
Cutthroat Trout Virus—Towards a Virus Model to Support Hepatitis E Research

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Propagation
2.2. Quantification of CTV RNA by Reverse Transcription-Quantitative Polymerase Chain Reaction
2.3. Infectivity Assay
2.4. Antibodies
2.5. Dot Blot
2.6. Western Blotting
2.7. Immunofluorescence
2.8. Iodixanol Density Gradient Centrifugation
3. Results
3.1. CTV Replication in Cell Culture
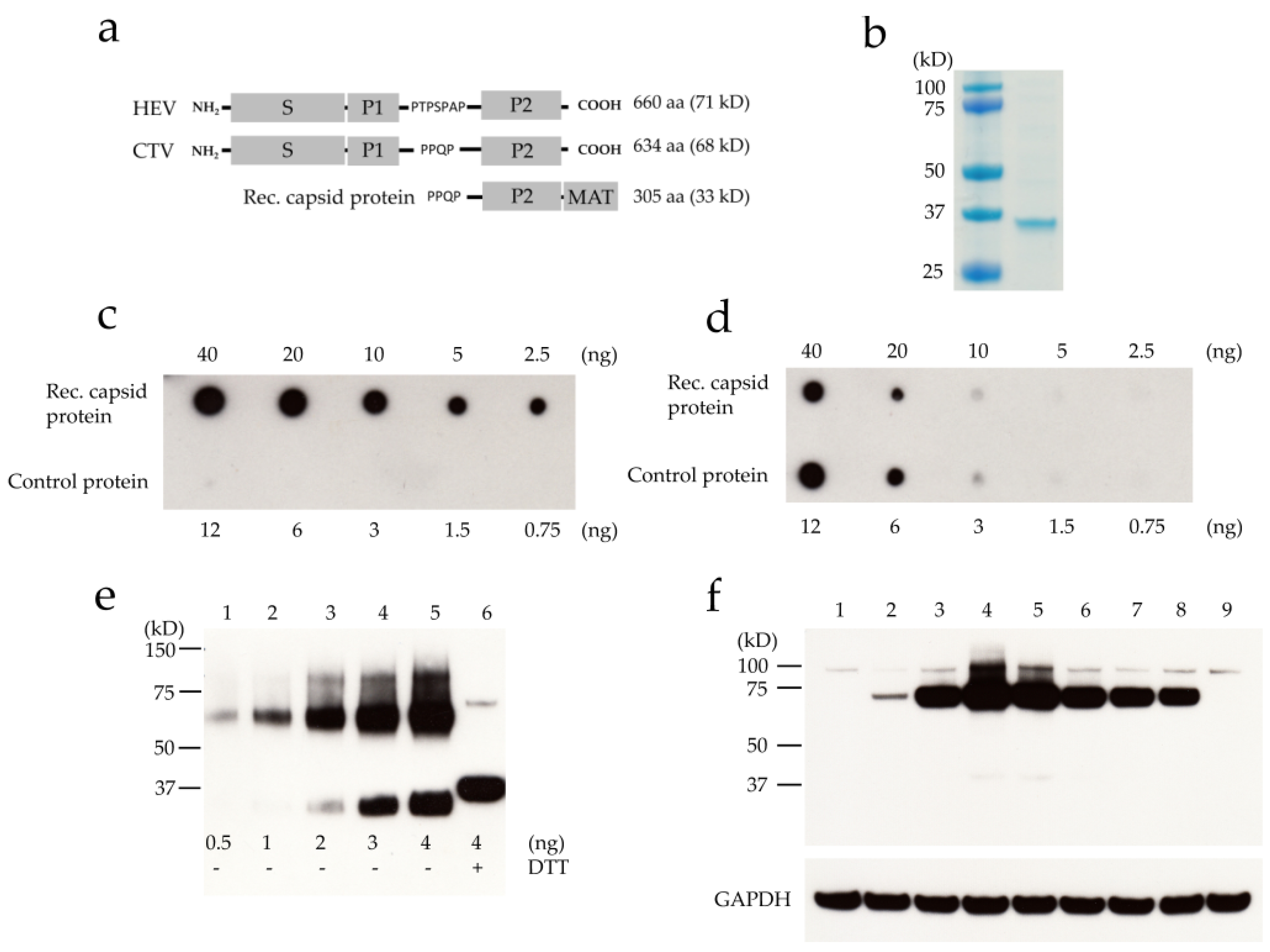
3.2. Protein Expression of ORF2-Encoded Protein
3.3. Immunofluorescence Detection of ORF2-Encoded Protein and dsRNA
3.4. Association of CTV with Lipids
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hedrick, R.P.; Yun, S.; Wingfield, W.H. A small RNA virus isolated from salmonid fishes in California, USA. Can. J. Fish. Aquat. Sci. 1991, 48, 99–104. [Google Scholar] [CrossRef]
- Batts, W.; Yun, S.; Hedrick, R.; Winton, J. A novel member of the family hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res. 2011, 158, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family hepeviridae. J. Gen. Virol. 2015, 96, 1191–1192. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch. Virol. 2014, 159, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, R.P.; Lapatra, S.E.; Yun, S.; Lauda, K.A.; Jones, G.R.; Congleton, J.L.; Dekinkelin, P. Induction of protection from infectious hematopoietic necrosis virus in rainbow-trout Oncorhynchus-mykiss by preexposure to the avirulent cutthroat trout virus (CTV). Dis. Aquat. Organ. 1994, 20, 111–118. [Google Scholar] [CrossRef]
- Debing, Y.; Winton, J.; Neyts, J.; Dallmeier, K. Cutthroat trout virus as a surrogate in vitro infection model for testing inhibitors of hepatitis E virus replication. Antivir. Res. 2013, 100, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S. Study of an epidemic of non-A, non-B hepatitis. Possibility of another human hepatitis virus distinct from post-transfusion non-A, non-B type. Am. J. Med. 1980, 68, 818–824. [Google Scholar] [CrossRef]
