
Characterization of Hantavirus N Protein Intracellular Dynamics and Localization

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Fluorescently Labelled Hantavirus N Protein
2.2. Cell Culture and Transfection
2.3. Immunofluorescence Staining
2.4. Fluorescence Microscopy
2.5. Image Analysis
2.6. Number and Brightness Analysis
2.7. Statistical Test
3. Results
3.1. Generation and Characterization of Fluorescently Tagged N Protein Constructs
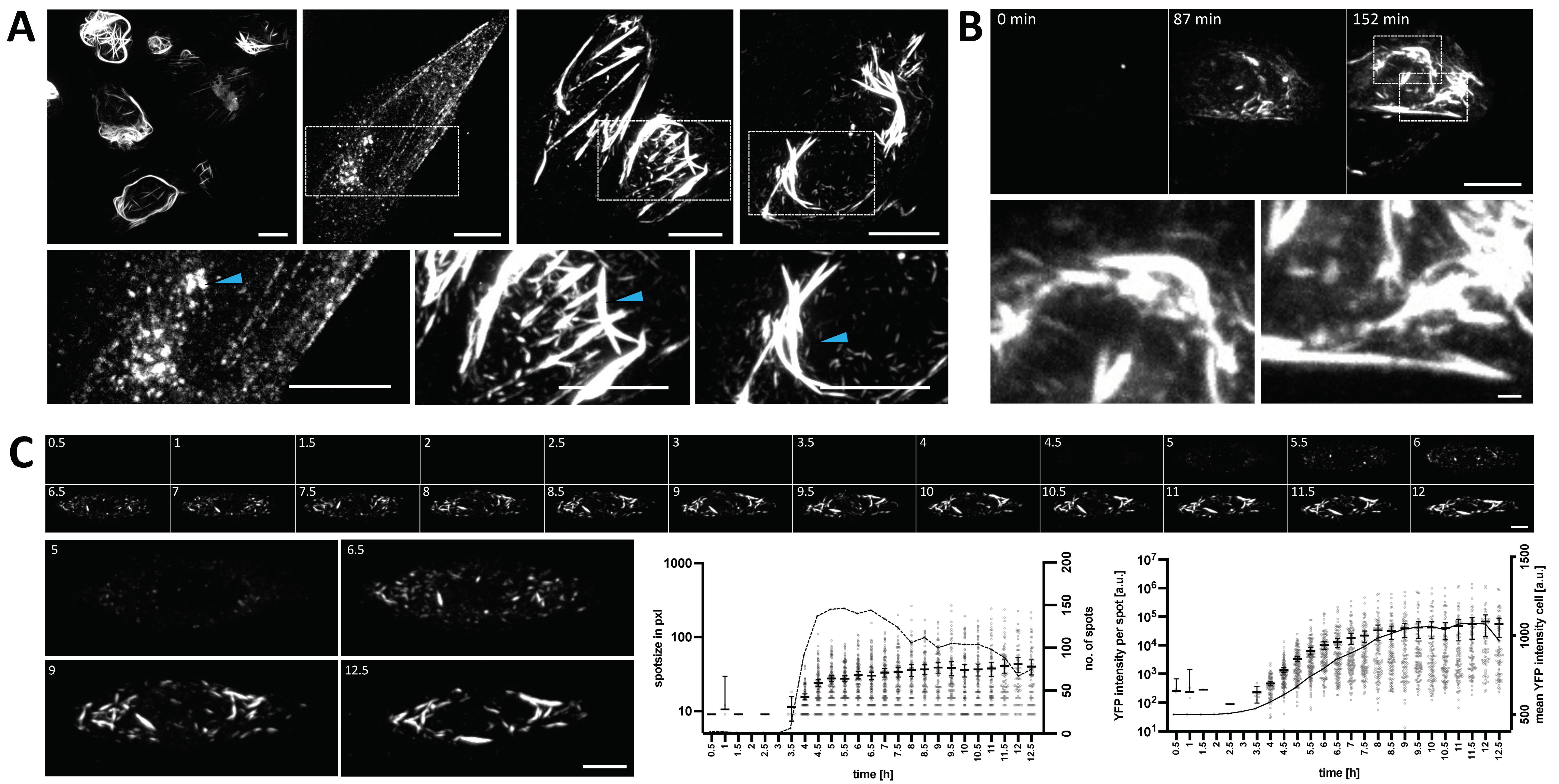
3.2. Dynamics of YFP-N Aggregate Formation
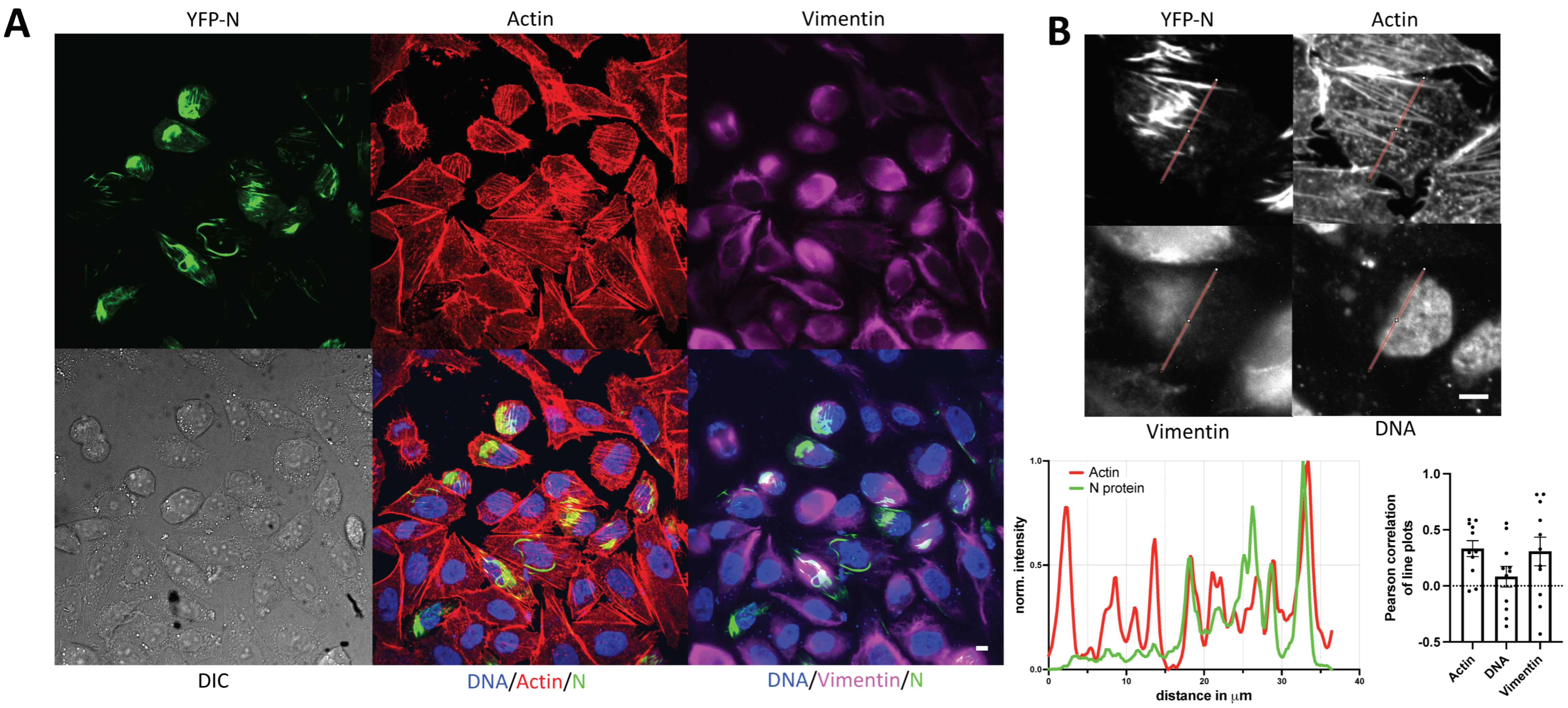
3.3. YFP-N Co-Localizes with Vimentin and Actin Fibers
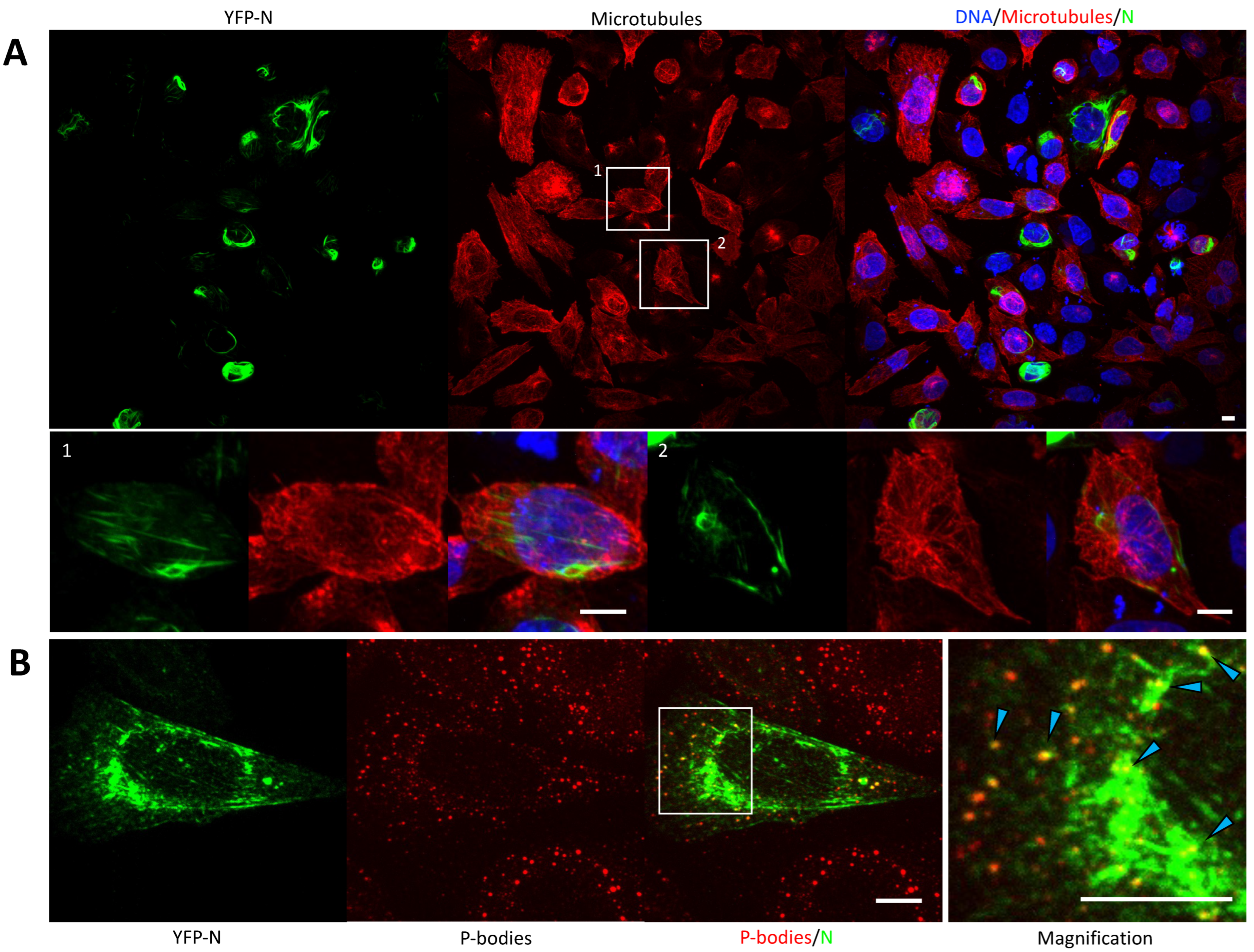
3.4. YFP-N Does Not Co-Localize with Microtubules
3.5. YFP-N Puncta Heavily Associated with P-Bodies
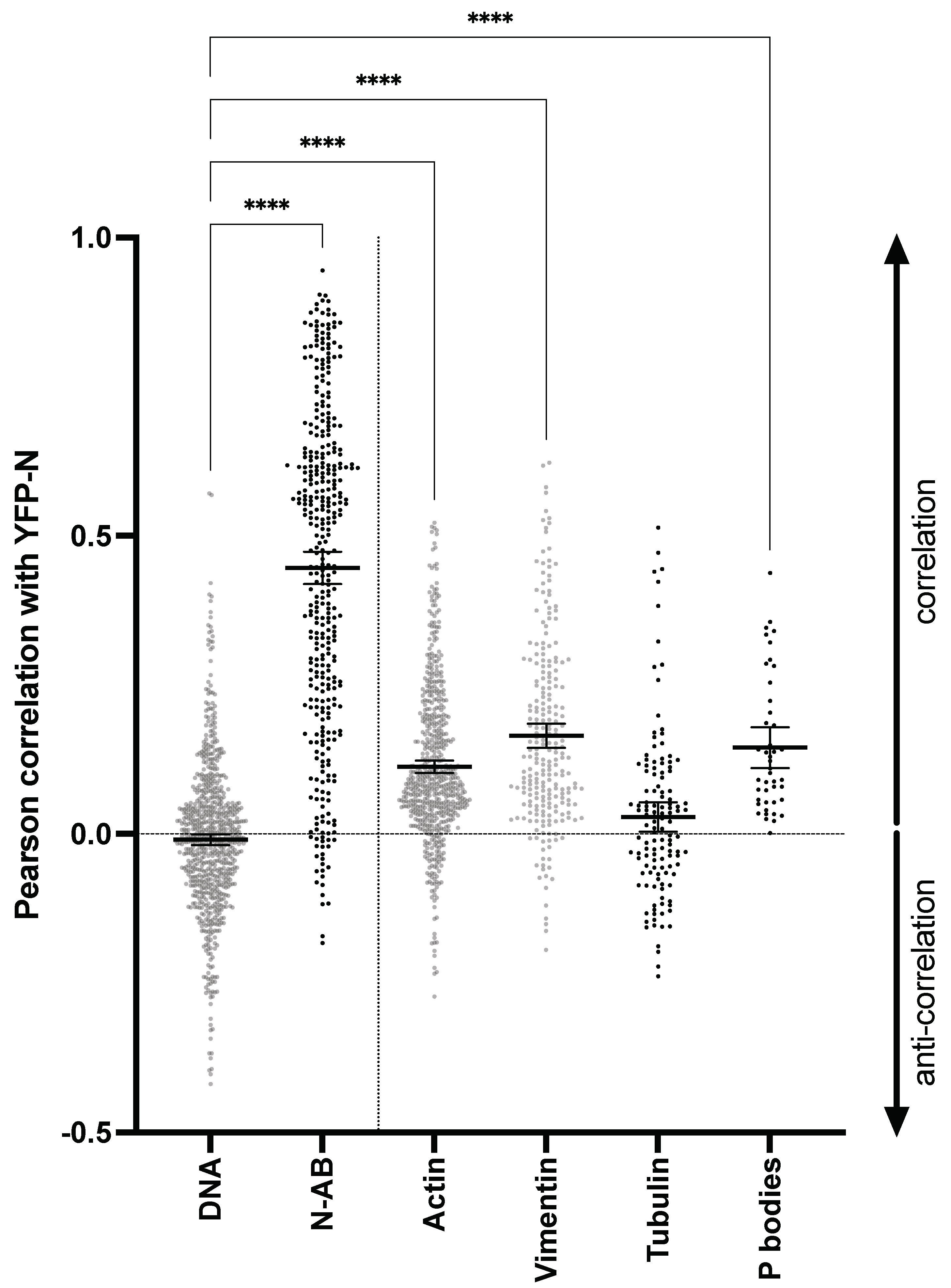
3.6. Unsupervised, Quantitative Image Analysis Corroborates YFP-N Co-Localization with PBs, Actin, and Vimentin
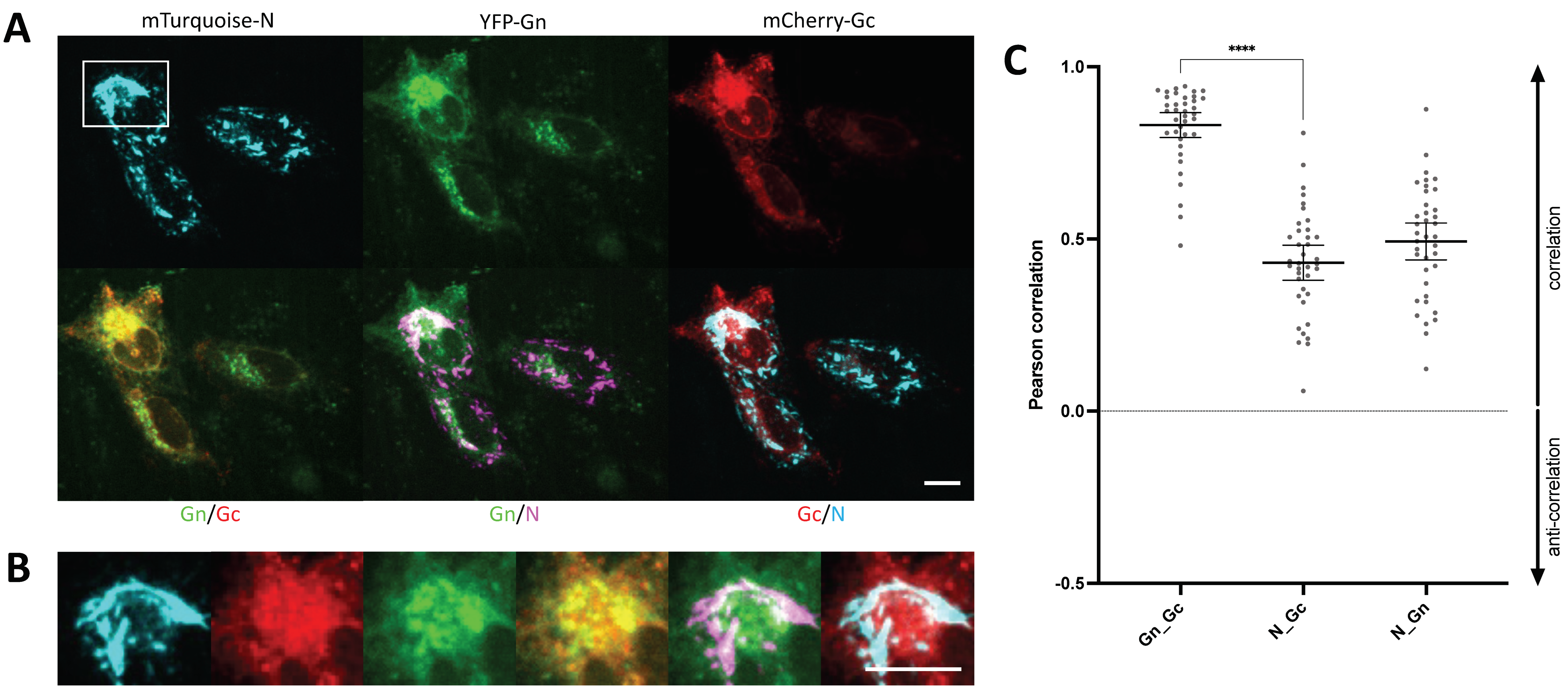
3.7. YFP-N Co-Localizes with Gc and Gn
3.8. N protein Multimerization Is Dose-Dependent and Independent of Hantavirus Glycoproteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonsson, C.B.; Figueiredo, L.T.M.; Vapalahti, O. A Global Perspective on Hantavirus Ecology, Epidemiology, and Disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, D.H.; Figueiredo, L.T.M.; Song, J.-W.; Klempa, B. Hantaviruses—Globally Emerging Pathogens. J. Clin. Virol. 2015, 64, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulou, C.F. Molecular Biology of Hantavirus Infection. In Bunyaviridae: Molecular and Cellular Biology; Plyusnin, A., Elliott, R., Eds.; Caister Academic Press: Poole, UK, 2011; pp. 41–60. ISBN 978-1-904455-90-5. [Google Scholar]
