
Tryptophan Residues Are Critical for Portal Protein Assembly and Incorporation in Bacteriophage P22


Abstract
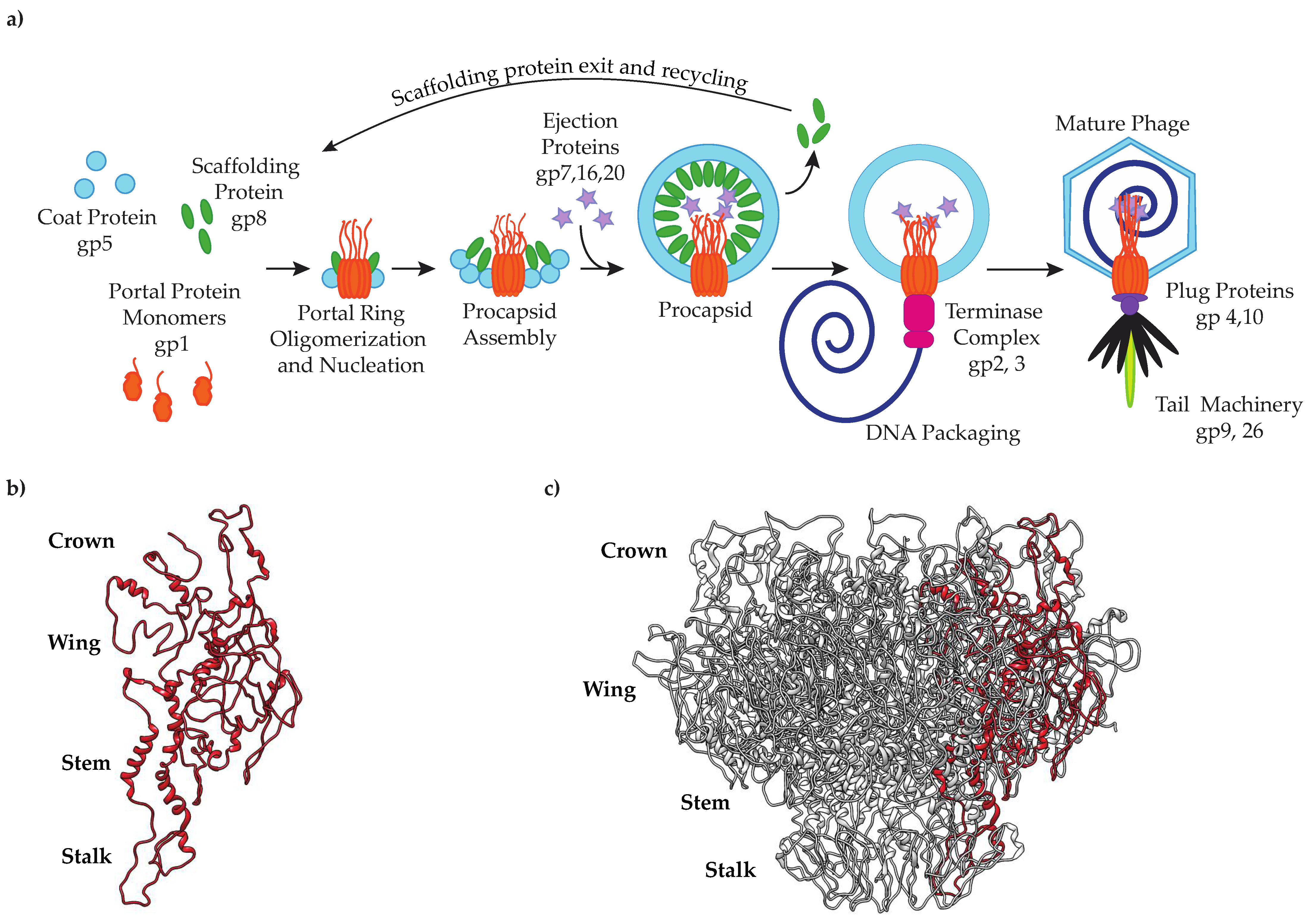
:1. Introduction
2. Materials and Methods
2.1. Subcloning Portal Protein Gene into Twin-Strep-Tag Vector
2.2. Generation of His6-Tagged and Twin-Strep-Tagged Portal Protein Variants
2.3. Efficiency of Plating (EOP) Assay
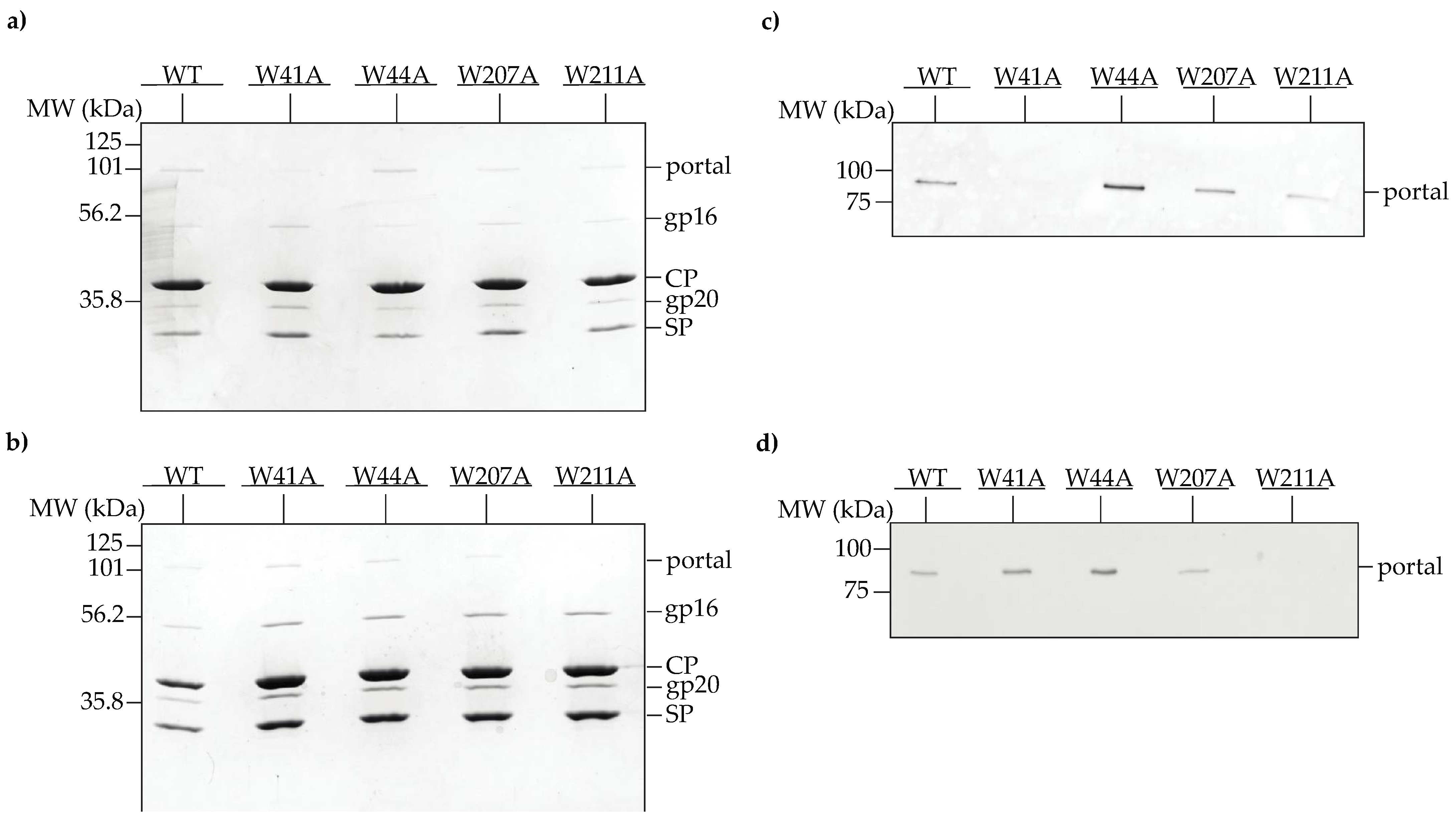
2.4. In Vivo Portal Protein Incorporation into PCs
2.5. Negative-Stain Transmission Electron Microscopy (TEM)
2.6. In Vivo Production of Procapsids
2.7. In Vitro Capsid Maturation and Stability Assays
2.8. Fluorescent Western Blot
2.9. Portal Expression and Purification
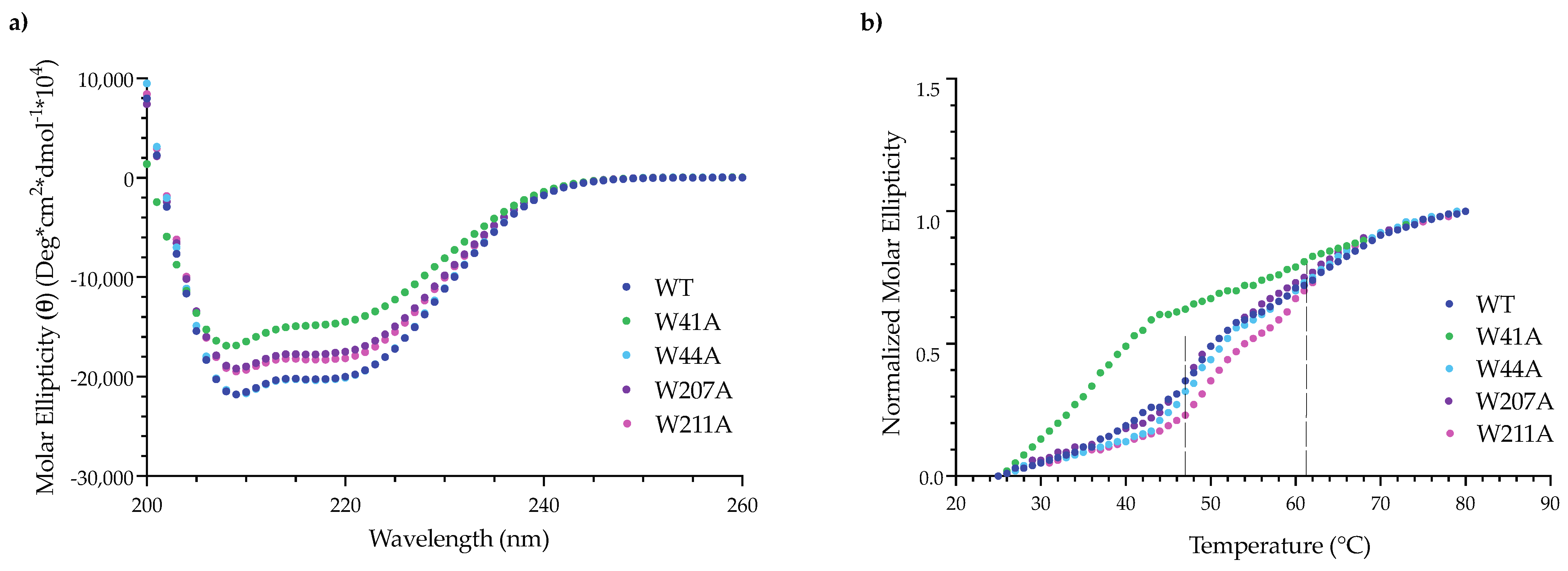
2.10. Circular Dichroism (CD) of PMs and PC Portal Rings
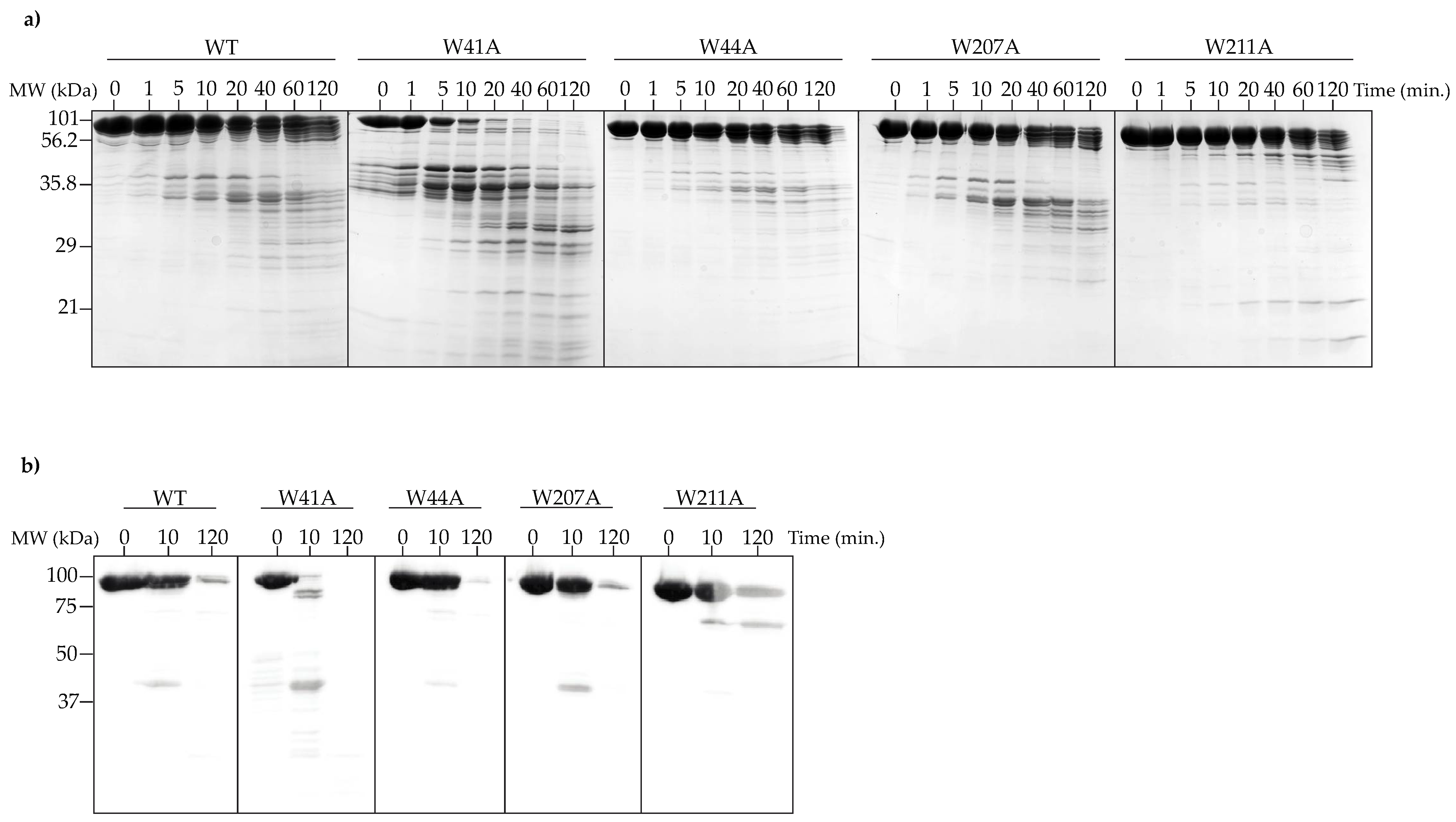
2.11. Proteolytic Digest
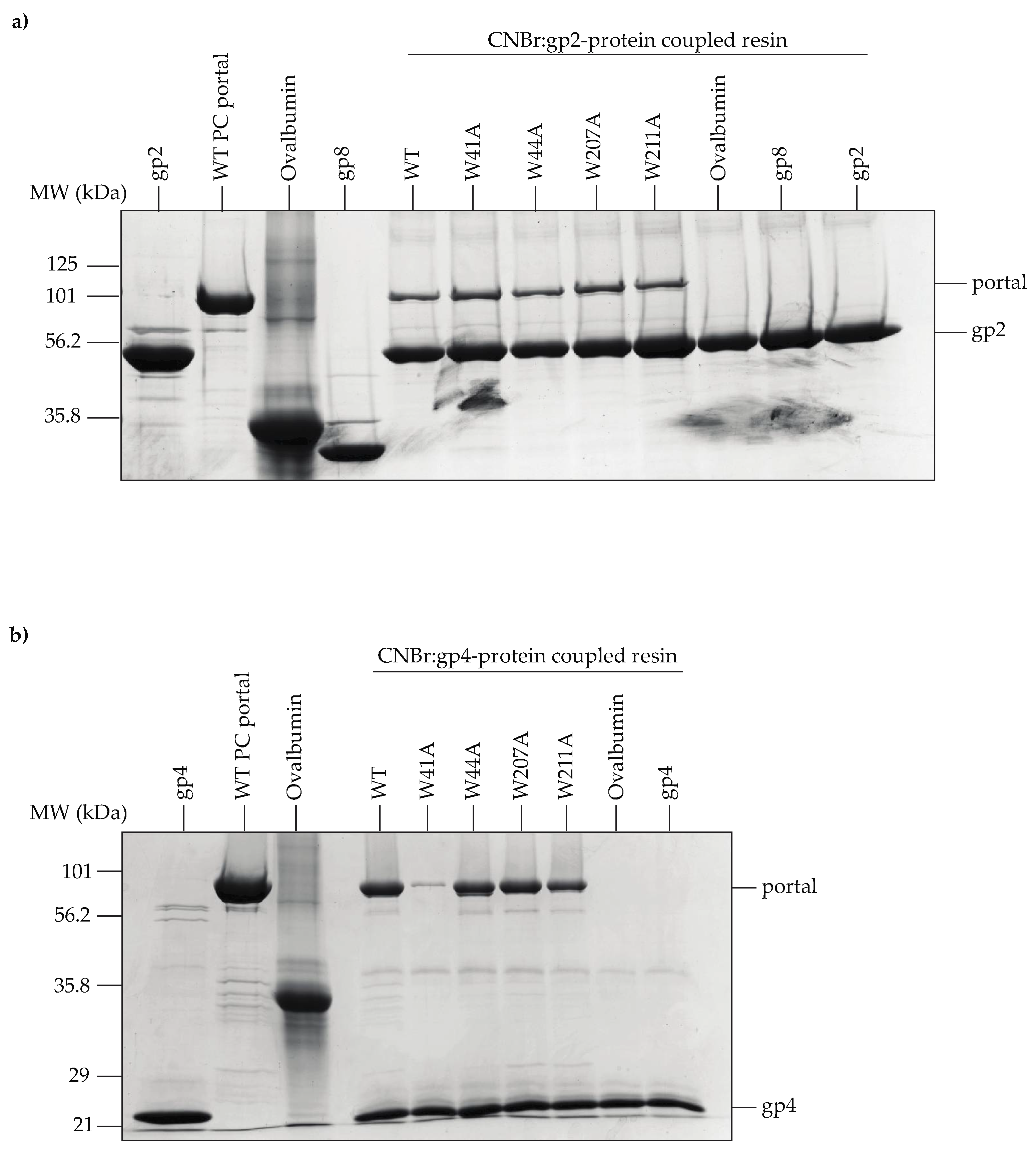
2.12. Binding of Purified PC Portal Rings to L-Terminase and gp4
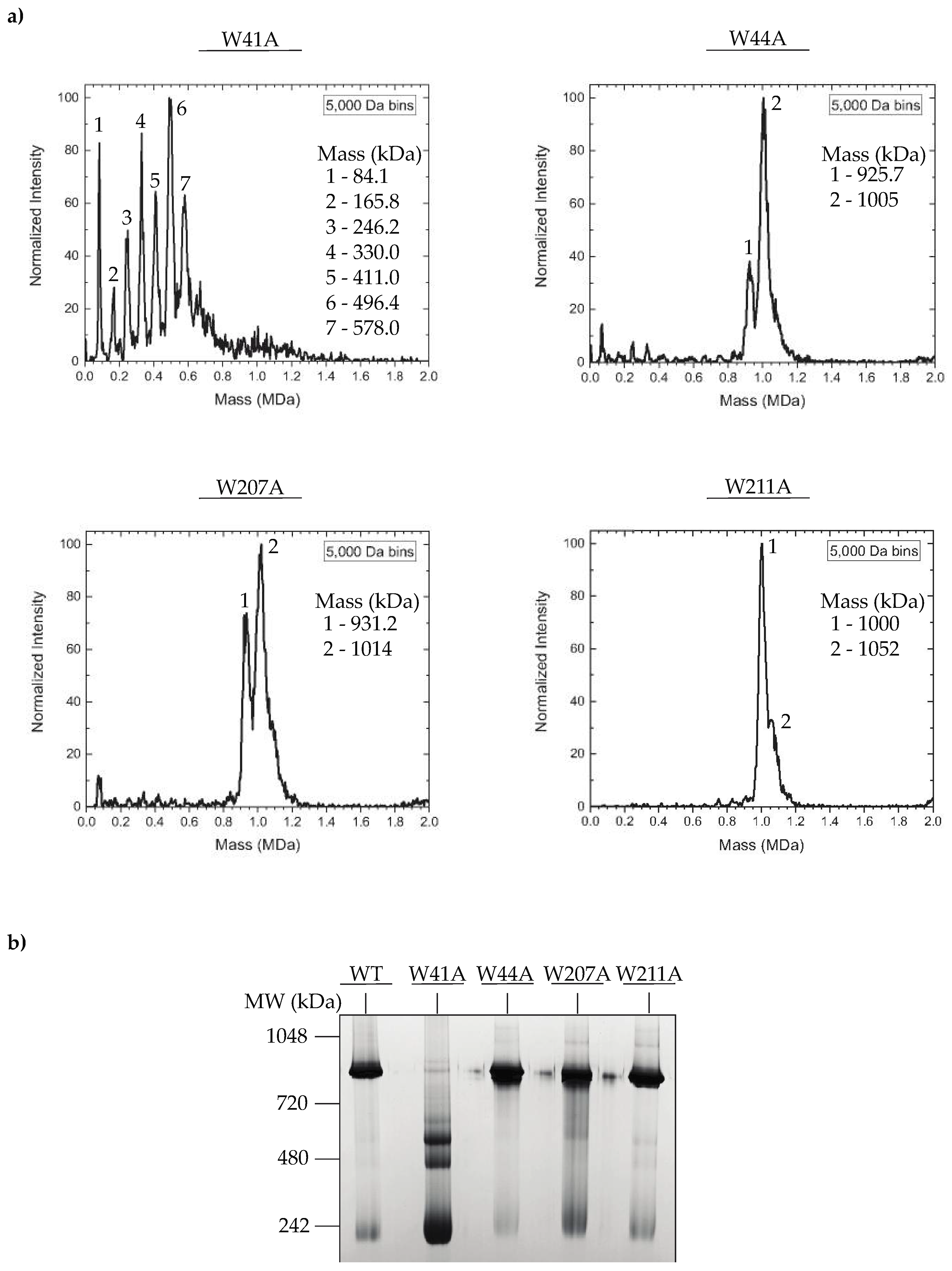
2.13. CDMS
3. Results
3.1. Residues in Portal Protein Tryptophan Belt Are Required to Generate Infectious Virions
3.2. Some Tryptophan Belt Residues Are Important for Portal Protein Incorporation into PCs In Vivo
3.3. In Vitro Assembled PC Portal Rings Are Primarily Dodecameric, except for the W41A Variant
3.4. Alanine Substitutions Affect Portal Protein Folding into PC Portal Rings
3.5. Portal Protein Variants Bind to L-Terminase and the Plug Protein, gp4
4. Discussion
4.1. The Tryptophan Belt Residues Are Not Critical for Binding to Scaffolding Protein but Are Essential for Viral Maturation
4.2. In Vitro Characterization of Variant Portal Protein Rings Reveals Oligomerization Defect
4.3. DNA Packaging Is Affected by Substitutions in the Tryptophan Belt
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, J.; Casjens, S. Catalytic Head Assembling Protein in Virus Morphogenesis. Nature 1974, 251, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Homa, F.L.; Thomsen, D.R.; Trus, B.L.; Cheng, N.; Steven, A.; Booy, F.; Brown, J.C. Assembly of the Herpes Simplex Virus Procapsid from Purified Components and Identification of Small Complexes Containing the Major Capsid and Scaffolding Proteins. J. Virol. 1999, 73, 4239–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casjens, S.; Wyckoff, E.; Hayden, M.; Sampson, L.; Eppler, K.; Randall, S.; Moreno, E.T.; Serwer, P. Bacteriophage P22 Portal Protein Is Part of the Gauge That Regulates Packing Density of Intravirion DNA. J. Mol. Biol. 1992, 224, 1055–1074. [Google Scholar] [CrossRef]