- Kamar, N.; Bendall, R.; Legrand-Abravanel, F.; Xia, N.S.; Ijaz, S.; Izopet, J.; Dalton, H.R. Hepatitis E. Lancet 2012, 379, 2477–2488. [Google Scholar] [CrossRef]
- Lu, L.; Li, C.; Hagedorn, C.H. Phylogenetic analysis of global hepatitis E virus sequences: Genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 2006, 16, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.R.; Kotwal, V.; Chawla, S. Chronic hepatitis E: A brief review. World J. Hepatol. 2015, 7, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Viral Hepatitis—Hepatitis E Information. Available online: http://www.webcitation.org/6kT3FyDQK (accessed on 7 September 2016).
- Huang, R.T.; Li, D.R.; Wei, J.; Huang, X.R.; Yuan, X.T.; Tian, X. Isolation and identification of hepatitis-E virus in Xinjiang, China. J. Gen. Virol. 1992, 73, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for hepatitis E virus. J. Gen. Virol. 2007, 88, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Nakazono, N.; Ishii, K.; Li, D.R.; Kawamata, O.; Kawaguchi, R.; Tsukada, Y. I. Hepatitis-E virus (87A strain) propagated in A549 cells. J. Med. Virol. 1995, 47, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Li, D.R.; Wei, S.J.; Li, Q.H.; Yuan, X.T.; Geng, L.Q.; Li, X.Y.; Liu, M.X. Cell culture of sporadic hepatitis E virus in China. Clin. Diagn. Lab. Immunol. 1999, 6, 729–733. [Google Scholar] [PubMed]
- Yugo, D.M.; Cossaboom, C.M.; Meng, X.J. Naturally occurring animal models of human hepatitis E virus infection. ILAR J. 2014, 55, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Construction of an infectious cDNA clone of hepatitis E virus strain JE03–1760F that can propagate efficiently in cultured cells. J. Gen. Virol. 