- Mackow, E.R.; Gavrilovskaya, I.N.I. Hantavirus Regulation of Endothelial Cell Functions. Thromb. Haemost. 2009, 102, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Bauherr, S.; Larsberg, F.; Petrich, A.; Sperber, H.S.; Klose-Grzelka, V.; Luckner, M.; Azab, W.; Schade, M.; Höfer, C.T.; Lehmann, M.J.; et al. Macropinocytosis and Clathrin-Dependent Endocytosis Play Pivotal Roles for the Infectious Entry of Puumala Virus. J. Virol. 2020, 94, e00184-20. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the Mysteries of Hantavirus Infections. Nat. Rev. Genet. 2013, 11, 539–550. [Google Scholar] [CrossRef]
- Torriani, G.; Mayor, J.; Zimmer, G.; Kunz, S.; Rothenberger, S.; Engler, O. Macropinocytosis Contributes to Hantavirus Entry into Human Airway Epithelial Cells. Virology 2019, 531, 57–68. [Google Scholar] [CrossRef]
- Mir, M.A.; Duran, W.A.; Hjelle, B.L.; Ye, C.; Panganiban, A.T. Storage of Cellular 5′ mRNA Caps in P Bodies for Viral Cap-snatching. Proc. Natl. Acad. Sci. USA 2008, 105, 19294–19299. [Google Scholar] [CrossRef] [Green Version]
- Ravkov, E.V.; Nichol, S.T.; Peters, C.J.; Compans, R.W. Role of Actin Microfilaments in Black Creek Canal Virus Morphogenesis. J. Virol. 1998, 72, 2865–2870. [Google Scholar] [CrossRef] [Green Version]
- Plyusnin, A.; Elliott, R. Bunyaviridae: Molecular and Cellular Biology; Caister Academic Press: Poole, UK, 2011; ISBN 978-1-912530-52-6. [Google Scholar]
