Proteomic Methods of Detection and Quantification of Protein Toxins


1
Faculty of Military Health Sciences, University of Defense in Brno, Třebešská 1575, CZ-500 01 Hradec Králové, Czech Republic
2
Military Health Institute, Military Medical Agency, Tychonova 1, CZ-160 00 Prague 6, Czech Republic
*
Author to whom correspondence should be addressed.
Toxins 2018, 10(3), 99; https://doi.org/10.3390/toxins10030099
Submission received: 12 February 2018
/
Revised: 21 February 2018
/
Accepted: 23 February 2018
/
Published: 28 February 2018
Abstract
:Biological toxins are a heterogeneous group of compounds that share commonalities with biological and chemical agents. Among them, protein toxins represent a considerable, diverse set. They cover a broad range of molecular weights from less than 1000 Da to more than 150 kDa. This review aims to compare conventional detection methods of protein toxins such as in vitro bioassays with proteomic methods, including immunoassays and mass spectrometry-based techniques and their combination. Special emphasis is given to toxins falling into a group of selected agents, according to the Centers for Disease Control and Prevention, such as Staphylococcal enterotoxins, Bacillus anthracis toxins, Clostridium botulinum toxins, Clostridium perfringens epsilon toxin, ricin from Ricinus communis, Abrin from Abrus precatorius or control of trade in dual-use items in the European Union, including lesser known protein toxins such as Viscumin from Viscum album. The analysis of protein toxins and monitoring for biological threats, i.e., the deliberate spread of infectious microorganisms or toxins through water, food, or the air, requires rapid and reliable methods for the early identification of these agents.
Key Contribution: Protein toxins are considered as potential warfare and bio-terrorism agents and represent thus an important group from the analytical point of view. The review summarizes methods of detection of protein toxins with special regards to mass spectrometry-based techniques.
1. Introduction
Bacterial and plant protein toxins are among the most powerful poisons known. Protein toxins, especially those of bacterial origin, have harmful effects and often are considered as potential agents used for bio-terrorism and warfare [1]. Therefore, they are relevant in the health and food sector and in the security area. The threat posed by bioterrorism is still a serious concern, as the consequences of a large-scale biological attack would be devastating, causing significant social and economic problems, while being potentially available at relatively modest cost without the need for specific sophisticated technology [2]. According to their above-mentioned properties, biological toxins and their organisms of origin are classified as category A, B, or C priority pathogens depending on how easily they can be spread and the severity of illness they cause [3]. Similar lists exist for control of trade in dual-use items in the European Union [4].
Protein toxins are generally highly toxic to eukaryotic cells and operate via various mechanisms. Many bind to specific receptors on the membrane, most often glycoproteins or gangliosides, and penetrate the cell to achieve their intracellular target [5]. Likely because of this, many protein toxins require two or more subunits to achieve full biological activity, with one subunit binding to the receptor and the second having enzymatic activity (e.g., plant toxins: Ricin from Ricinus communis, abrin from Abrus precatorius; and bacterial toxins: iota toxin from Clostridium perfringens, Shiga toxin produced by Shigella dysenteriae, Shiga-like toxins produced by Escherichia coli, diphtheria toxin, Pseudomonas exotoxin A, and cholera toxin) [6]. Pore-forming toxins are the largest class of bacterial toxins (e.g., epsilon toxin from Clostridium perfringens, toxins from Bacillus anthracis) and belong to an ancient protein family also found in non-pathogenic bacteria [7].
Biological threats are difficult to predict and require specific preventive precautions. The response to bioterrorism attacks requires rapid methods that can quickly detect the biological agents used, enabling effective treatment and decontamination. As these agents are effective in very low quantities, detection methods must exhibit both a high degree of sensitivity and a high degree of selectivity to discriminate them from other biological and non-biological material (interference and contamination). One major complication associated with their detection is the complexity and diversity of the samples to be analyzed. In some instances, these agents can be violently delivered by contaminated matrices such as water, food and aerosol or distributed through the postal system (e.g., the anthrax incident of 2001 in the USA, in which ricin was delivered to U.S. government offices in 2004 and, more recently, to the U.S, president and other officials on 16 April 2013) [8]. To date, protein toxins have been identified using various conventional methods involving bioassays or analytical methods, including molecular biology techniques, such as nucleic acid-based assays, i.e., polymerase chain reaction (PCR), and/or immunological techniques, such as enzyme-linked immunosorbent assays (ELISA) or western blotting, for both protein toxins and their producers [9]. These methods are valuable for rapid preliminary screening but may have analytical limitations. Although immunoassays are very sensitive methods compared to mass spectrometry, they may suffer from a lack of specificity and therefore pose a risk of false-positives due to cross-reactions with similar molecules. However, PCR assays are rapid, sensitive and specific but cannot be used for classification of unknown samples or in cases where an isolated toxin is to be detected in the absence of its producer. When there is a higher demand for rapid analysis and a tendency to reach the lowest possible detection limit, these methods are not amenable to absolute protein identification and quantification. Unequivocal detection of potential sources of contamination and the protein toxin itself can be achieved using mass spectrometry (MS). MS is an analytical technique that can provide fast, sensitive and specific analysis in a single technique. MS-based analyses have become a powerful tool in relative/absolute and targeted/untargeted proteomics, also thanks to the continuous development of MS technologies (high resolution, accurate mass HR/AM instruments, hybrid configurations). They are suitable for various analytes, ranging from small organic molecules to large biomolecules and including bioterrorist agents [10]. In addition, MS is a preferred technique according to the Organisation for the Prohibition of Chemical Weapons [11].