2009, 90, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. Hepatitis E virus cell culture models. Virus Res. 2011, 161, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Guu, T.S.; Liu, Z.; Ye, Q.; Mata, D.A.; Li, K.; Yin, C.; Zhang, J.; Tao, Y.J. Structure of the hepatitis E virus-like particle suggests mechanisms for virus assembly and receptor binding. Proc. Natl. Acad. Sci. USA 2009, 106, 12992–12997. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Membranous replication factories induced by plus-strand RNA viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Karpala, A.J.; Doran, T.J.; Bean, A.G. Immune responses to dsRNA: Implications for gene silencing technologies. Immunol. Cell Biol. 2005, 83, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; Binn, L.N. Incomplete neutralization of hepatitis-A virus invitro due to lipid-associated virions. J. Gen. Virol. 1985, 66, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.D.; Hensley, L.; McKnight, K.L.; Hu, F.Y.; Madden, V.; Ping, L.F.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, K.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch. Virol. 2008, 153, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, F.; Zhang, L.; Harrison, T.J.; Huang, W.J.; Zhao, C.Y.; Kong, W.; Jiang, C.L.; Wang, Y.C. Hepatitis E virus produced from cell culture has a lipid envelope. PLoS ONE 2015, 10, e0132503. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; Kusano, E.; et al. Hepatitis E virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
 ) and cellular fractions (
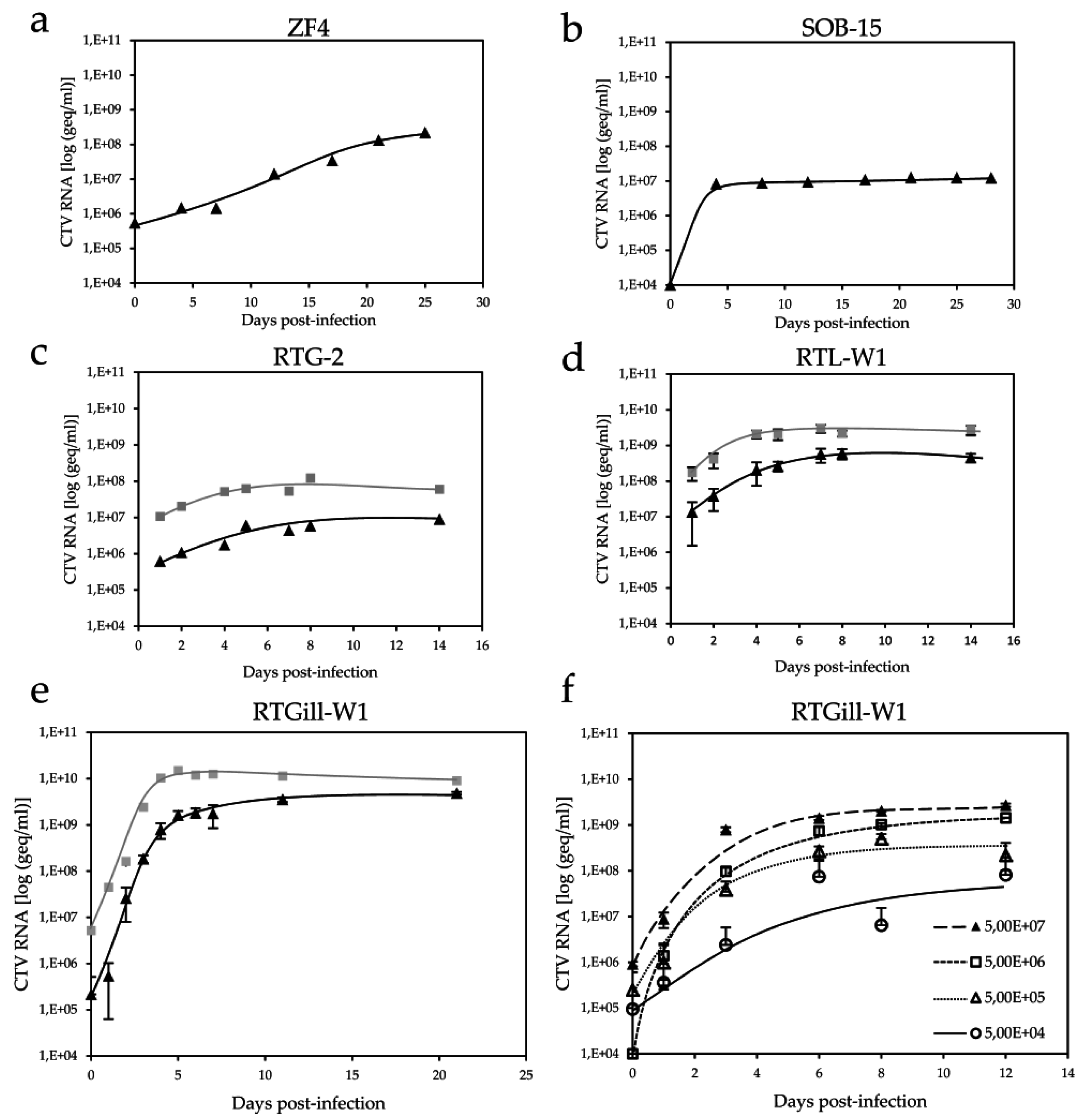
) and cellular fractions (  ) by reverse transcription-polymerase chain reaction (RT-PCR). (a) ZF4; (b) SOB-15; (c) RTG-2; (d) duplicate samples of RTL-W1; and (e) triplicate samples of RTGill-W1 cells; (f) Comparison of viral RNA in duplicates in cell culture SN using 10-fold dilutions of input CTV, as indicated.