- Reuter, M.; Krüger, D.H. The Nucleocapsid Protein of Hantaviruses: Much More than a Genome-Wrapping Protein. Virus Genes 2017, 54, 5–16. [Google Scholar] [CrossRef]
- Hepojoki, J.; Strandin, T.; Wang, H.; Vapalahti, O.; Vaheri, A.; Lankinen, H. Cytoplasmic Tails of Hantavirus Glycoproteins Interact with the Nucleocapsid Protein. J. Gen. Virol. 2010, 91, 2341–2350. [Google Scholar] [CrossRef]
- Strandin, T.; Hepojoki, J.; Wang, H.; Vaheri, A.; Lankinen, H. The Cytoplasmic Tail of Hantavirus Gn Glycoprotein Interacts with RNA. Virology 2011, 418, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.; Suszko, J.W.; Pekosz, A. Roles for the Recycling Endosome, Rab8, and Rab11 in Hantavirus Release from Epithelial Cells. Virology 2008, 382, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, H.N.; Jonsson, C. New and Old World Hantaviruses Differentially Utilize Host Cytoskeletal Components during Their Life Cycles. Virology 2008, 374, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Hägele, S.; Müller, A.; Nusshag, C.; Reiser, J.; Zeier, M.; Krautkrämer, E. Virus- and Cell Type-Specific Effects in Orthohantavirus Infection. Virus Res. 2018, 260, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Binder, F.; Lenk, M.; Weber, S.; Stoek, F.; Dill, V.; Reiche, S.; Riebe, R.; Wernike, K.; Hoffmann, D.; Ziegler, U.; et al. Common Vole (Microtus Arvalis) and Bank Vole (Myodes Glareolus) Derived Permanent Cell Lines Differ in Their Susceptibility and Replication Kinetics of Animal and Zoonotic Viruses. J. Virol. Methods 2019, 274, 113729. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image Analysis Software for Identifying and Quantifying Cell Phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Petazzi, R.A.; Koikkarah, A.A.; Tischler, N.D.; Chiantia, S. Detection of Envelope Glycoprotein Assembly from Old World Hantaviruses in the Golgi Apparatus of Living Cells. J. Virol. 2021, 95, e01238-20. [Google Scholar] [CrossRef]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the Number of Molecules and Brightness in the Laser Scanning Microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [Green Version]
- Sperber, H.S.; Welke, R.-W.; Petazzi, R.A.; Bergmann, R.; Schade, M.; Shai, Y.; Chiantia, S.; Herrmann, A.; Schwarzer, R. Self-Association and Subcellular Localization of Puumala Hantavirus Envelope Proteins. Sci. Rep. 2019, 9, 707. [Google Scholar] [CrossRef]
- Mayer, M.C.; Schauenburg, L.; Thompson-Steckel, G.; Dunsing, V.; Kaden, D.; Voigt, P.; Schaefer, M.; Chiantia, S.; Kennedy, T.E.; Multhaup, G. Amyloid Precursor-Like Protein 1 (APLP1) Exhibits Stronger Zinc-Dependent Neuronal Adhesion than Amyloid Precursor Protein and APLP2. J. Neurochem. 2016, 137, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Dunsing, V.; Chiantia, S. A Fluorescence Fluctuation Spectroscopy Assay of Protein-Protein Interactions at Cell-Cell Contacts. J. Vis. Exp. 2018, 142, e58582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Wang, W.; Sun, Y.; Ma, C.; Wang, X.; Wang, X.; Liu, P.; Shen, S.; Li, B.; Lin, J.; et al. Crystal Structure of the Core Region of Hantavirus Nucleocapsid Protein Reveals the Mechanism for Ribonucleoprotein Complex Formation. J. Virol. 