- Motwani, T.; Teschke, C.M. Architect of Virus Assembly: The Portal Protein Nucleates Procapsid Assembly in Bacteriophage P22. J. Virol. 2019, 93, e00187-19. [Google Scholar] [CrossRef] [Green Version]
- Yap, N.L.; Rao, V.B. Novel Mutants in the 5′ Upstream Region of the Portal Protein Gene 20 Overcome a Gp40-Dependent Prohead Assembly Block in Bacteriophage T4. J. Mol. Biol. 1996, 263, 539–550. [Google Scholar] [CrossRef]
- Kato, H.; Baschong, C. Isolation of a Gp20-Complex and Its Role in in Vitro Assembly of Both Prohead and Core of Bacteriophage T4. Virology 1997, 227, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Droge, A.; Santos, M.A.; Stiege, A.C.; Alonso, J.C.; Lurz, R.; Trautner, T.A.; Tavares, P. Shape and DNA Packaging Activity of Bacteriophage SPP1 Procapsid: Protein Components and Interactions during Assembly. J. Mol. Biol. 2000, 296, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Murialdo, H.; Becker, A. A Genetic Analysis of Bacteriophage Lambda Prohead Assembly in Vitro. J. Mol. Biol. 1978, 125, 57–74. [Google Scholar] [CrossRef]
- Fu, C.; Prevelige, P.E. In Vitro Incorporation of the Phage Phi29 Connector Complex. Virology 2009, 394, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Erickson, S.; Xu, W.; Baker, S. Regulation of the Phage Phi29 Prohead Shape and Size by the Portal Vertex. Virology 1991, 183, 366–373. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Homa, F.L.; Brown, J.C. Involvement of the Portal at an Early Step in Herpes Simplex Virus Capsid Assembly. J. Virol. 2005, 79, 10540–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, K.; Schrad, J.; Cingolani, G. Breaking Symmetry in Viral Icosahedral Capsids as Seen through the Lenses of X-Ray Crystallography and Cryo-Electron Microscopy. Viruses 2018, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinder, N.D.; Lederberg, J. Genetic Exchange in Salmonella. J. Bacteriol. 1952, 64, 679–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susskind, M.M.; Botstein, D. Molecular Genetics of Bacteriophage P22. Microbiol. Rev. 1978, 42, 385–413. [Google Scholar] [CrossRef] [PubMed]
