immunogenicity of papaya mosaic virus-like particles fused to a hepatitis C virus epitope: Evidence for the critical function of multimerization. Virology 2007, 363, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wang, X.; Lou, P.; Hu, Z.; Qu, P.; Li, D.; Li, Q.; Xu, Y.; Niu, J.; He, Y.; et al. A nanoparticle-based HCV vaccine with enhanced potency. J. Infect. Dis. 2019, 221, 1304–1314. [Google Scholar] [CrossRef]
- He, L.; Tzarum, N.; Lin, X.; Shapero, B.; Sou, C.; Mann, C.J.; Stano, A.; Zhang, L.; Nagy, K.; Giang, E.; et al. Proof of concept for rational design of hepatitis C virus E2 core nanoparticle vaccines. Sci. Adv. 2020, 6, eaaz6225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstrepen, B.E.; Verschoor, E.J.; Fagrouch, Z.C.; Mooij, P.; de Groot, N.G.; Bontrop, R.E.; Bogers, W.M.; Heeney, J.L.; Koopman, G. Strong vaccine-induced CD8 T-cell responses have cytolytic function in a chimpanzee clearing HCV infection. PLoS ONE 2014, 9, e95103. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Li, X.-D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadi, F.; Rahimi, P.; Modarressi, M.H.; Bolhassani, A.; Ardestani, M.S.; Sadat, S.M.; Aghasadeghi, M.R. Evaluation of Truncated HCV-NS3 Protein for Potential Applications in Immunization and Diagnosis. Clin. Lab. 2016, 62, 1271–1278. [Google Scholar] [CrossRef]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Flynn, J.K.; Dore, G.J.; Hellard, M.; Yeung, B.; Rawlinson, W.D.; White, P.A.; Kaldor, J.M.; Lloyd, A.R.; Ffrench, R.A. Maintenance of Th1 HCV-specific responses in individuals with acute HCV who achieve sustained virological clearance after treatment. J. Gastroenterol. Hepatol. 2013, 28, 1770–1781. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hao, C.Q.; Miao, L.; Dou, X.G. Role of Th1/Th2 cytokines in serum on the pathogenesis of chronic hepatitis C and the outcome of interferon therapy. Genet. Mol. Res. GMR 2014, 13, 9747–9755. [Google Scholar] [CrossRef]
- Sofian, M.; Aghakhani, A.; Farazi, A.A.; Banifazl, M.; Eslamifar, A.; Rashidi, N.; Khadem Sadegh, A.; Ramezani, A. Serum Profile of T Helper 1 and T Helper 2 Cytokines in Hepatitis C Virus Infected Patients. Hepat. Mon. 2012, 12, e6156. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.-Q.; Huang, T.; Deng, Y.-Z.; Zhu, G.-Z. Expression profile and kinetics of cytokines and chemokines in patients with chronic hepatitis C. Int. J. Clin. Exp. Med. 2015, 8, 17995–18003. [Google Scholar]
- Najafizadeh, M.; Farhadi, N.; Sarkari, B. Th1 cytokine profiles in Hepatitis C virus infected patients and their contribution to inflammatory responses. Shiraz E Med. J. 2007, 8, 22–27. [Google Scholar]
- Wright, H.; Alex, P.; Nguyen, T.; Bader, T.; Gurakar, A.; Sebastian, A.; Gonzales, L.; Wallis, G.; Naylor, M.; Dozmorov, I.; et al. Multiplex cytokine profiling of initial therapeutic response in patients with chronic hepatitis C virus infection. Dig. Dis. Sci. 2005, 50, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Alhetheel, A.; Albarrag, A.; Shakoor, Z.; Alswat, K.; Abdo, A.; Al-Hamoudi, W. Assessment of pro-inflammatory cytokines in sera of patients with hepatitis C virus infection before and after anti-viral therapy. J. Infect. Dev. Ctries. 2016, 10, 1093–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osburn, W.O.; Levine, J.S.; Chattergoon, M.A.; Thomas, D.L.; Cox, A.L. Anti-inflammatory cytokines, pro-fibrogenic chemokines and persistence of acute HCV infection. J. Viral Hepat. 2013, 20, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824. [Google Scholar] [CrossRef]
- Bazzill, J.D.; Ochyl, L.J.; Giang, E.; Castillo, S.; Law, M.; Moon, J.J. Interrogation of Antigen Display on Individual Vaccine Nanoparticles for Achieving Neutralizing Antibody Responses against Hepatitis C Virus. Nano Lett. 2018, 18, 7832–7838. [Google Scholar] [CrossRef]
- Ruwona, T.B.; Giang, E.; Nieusma, T.; Law, M. Fine mapping of murine antibody responses to immunization with a novel soluble form of hepatitis C virus envelope glycoprotein complex. J. Virol. 2014, 88, 10459–10471. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Nagy, K.; Chavez, D.; Willis, S.; McBride, R.; Giang, E.; Honda, A.; Bukh, J.; Ordoukhanian, P.; Zhu, J.; et al. Antibody Responses to Immunization with HCV Envelope Glycoproteins as a Baseline for B-Cell-Based Vaccine Development. Gastroenterology 2020, 158, 1058–1071. [Google Scholar] [CrossRef]
- Beaumont, E.; Roch, E.; Chopin, L.; Roingeard, P. Hepatitis C Virus E1 and E2 Proteins Used as Separate Immunogens Induce Neutralizing Antibodies with Additive Properties. PLoS ONE 2016, 11, e0151626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 VACCINES. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, L.; Yang, Y.R.; Tran, K.; Wang, Y.; Chiang, C.-I.; Ozorowski, G.; Xiao, Y.; Ward, A.B.; Wyatt, R.T.; Li, Y. The HIV-1 Envelope Glycoprotein C3/V4 Region Defines a Prevalent Neutralization Epitope following Immunization. Cell Rep. 2019, 27, 586–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marín, M.Q.; Sliepen, K.; García-Arriaza, J.; Koekkoek, S.M.; Pérez, P.; Sorzano, C.Ó.S.; Gómez, C.E.; Sanders, R.W.; Esteban, M. Optimized Hepatitis C Virus (HCV) E2 Glycoproteins and their Immunogenicity in Combination with MVA-HCV. Vaccines 2020, 8, 440. https://doi.org/10.3390/vaccines8030440
Marín MQ, Sliepen K, García-Arriaza J, Koekkoek SM, Pérez P, Sorzano CÓS, Gómez CE, Sanders RW, Esteban M. Optimized Hepatitis C Virus (HCV) E2 Glycoproteins and their Immunogenicity in Combination with MVA-HCV. Vaccines. 2020; 8(3):440. https://doi.org/10.3390/vaccines8030440
Chicago/Turabian StyleMarín, María Q., Kwinten Sliepen, Juan García-Arriaza, Sylvie M. Koekkoek, Patricia Pérez, Carlos Óscar S. Sorzano, Carmen E. Gómez, Rogier W. Sanders, and Mariano Esteban. 2020. "Optimized Hepatitis C Virus (HCV) E2 Glycoproteins and their Immunogenicity in Combination with MVA-HCV" Vaccines 8, no. 3: 440. https://doi.org/10.3390/vaccines8030440