Expression, Purification and Refolding of a Human NaV1.7 Voltage Sensing Domain with Native-like Toxin Binding Properties

 and
and
Abstract
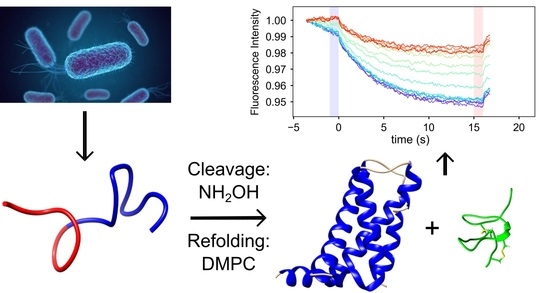
:
1. Introduction
2. Results
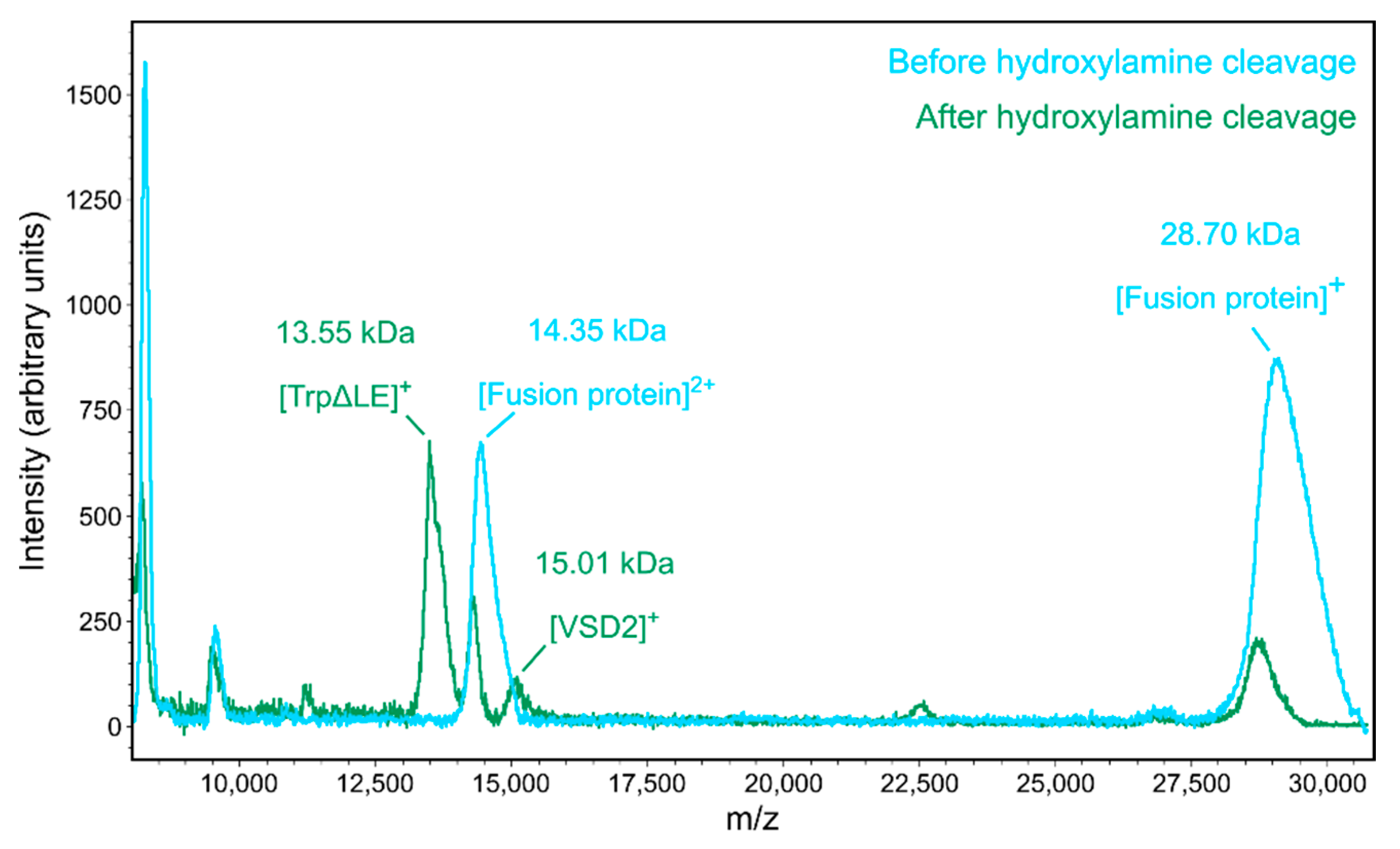
2.1. Expression and Purification of NaV1.7 VSD2
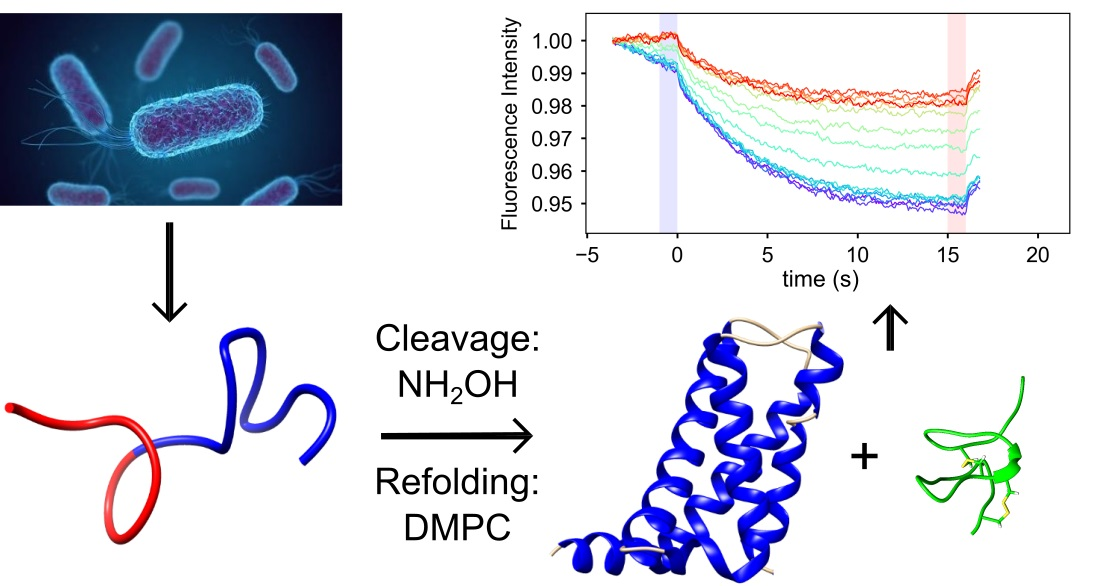
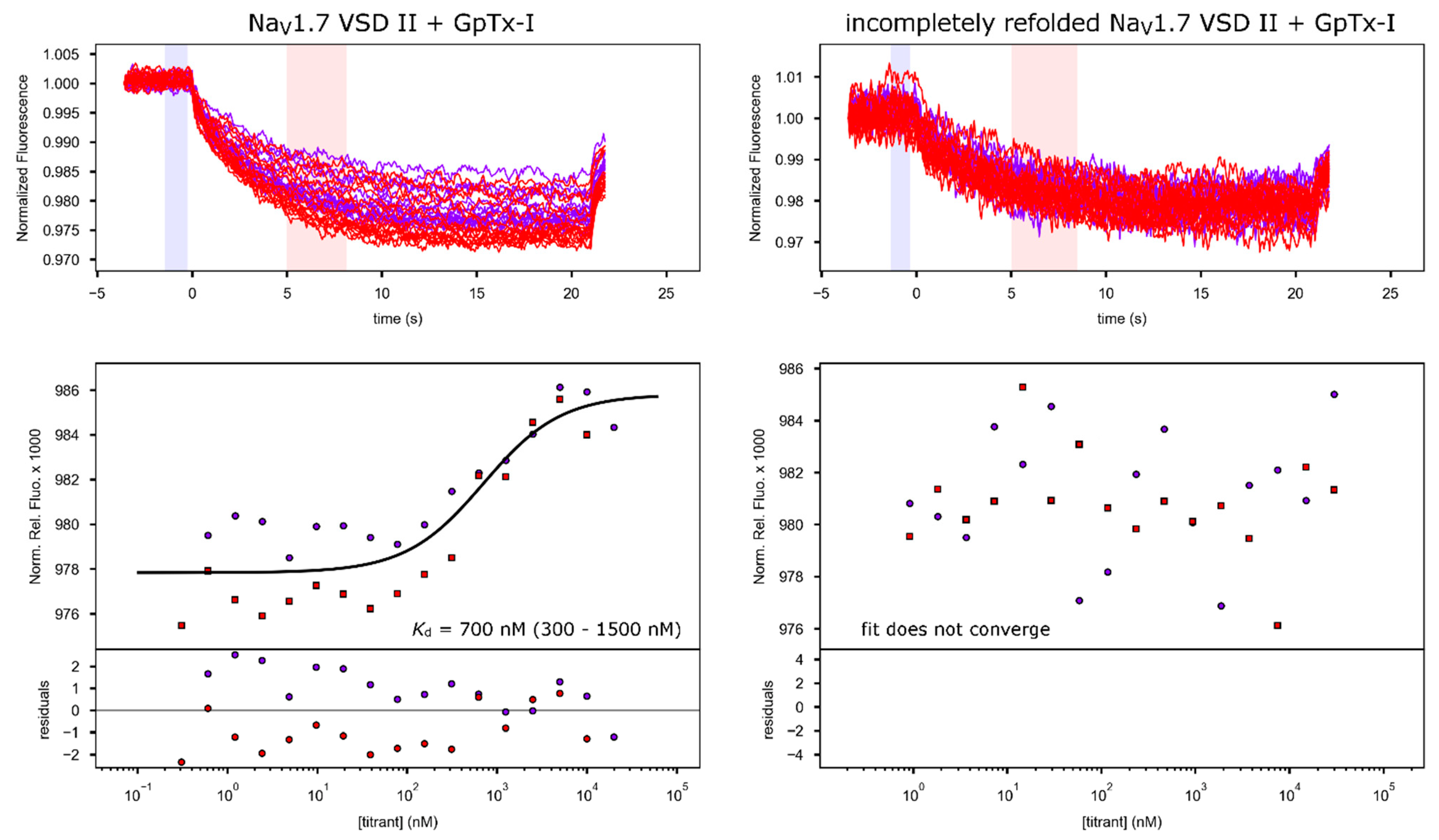
2.2. Toxin Binding Studies
3. Discussion
4. Materials and Methods
4.1. Cloning and Expression of VSD2
4.2. Cleavage and Reconstitution of the VSD
4.3. Mass Spectrometry
4.4. Circular Dichroism Spectroscopy
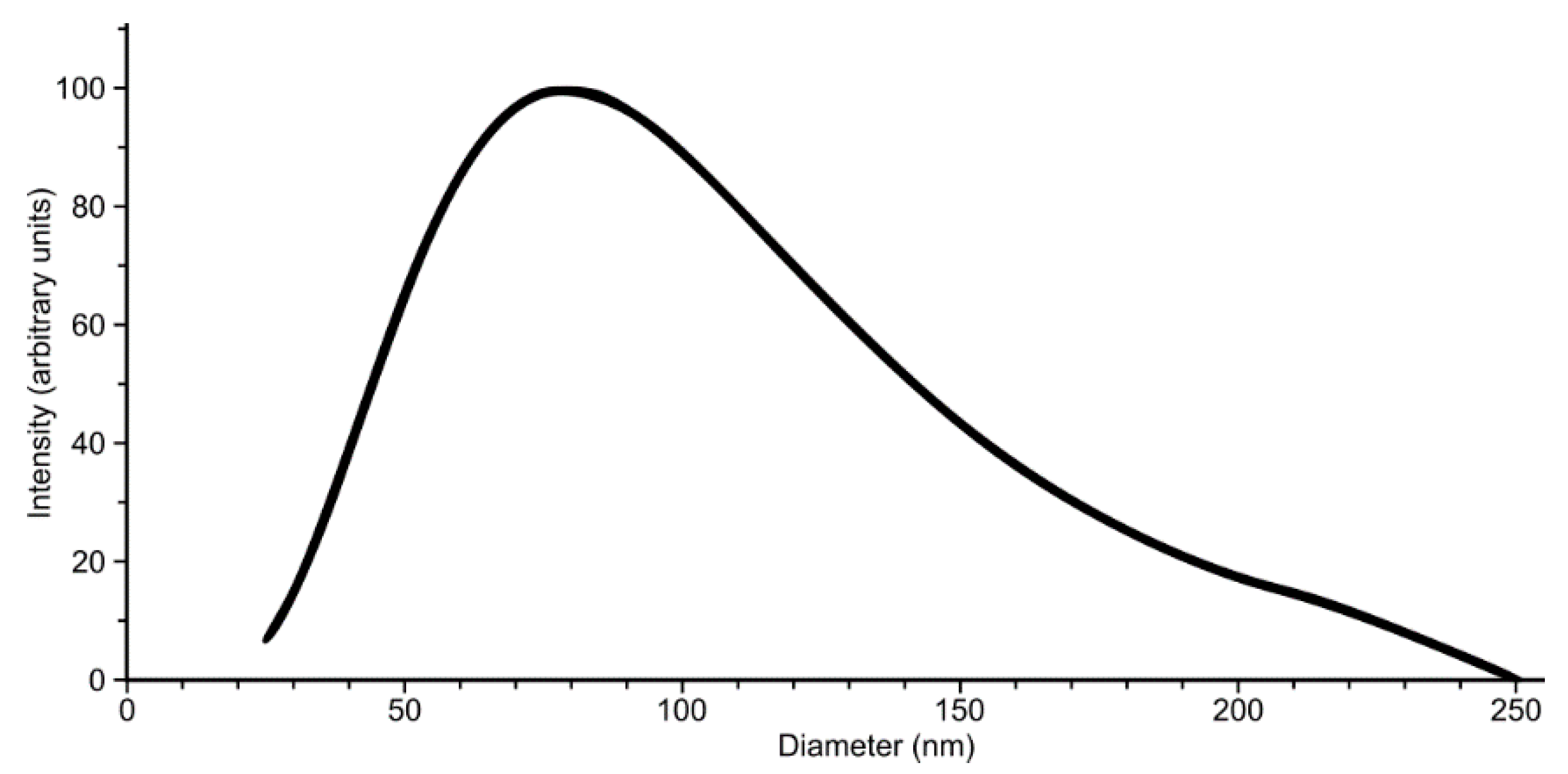
4.5. Dynamic Light Scattering
4.6. Recombinant Production of ProTx-II
4.7. FMOC Peptide Synthesis and Refolding of GpTx-I
4.8. Microscale Thermophoresis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clare, J.J.; Tate, S.N.; Nobbs, M.; Romanos, M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 2000, 5, 506–520. [Google Scholar] [CrossRef]
- Catterall, W.A. From Ionic Currents to Molecular Mechanisms. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Carlin, K.P.; Wu, G.; Ilyin, V.I.; Musza, L.L.; Blake, P.R.; Kyle, D.J. Studies Examining the Relationship between the Chemical Structure of Protoxin II and Its Activity on Voltage Gated Sodium Channels. J. Med. Chem. 2014, 57, 6623–6631. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, T.; Rohou, A.; Arthur, C.P.; Tzakoniati, F.; Wong, E.; Estevez, A.; Kugel, C.; Franke, Y.; Chen, J.; et al. Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 2019, 176, 702–715.e14. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering Potent and Selective Analogues of GpTx-1, a Tarantula Venom Peptide Antagonist of the NaV1.7 Sodium Channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef]
- Deuis, J.R.; Wingerd, J.S.; Winter, Z.; Durek, T.; Dekan, Z.; Sousa, S.R.; Zimmermann, K.; Hoffmann, T.; Weidner, C.; Nassar, M.A.; et al. Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins 2016, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Devaraneni, P.K.; Devereaux, J.J.; Valiyaveetil, F.I. In Vitro Folding of KvAP, a Voltage-Gated K+ Channel. Biochemistry 2011, 50, 10442–10450. [Google Scholar] [CrossRef] [Green Version]
- Myshkin, M.Y.; Männikkö, R.; Krumkacheva, O.A.; Kulbatskii, D.S.; Chugunov, A.O.; Berkut, A.A.; Paramonov, A.S.; Shulepko, M.A.; Fedin, M.; Hanna, M.G.; et al. Cell-Free Expression of Sodium Channel Domains for Pharmacology Studies. Noncanonical Spider Toxin Binding Site in the Second Voltage-Sensing Domain of Human Nav1.4 Channel. Front. Pharmacol. 2019, 10, 953. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.P.; Kim, P.S. Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 1994, 3, 1822–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naider, F.; Estephan, R.; Englander, J.; Babu, V.V.S.; Arevalo, E.; Samples, K.; Becker, J.M. Sexual conjugation in yeast: A paradigm to study G-protein-coupled receptor domain structure. Biopolymers 2004, 76, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, P.; Balian, G. The specific nonenzymatic cleavage of bovine ribonuclease with hydroxylamine. J. Biol. Chem. 1970, 245, 4854–4856. [Google Scholar] [CrossRef]
- Fujii, T.; Ohkuri, T.; Onodera, R.; Ueda, T. Stable Supply of Large Amounts of Human Fab from the Inclusion Bodies in E. coli. J. Biochem. 2007, 141, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Szoka, F.; Papahadjopoulos, D. Comparative Properties and Methods of Preparation of Lipid Vesicles (Liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Cummins, T.R.; Alphy, S.; Blumenthal, K.M. Molecular interactions of the gating modifier toxin ProTx-II with Nav1.5: Implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 2007, 282, 12687–12697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuermann, T.H.; Padrick, S.; Gardner, K.; Brautigam, C.A. On the acquisition and analysis of microscale thermophoresis data. Anal. Biochem. 2016, 496, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. ProTx-II, a Selective Inhibitor of NaV1.7 Sodium Channels, Blocks Action Potential Propagation in Nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Barbet, J.; Huclier-Markai, S. Equilibrium, affinity, dissociation constants, IC5O: Facts and fantasies. Pharm. Stat. 2019, 18, 513–525. [Google Scholar] [CrossRef]