- Motwani, T.; Lokareddy, R.K.; Dunbar, C.A.; Cortines, J.R.; Jarrold, M.F.; Cingolani, G.; Teschke, C.M. A Viral Scaffolding Protein Triggers Portal Ring Oligomerization and Incorporation during Procapsid Assembly. Sci. Adv. 2017, 3, e1700423. [Google Scholar] [CrossRef] [Green Version]
- Teschke, C.M.; Parent, K.N. ‘Let the Phage Do the Work’: Using the Phage P22 Coat Protein Structures as a Framework to Understand Its Folding and Assembly Mutants. Virology 2010, 401, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Cingolani, G. Structure of P22 Headful Packaging Nuclease. J. Biol. Chem. 2012, 287, 28196–28205. [Google Scholar] [CrossRef] [Green Version]
- Olia, A.S.; Prevelige, P.E.; Johnson, J.E.; Cingolani, G. Three-Dimensional Structure of a Viral Genome-Delivery Portal Vertex. Nat. Struct. Mol. Biol. 2011, 18, 597–603. [Google Scholar] [CrossRef]
- Lokareddy, R.K.; Sankhala, R.S.; Roy, A.; Afonine, P.V.; Motwani, T.; Teschke, C.M.; Parent, K.N.; Cingolani, G. Portal Protein Functions Akin to a DNA-Sensor That Couples Genome-Packaging to Icosahedral Capsid Maturation. Nat. Commun. 2017, 8, 14310. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural Assembly of the Tailed Bacteriophage Φ29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-T.; Jih, J.; Dai, X.; Bi, G.-Q.; Zhou, Z.H. Cryo-EM Structures of Herpes Simplex Virus Type 1 Portal Vertex and Packaged Genome. Nature 2019, 570, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 Bacteriophage Portal and Tail Suggest a Viral DNA Retention and Ejection Mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machón, C.; Fàbrega-Ferrer, M.; Zhou, D.; Cuervo, A.; Carrascosa, J.L.; Stuart, D.I.; Coll, M. Atomic Structure of the Epstein-Barr Virus Portal. Nat. Commun. 2019, 10, 3891. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhang, X.; Gao, S.; Rao, P.A.; Padilla-Sanchez, V.; Chen, Z.; Sun, S.; Xiang, Y.; Subramaniam, S.; Rao, V.B.; et al. Cryo-EM Structure of the Bacteriophage T4 Portal Protein Assembly at near-Atomic Resolution. Nat. Commun. 2015, 6, 7548. [Google Scholar] [CrossRef] [Green Version]