2016, 90, 1048–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olal, D.; Daumke, O. Structure of the Hantavirus Nucleoprotein Provides Insights into the Mechanism of RNA Encapsidation. Cell Rep. 2016, 14, 2092–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; Love, Z.; Arvidson, B.; Seeds, J.; Willey, J.; Barklis, E. Hantavirus Nucleocapsid Protein Oligomerization. J. Virol. 2001, 75, 2019–2023. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Steel, E.; Finlay, L.; Bächinger, H.P.; Barklis, E. Hantavirus Nucleocapsid Protein Coiled-Coil Domains. J. Biol. Chem. 2002, 277, 27103–27108. [Google Scholar] [CrossRef] [Green Version]
- Kaukinen, P.; Koistinen, V.; Vapalahti, O.; Vaheri, A.; Plyusnin, A. Interaction between Molecules of Hantavirus Nucleocapsid Protein. J. Gen. Virol. 2001, 82, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Safronetz, D.; Ebihara, H.; Feldmann, H.; Hooper, J.W. The Syrian Hamster Model of Hantavirus Pulmonary Syndrome. Antivir. Res. 2012, 95, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, K.; Scott, D.; Safronetz, D.; Brining, D.L.; Ebihara, H.; Feldmann, H.; LaCasse, R.A. Laguna Negra Virus Infection Causes Hantavirus Pulmonary Syndrome in Turkish Hamsters (Mesocricetus Brandti). Vet. Pathol. 2015, 53, 182–189. [Google Scholar] [CrossRef]