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar] [CrossRef]
- Agwa, A.J.; Peigneur, S.; Chow, C.Y.; Lawrence, N.; Craik, D.J.; Tytgat, J.; King, G.F.; Henriques, S.T.; Schroeder, C.I. Gating modifier toxins isolated from spider venom: Modulation of voltage-gated sodium channels and the role of lipid membranes. J. Biol. Chem. 2018, 293, 9041–9052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruta, V.; MacKinnon, R. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry 2004, 43, 10071–10079. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; King, G.F.; Mobli, M. Molecular basis of the interaction between gating modifier spider toxins and the voltage sensor of voltage-gated ion channels. Sci. Rep. 2016, 6, srep34333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, S.; Kimura, T.; Nozaki, T.; Harada, H.; Shimada, I.; Osawa, M. Structural basis for the inhibition of volt-age-dependent K+ channel by gating modifier toxin. Sci. Rep. 2015, 5, 14226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Männikkö, R.; Shenkarev, Z.O.; Thor, M.G.; Berkut, A.A.; Myshkin, M.Y.; Paramonov, A.S.; Kulbatskii, D.S.; Kuzmin, D.A.; Castañeda, M.S.; King, L.; et al. Spider toxin inhibits gating pore currents underlying periodic paralysis. Proc. Natl. Acad. Sci. USA 2018, 115, 4495–4500. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.; Sindhikara, D.; DiMattia, M.; Leffler, A.E. Potency-Enhancing Mutations of Gating Modifier Toxins for the Voltage-Gated Sodium Channel NaV1.7 Can be Predicted Using Accurate Free-Energy Calculations. Toxins 2021, 13, 193. [Google Scholar] [CrossRef]
- Napolitano, L.M.R.; Torre, V.; Marchesi, A. CNG channel structure, function, and gating: A tale of conformational flexibility. Pflug. Arch.-Eur. J. Physiol. 2021, 473, 1423–1435. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J. Over-production of Proteins inEscherichia coli: Mutant Hosts that Allow Synthesis of some Membrane Proteins and Globular Proteins at High Levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Cadene, M.; Chait, B.T. A Robust, Detergent-Friendly Method for Mass Spectrometric Analysis of Integral Membrane Proteins. Anal. Chem. 2000, 72, 5655–5658. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Blumenthal, K.; Jackson, J.O.; Liang, S.; Cummins, T.R. The Tarantula Toxins ProTx-II and Huwentoxin-IV Differentially Interact with Human Nav1.7 Voltage Sensors to Inhibit Channel Activation and Inactivation. Mol. Pharmacol. 2010, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Kd (nM) | Conf. Int. (nM) | Repeats | FB * (‰) | FAB * (‰) | rmsd (‰) |