- McElwee, M.; Vijayakrishnan, S.; Rixon, F.; Bhella, D. Structure of the Herpes Simplex Virus Portal-Vertex. PLoS Biol. 2018, 16, e2006191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayfield, O.W.; Steven, A.C.; Antson, A.A. Cryo-EM Structure in Situ Reveals a Molecular Switch That Safeguards Virus against Genome Loss. eLife 2020, 9, e55517. [Google Scholar] [CrossRef]
- Prevelige, P.E.; Cortines, J.R. Phage Assembly and the Special Role of the Portal Protein. Curr. Opin. Virol. 2018, 31, 66–73. [Google Scholar] [CrossRef]
- Bedwell, G.J.; Prevelige, P.E. Targeted Mutagenesis of the P22 Portal Protein Reveals the Mechanism of Signal Transmission during DNA Packaging. Virology 2017, 505, 127–138. [Google Scholar] [CrossRef]
- Buch, M.H.C.; Newcomb, W.W.; Winkler, D.C.; Steven, A.C.; Heymann, J.B. Cryo-Electron Tomography of the Herpesvirus Procapsid Reveals Interactions of the Portal with the Scaffold and a Shift on Maturation. mBio 2021, 12, e03575-20. [Google Scholar] [CrossRef]
- Fang, Q.; Tang, W.-C.; Tao, P.; Mahalingam, M.; Fokine, A.; Rossmann, M.G.; Rao, V.B. Structural Morphing in a Symmetry-Mismatched Viral Vertex. Nat. Commun. 2020, 11, 1713. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Lander, G.C.; Olia, A.; Li, R.; Casjens, S.; Prevelige, P.; Cingolani, G.; Baker, T.S.; Johnson, J.E. Peering Down the Barrel of a Bacteriophage Portal: The Genome Packaging and Release Valve in P22. Structure 2011, 19, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, X.; Dong, L.; Pang, J.; Xu, M.; Zhong, Q.; Zeng, M.-S.; Yu, X. CryoEM Structure of the Tegumented Capsid of Epstein-Barr Virus. Cell. Res. 2020, 30, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Baines, J.D. Tryptophan Residues in the Portal Protein of Herpes Simplex Virus 1 Critical to the Interaction with Scaffold Proteins and Incorporation of the Portal into Capsids. J. Virol. 2009, 83, 11726–11733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeo, C.L.; Cingolani, G.; Teschke, C.M. Portal Protein: The Orchestrator of Capsid Assembly for the DsDNA Tailed Bacteriophages and Herpesviruses. Annu. Rev. Virol. 2019, 6, 141–160. [Google Scholar] [CrossRef]