- Chen, Y.; Johnson, J.; Macdonald, P.; Wu, B.; Mueller, J.D. Observing Protein Interactions and Their Stoichiometry in Living Cells by Brightness Analysis of Fluorescence Fluctuation Experiments. Methods Enzymol. 2010, 472, 345–363. [Google Scholar] [CrossRef]
- Gallo, G.; Caignard, G.; Badonnel, K.; Chevreux, G.; Terrier, S.; Szemiel, A.; Roman-Sosa, G.; Binder, F.; Gu, Q.; Filipe, A.D.S.; et al. Interactions of Viral Proteins from Pathogenic and Low or Non-Pathogenic Orthohantaviruses with Human Type I Interferon Signaling. Viruses 2021, 13, 140. [Google Scholar] [CrossRef]
- Davies, K.A.; Chadwick, B.; Hewson, R.; Fontana, J.; Mankouri, J.; Barr, J.N. The RNA Replication Site of Tula Orthohantavirus Resides within a Remodelled Golgi Network. Cells 2020, 9, 1569. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Kramer, S.; Herrlinger, K.R.; Jeske, K.; Kuhns, M.; Weiss, S.; Ulrich, R.G.; Krüger, D.H. Tula Virus as Causative Agent of Hantavirus Disease in Immunocompetent Person, Germany. Emerg. Infect. Dis. 2021, 27, 1234–1237. [Google Scholar] [CrossRef]
- Ravkov, E.V.; Compans, R.W. Hantavirus Nucleocapsid Protein Is Expressed as a Membrane-Associated Protein in the Perinuclear Region. J. Virol. 2001, 75, 1808–1815. [Google Scholar] [CrossRef] [Green Version]
- Kallio-Kokko, H.; Leveelahti, R.; Brummer-Korvenkontio, M.; Vaheri, A.; Vapalahti, O. Human Immune Response to Puumala Virus Glycoproteins and Nucleocapsid Protein Expressed in Mammalian Cells. J. Med. Virol. 