|---|---|---|---|---|---|---|
| NaV1.7 VSD2 in DMPC + ProTx-II | 200 | 160–250 | 2 | 956.6 | 981.9 | 1.283 |
| DMPC control + ProTx-II | 50,000 * | 10,000–∞ | 2 | 974.5 | 984 | 1.318 |
| NaV1.7 VSD2 in DMPC + GpTx-I | 700 | 300–1500 | 2 | 977.8 | 985.8 | 1.369 |
| Incompletely refolded NaV1.7 VSD II + GpTx-I | n/a | n/a | 2 | n/a | n/a | n/a |
| F813A NaV1.7 VSD2 in DMPC + GpTx-I | 1300 | 600–3000 | 1 | 986.1 | 988.5 | 0.225 |
| D816A NaV1.7 VSD2 in DMPC + GpTx-I | 5000 | 3000–15,000 | 1 | 981.7 | 993 | 0.636 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroder, R.V.; Cohen, L.S.; Wang, P.; Arizala, J.D.; Poget, S.F. Expression, Purification and Refolding of a Human NaV1.7 Voltage Sensing Domain with Native-like Toxin Binding Properties. Toxins 2021, 13, 722. https://doi.org/10.3390/toxins13100722
Schroder RV, Cohen LS, Wang P, Arizala JD, Poget SF. Expression, Purification and Refolding of a Human NaV1.7 Voltage Sensing Domain with Native-like Toxin Binding Properties. Toxins. 2021; 13(10):722. https://doi.org/10.3390/toxins13100722
Chicago/Turabian StyleSchroder, Ryan V., Leah S. Cohen, Ping Wang, Joekeem D. Arizala, and Sébastien F. Poget. 2021. "Expression, Purification and Refolding of a Human NaV1.7 Voltage Sensing Domain with Native-like Toxin Binding Properties" Toxins 13, no. 10: 722. https://doi.org/10.3390/toxins13100722