- Guasch, A.; Pous, J.; Ibarra, B.; Gomis-Rüth, F.X.; Valpuesta, J.M.; Sousa, N.; Carrascosa, J.L.; Coll, M. Detailed Architecture of a DNA Translocating Machine: The High-Resolution Structure of the Bacteriophage Φ29 Connector Particle 1 1Edited by R. Huber. J. Mol. Biol. 2002, 315, 663–676. [Google Scholar] [CrossRef]
- Chen, D.-H.; Baker, M.L.; Hryc, C.F.; DiMaio, F.; Jakana, J.; Wu, W.; Dougherty, M.; Haase-Pettingell, C.; Schmid, M.F.; Jiang, W.; et al. Structural Basis for Scaffolding-Mediated Assembly and Maturation of a DsDNA Virus. Proc. Natl. Acad. Sci. USA 2011, 108, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Weigele, P.R.; Sampson, L.; Winn-Stapley, D.; Casjens, S.R. Molecular Genetics of Bacteriophage P22 Scaffolding Protein’s Functional Domains. J. Mol. Biol. 2005, 348, 831–844. [Google Scholar] [CrossRef]
- Greene, B.; King, J. Scaffolding Mutants Identifying Domains Required for P22 Procapsid Assembly and Maturation. Virology 1996, 225, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.D.; Prevelige, P.E. A P22 Scaffold Protein Mutation Increases the Robustness of Head Assembly in the Presence of Excess Portal Protein. J. Virol. 2002, 76, 10245–10255. [Google Scholar] [CrossRef] [Green Version]
- Winston, F.; Botstein, D.; Miller, J.H. Characterization of Amber and Ochre Suppressors in Salmonella Typhimurium. J. Bacteriol. 1979, 137, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Asija, K.; Teschke, C.M. Of Capsid Structure and Stability: The Partnership between Charged Residues of E-Loop and P-Domain of the Bacteriophage P22 Coat Protein. Virology 2019, 534, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, K.; Olia, A.S.; Uetrecht, C.; Cingolani, G.; Heck, A.J.R. Determination of Stoichiometry and Conformational Changes in the First Step of the P22 Tail Assembly. J. Mol. Biol. 2008, 379, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.D.; Prevelige, P.E. Structural Transformations Accompanying the Assembly of Bacteriophage P22 Portal Protein Rings in Vitro. J. Biol. Chem. 2001, 276, 6779–6788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuerstenau, S.D.; Benner, W.H. Molecular Weight Determination of Megadalton DNA Electrospray Ions Using Charge Detection Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 1995, 9, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Keifer, D.Z.; Pierson, E.E.; Jarrold, M.F. Charge Detection Mass Spectrometry: Weighing Heavier Things. Analyst 2017, 142, 1654–1671. [Google Scholar] [CrossRef]