2001, 65, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Ogino, M.; Yoshimatsu, K.; Ebihara, H.; Lee, B.; Okumura, M.; Arikawa, J.; Araki, K. Cell Fusion Activities of Hantaan Virus Envelope Glycoproteins Cell Fusion Activities of Hantaan Virus Envelope Glycoproteins. J. Virol. 2004, 78, 10776–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiropoulou, C.; Goldsmith, C.; Shoemaker, T.; Peters, C.; Compans, R. Sin Nombre Virus Glycoprotein Trafficking. Virology 2003, 308, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Hepojoki, J.; Strandin, T.; Lankinen, H.; Vaheri, A. Hantavirus Structure–Molecular Interactions behind the Scene. J. Gen. Virol. 2012, 93, 1631–1644. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, H.N.; Chung, D.-H.; Plane, S.J.; Sztul, E.; Chu, Y.-K.; Guttieri, M.C.; McDowell, M.; Ali, G.; Jonsson, C. Dynein-Dependent Transport of the Hantaan Virus Nucleocapsid Protein to the Endoplasmic Reticulum-Golgi Intermediate Compartment. J. Virol. 2007, 81, 8634–8647. [Google Scholar] [CrossRef] [Green Version]
- Cheng, E.; Mir, M.A. Signatures of Host mRNA 5′ Terminus for Efficient Hantavirus Cap Snatching. J. Virol. 2012, 86, 10173–10185. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Manufacturer |
| Hantavirus (Puumala) N protein antibody | Fitzgerald, UK (cat. 10R-2502) |
| Anti-Vimentin antibody | Abcam, Cambridge, UK (cat. ab45939) |
| Anti-Dcp1a antibody | Abcam, Cambridge, UK (cat. ab57654) |
| Anti-tubulin (clone B512) | Sigma-Aldrich, Munich, Germany (cat. T5168) |
| Fluorescently conjugated phalloidin | Thermo Fisher Scientific, Waltham, MA, USA (cat. R415) |