) and cellular fractions ( ) by reverse transcription-polymerase chain reaction (RT-PCR). (a) ZF4; (b) SOB-15; (c) RTG-2; (d) duplicate samples of RTL-W1; and (e) triplicate samples of RTGill-W1 cells; (f) Comparison of viral RNA in duplicates in cell culture SN using 10-fold dilutions of input CTV, as indicated.
) by reverse transcription-polymerase chain reaction (RT-PCR). (a) ZF4; (b) SOB-15; (c) RTG-2; (d) duplicate samples of RTL-W1; and (e) triplicate samples of RTGill-W1 cells; (f) Comparison of viral RNA in duplicates in cell culture SN using 10-fold dilutions of input CTV, as indicated.
) and cellular fractions ( ) by reverse transcription-polymerase chain reaction (RT-PCR). (a) ZF4; (b) SOB-15; (c) RTG-2; (d) duplicate samples of RTL-W1; and (e) triplicate samples of RTGill-W1 cells; (f) Comparison of viral RNA in duplicates in cell culture SN using 10-fold dilutions of input CTV, as indicated.



 ) was quantified by RT-qPCR and the density (
) was quantified by RT-qPCR and the density (  ) of each fraction was measured by refractometry. (a) Iodixanol density gradient of CTV from cell culture SN; (b) Treatment of cell culture SN with 1% NP40; (c) Fractions 10 and 11 of the low-density fraction from (a) were pooled and re-centrifuged; (d) The same fraction used for (c) was pretreated with 1% NP40 and re-centrifuged.
) was quantified by RT-qPCR and the density ( ) of each fraction was measured by refractometry. (a) Iodixanol density gradient of CTV from cell culture SN; (b) Treatment of cell culture SN with 1% NP40; (c) Fractions 10 and 11 of the low-density fraction from (a) were pooled and re-centrifuged; (d) The same fraction used for (c) was pretreated with 1% NP40 and re-centrifuged.
) of each fraction was measured by refractometry. (a) Iodixanol density gradient of CTV from cell culture SN; (b) Treatment of cell culture SN with 1% NP40; (c) Fractions 10 and 11 of the low-density fraction from (a) were pooled and re-centrifuged; (d) The same fraction used for (c) was pretreated with 1% NP40 and re-centrifuged.
) was quantified by RT-qPCR and the density ( ) of each fraction was measured by refractometry. (a) Iodixanol density gradient of CTV from cell culture SN; (b) Treatment of cell culture SN with 1% NP40; (c) Fractions 10 and 11 of the low-density fraction from (a) were pooled and re-centrifuged; (d) The same fraction used for (c) was pretreated with 1% NP40 and re-centrifuged.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Nordheim, M.; Boinay, M.; Leisi, R.; Kempf, C.; Ros, C. Cutthroat Trout Virus—Towards a Virus Model to Support Hepatitis E Research. Viruses 2016, 8, 289. https://doi.org/10.3390/v8100289
Von Nordheim M, Boinay M, Leisi R, Kempf C, Ros C. Cutthroat Trout Virus—Towards a Virus Model to Support Hepatitis E Research. Viruses. 2016; 8(10):289. https://doi.org/10.3390/v8100289
Chicago/Turabian StyleVon Nordheim, Marcus, Michel Boinay, Remo Leisi, Christoph Kempf, and Carlos Ros. 2016. "Cutthroat Trout Virus—Towards a Virus Model to Support Hepatitis E Research" Viruses 8, no. 10: 289. https://doi.org/10.3390/v8100289