- Jarrold, M.F. Applications of Charge Detection Mass Spectrometry in Molecular Biology and Biotechnology. Chem. Rev. 2021, 122, 7415–7441. [Google Scholar] [CrossRef]
- Contino, N.C.; Jarrold, M.F. Charge Detection Mass Spectrometry for Single Ions with a Limit of Detection of 30 Charges. Int. J. Mass Spectrom. 2013, 345–347, 153–159. [Google Scholar] [CrossRef]
- Keifer, D.Z.; Shinholt, D.L.; Jarrold, M.F. Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Anal. Chem. 2015, 87, 10330–10337. [Google Scholar] [CrossRef]
- Draper, B.E.; Anthony, S.N.; Jarrold, M.F. The FUNPET—A New Hybrid Ion Funnel-Ion Carpet Atmospheric Pressure Interface for the Simultaneous Transmission of a Broad Mass Range. J. Am. Soc. Mass Spectrom. 2018, 29, 2160–2172. [Google Scholar] [CrossRef]
- Draper, B.E.; Jarrold, M.F. Real-Time Analysis and Signal Optimization for Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 898–904. [Google Scholar] [CrossRef]
- Poliakov, A.; van Duijn, E.; Lander, G.; Fu, C.; Johnson, J.E.; Prevelige, P.E.; Heck, A.J.R. Macromolecular Mass Spectrometry and Electron Microscopy as Complementary Tools for Investigation of the Heterogeneity of Bacteriophage Portal Assemblies. J. Struct. Biol. 2007, 157, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Poteete, A.R.; King, J. Functions of Two New Genes in Salmonella Phage P22 Assembly. Virology 1977, 76, 725–739. [Google Scholar] [CrossRef]
- Olia, A.S.; Al-Bassam, J.; Winn-Stapley, D.A.; Joss, L.; Casjens, S.R.; Cingolani, G. Binding-Induced Stabilization and Assembly of the Phage P22 Tail Accessory Factor Gp4. J. Mol. Biol. 2006, 363, 558–576. [Google Scholar] [CrossRef]
- Grimaud, R. Bacteriophage Mu Head Assembly. Virology 1996, 217, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Uetrecht, C.; Kang, S.; Morais, M.C.; Heck, A.J.R.; Walter, M.R.; Prevelige, P.E. A Docking Model Based on Mass Spectrometric and Biochemical Data Describes Phage Packaging Motor Incorporation. Mol. Cell. Proteom. 2010, 9, 1764–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Driel, R.; Couture, E. Assembly of the Scaffolding Core of Bacteriophage T4 Preheads. J. Mol. Biol. 1978, 123, 713–719. [Google Scholar] [CrossRef]