| Secondary Antibodies | Manufacturer |
| AlexaFluor 488 goat anti rabbit IgG (H + L) | Invitrogen, Carlsbad, CA, USA (cat. A-11008) |
| AlexaFluor 488 goat anti mouse IgG (H + L) | Abcam, Cambridge, UK (cat. ab150117) |
| AlexaFluor 568 goat anti mouse IgG (H + L) | Invitrogen, Carlsbad, CA, USA (cat. A-11004) |
| Alexa fluor 594 goat anti rabbit IgG (H + L) | Invitrogen, Carlsbad, CA, USA (cat. A-11012) |
| Alexa fluor 647 goat anti mouse IgG (H + L) | Invitrogen, Carlsbad, CA, USA (cat. A-21235) |
| Alexa fluor 647 goat anti rabbit IgG (H + L) | Abcam, Cambridge, UK (cat. ab150087) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welke, R.-W.; Sperber, H.S.; Bergmann, R.; Koikkarah, A.; Menke, L.; Sieben, C.; Krüger, D.H.; Chiantia, S.; Herrmann, A.; Schwarzer, R. Characterization of Hantavirus N Protein Intracellular Dynamics and Localization. Viruses 2022, 14, 457. https://doi.org/10.3390/v14030457
Welke R-W, Sperber HS, Bergmann R, Koikkarah A, Menke L, Sieben C, Krüger DH, Chiantia S, Herrmann A, Schwarzer R. Characterization of Hantavirus N Protein Intracellular Dynamics and Localization. Viruses. 2022; 14(3):457. https://doi.org/10.3390/v14030457
Chicago/Turabian StyleWelke, Robert-William, Hannah Sabeth Sperber, Ronny Bergmann, Amit Koikkarah, Laura Menke, Christian Sieben, Detlev H. Krüger, Salvatore Chiantia, Andreas Herrmann, and Roland Schwarzer. 2022. "Characterization of Hantavirus N Protein Intracellular Dynamics and Localization" Viruses 14, no. 3: 457. https://doi.org/10.3390/v14030457