- Rizzo, A.A.; Suhanovsky, M.M.; Baker, M.L.; Fraser, L.C.R.; Jones, L.M.; Rempel, D.L.; Gross, M.L.; Chiu, W.; Alexandrescu, A.T.; Teschke, C.M. Multiple Functional Roles of the Accessory I-Domain of Bacteriophage P22 Coat Protein Revealed by NMR Structure and CryoEM Modeling. Structure 2014, 22, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.; Oram, M.; Ma, J.; Black, L.W. Portal Control of Viral Prohead Expansion and DNA Packaging. Virology 2009, 391, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, A.; Ray, K.; Lakowicz, J.R.; Black, L.W. Dynamics of the T4 Bacteriophage DNA Packasome Motor. J. Biol. Chem. 2011, 286, 18878–18889. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Casado, A.; Moore, S.D.; Prevelige, P.E.; Thomas, G.J. Structure of Bacteriophage P22 Portal Protein in Relation to Assembly: Investigation by Raman Spectroscopy. Biochemistry 2001, 40, 13583–13591. [Google Scholar] [CrossRef]
- Rodríguez-Casado, A.; Thomas, G.J. Structural Roles of Subunit Cysteines in the Folding and Assembly of the DNA Packaging Machine (Portal) of Bacteriophage P22. Biochemistry 2003, 42, 3437–3445. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Overman, S.A.; Thomas, G.J. Impact of in Vitro Assembly Defects on in Vivo Function of the Phage P22 Portal. Virology 2007, 365, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, L.W. Old, New, and Widely True: The Bacteriophage T4 DNA Packaging Mechanism. Virology 2015, 479–480, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, R.G.; Mullaney, J.; Black, L.W. Portal Fusion Protein Constraints on Function in DNA Packaging of Bacteriophage T4: Portal Fusion Protein Constraints on DNA Packaging. Mol. Microbiol. 2006, 61, 16–32. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Portal Protein Variant | ~Fold Decrease in Titer Relative to Complementation with WT Portal Protein at 37 °C a |
|---|---|
| W41A | 10,000 |
| W41H | 300 |
| W41F | 100 |
| W41Y | 100 |
| W44A | 10 |
| W44H | 15 |
| W44F | 15 |
| W44Y | 15 |
| W207A | 10 |
| W207H | 5 (35-fold decrease at 41 °C) |
| W207F | 5 (50-fold decrease at 41 °C) |
| W207Y | 200 |
| W211A | 100 |
| W211H | 70 |
| W211F | 30 |
| W211Y | 150 |
| Portal Protein Variant | Incorporation of Portal Protein into Procapsids In Vivo at 37 °C a |
|---|---|
| W41H | Yes |
| W41F | No |
| W41Y | Yes |
| W44H | Yes |
| W44F | Yes |
| W44Y | Yes |
| W207H | Yes |
| W207F | Yes |
| W207Y | Yes |
| W211H | Yes |
| W211F | Yes |
| W211Y | Yes |
| Portal Protein Variant | Relative Fold Change in Titer for the Following Temperatures a: | |
|---|---|---|
| 30 °C | 37 °C | |
| WT | 1.0 ± 0.00 | 1.86 ± 0.29 |
| W41A | 1.31 ± 0.29 | 1.07 ± 0.45 |
| W44A | 2.06 ± 0.43 | 2.01 ± 0.31 |
| W207A | 0.93 ± 0.91 | 1.03 ± 0.83 |
| W211A | 0.98 ± 0.56 | 1.03 ± 0.40 |
| WT | W41A | W44A | W207A | W211A | |
|---|---|---|---|---|---|
| Portal protein incorporated into PCs in vivo at 30 °C, indicating ability to interact with scaffolding protein | Yes | Very little | Yes | Yes | Yes |
| Portal protein incorporated into PCs in vivo at 37 °C, indicating ability to interact with scaffolding protein | Yes | Yes | Yes | Yes | No |
| In vitro PC portal ring oligomeric state | 12-mers + some monomers | Monomers + small oligomers | 11- and 12-mers | 11- and 12-mers | 12-mers |
| Change in stability by circular dichroism temperature melts relative to WT PC portal rings | -- | Destabilized, melt similar to WT PMs [43] | WT-like | WT-like | More stable |
| PC portal ring sensitivity to α-chymotrypsin | -- | More digestion than WT | WT-like | WT-like | Less digestion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodbury, B.M.; Motwani, T.; Leroux, M.N.; Barnes, L.F.; Lyktey, N.A.; Banerjee, S.; Dedeo, C.L.; Jarrold, M.F.; Teschke, C.M. Tryptophan Residues Are Critical for Portal Protein Assembly and Incorporation in Bacteriophage P22. Viruses 2022, 14, 1400. https://doi.org/10.3390/v14071400
Woodbury BM, Motwani T, Leroux MN, Barnes LF, Lyktey NA, Banerjee S, Dedeo CL, Jarrold MF, Teschke CM. Tryptophan Residues Are Critical for Portal Protein Assembly and Incorporation in Bacteriophage P22. Viruses. 2022; 14(7):1400. https://doi.org/10.3390/v14071400
Chicago/Turabian StyleWoodbury, Brianna M., Tina Motwani, Makayla N. Leroux, Lauren F. Barnes, Nicholas A. Lyktey, Sanchari Banerjee, Corynne L. Dedeo, Martin F. Jarrold, and Carolyn M. Teschke. 2022. "Tryptophan Residues Are Critical for Portal Protein Assembly and Incorporation in Bacteriophage P22" Viruses 14, no. 7: 1400. https://doi.org/10.3390/v14071400