2. Methods of Detection of Protein Toxins
Early methods for the detection of protein toxins are based on in vivo (e.g., mouse bioassay) or in vitro techniques (e.g., tissue culture). Later, immunological tests were developed together with advanced mass spectrometric (MS) detection, which can give results much faster and are simpler to perform than bioassays. Thus, proteomic methods have brought new views and approaches for the detection of protein-based toxins. These methods involve techniques based on antigen-antibody interactions and sophisticated mass spectrometric detection. Molecular biology methods, such as PCR and its modifications, play an important role in the detection of toxins via indirect methods (detection of DNA as a potential source of contamination). PCR brings more sophisticated approaches (compared to conventional techniques), is much faster and can be applied to detect nucleic acids in most types of environmental matrices [12]. However, interference originating from target-cell lysis—which is necessary for nucleic acid extraction, e.g., nucleic acid degradation and/or direct inhibition of PCR—causes false negative PCR results. Despite the benefits that indirect molecular biology methods provide, in cases of suspected protein toxin contamination in the absence of the producing bacterium, confirmation by direct detection of the toxin is necessary. For this review, PCR-based detection methods are referred to only marginally in chapters covering particular toxins.
2.1. Biological Assays
Bioassays and related methods are still the test of choice for the detection of many toxins. These assays require purification of the toxin prior to testing. To perform bioassays, it is necessary to use live animals or plants (in vivo) or tissue cultures and cells (in vitro) to determine the biological activity of toxins. Many bioassay formats have been described, including whole animal tests (e.g., the mouse lethality test, monkey and kitten emesis tests, and rabbit and guinea-pig skin tests), part animal tests (e.g., ileal loop tests) and cell culture systems (e.g., Chinese hamster ovary cells) [13]. Despite enormous progress in the development of alternative in vitro methods, for botulinum neurotoxin (BoNT) detection [14,15], the mouse lethality assay remains the only accepted standard test to confirm active BoNT [16]. The mouse lethality assay has been used for the analysis of complex sample matrices ranging from bacterial cultures to serum, fecal, gastric, or wound samples, but examples of matrix interference with the assay are known, particularly when other substances or toxins present in the sample cause lethality, and it has some other substantial drawbacks [17,18]. Depending on the nature of the toxins to be detected, detection takes from days to months. This factor is limiting in situations when rapid detection and identification is required to protect populations from hazardous biological agents. Nevertheless, the assay is generally considered highly sensitive and specific. The other significant benefit is that it detects functionally active toxin, unlike the majority of immunological methods, which do not provide information about toxin activity.
2.2. Proteomic Methods
From a methodological point of view, proteomic methods can be divided into two main categories: methods using antibodies (immunoassays) and advanced proteomic methods based on mass spectrometry.
2.2.1. Immunological Assays
Since their establishment in the late 1960s, immunological assays have become a powerful tool for the detection of proteins. Immunological assays are obviously simpler than biological assays and are therefore widely used [19]. Traditionally, the detection of proteins has been achieved using the enzyme-linked immune sorbent assay (ELISA) [20,21,22] and radioimmunoassay (RIA) [23,24]. Many modified ELISA methods exist [25]. The most expensive and demanding step is to find and purchase an appropriate antibody. However, recent progress in antibody engineering will further support and facilitate the integration of antibodies or antibody fragments into new assay formats [26]. In RIA, the radioactive label is coupled to the antigen that reacts with the specific antibody. The amount of antigen combined with the antibody is then determined using a radioactivity counter. Although the RIA technique is extremely sensitive and specific, the requirement for specialized equipment makes it one of the most expensive methods. The use of RIA for the detection of protein toxins is presented in refs. [27,28,29,30]. Western blotting is another fundamental technique used for the detection and quantification of proteins in complex biological mixtures [31]. Various toxins have been identified according to the western blot results [32,33,34].
Although immunoassay-based methods are sensitive and widely used to measure protein toxins in various matrices, there is a need for methods that can directly confirm the molecular identity of a toxin in situations where immunoassay tests produce positive results. The problem of possible false positivity increases significantly with matrix complexity and requires a confirmatory method. Because immunoassay-based methods require high-quality antibodies due to the high sequence and structural similarity among protein toxins, very few highly specific antibodies are available.
2.2.2. Mass Spectrometry-Based Methods
A wide range of MS-based methods for protein toxins detection is available [8,10,35]. These methods are highly advantageous in complex matrices, where they profit from combination with various separation techniques, such as one-dimensional and two-dimensional polyacrylamide gel electrophoresis and gel-free approaches based on liquid chromatography. The above-mentioned antibody-based techniques have great advantages when used to concentrate the analyte of interest and reduce the complexity of the matrix prior to MS analysis. In combination with MS detection techniques, they represent a powerful tool in the detection and quantification of protein toxins. See Figure 1 for a summary of MS and separation methods used in protein toxin analysis.
The MS methods comprise two different approaches. In data-dependent experiments, all peptides present in the mixture are fragmented, and their MS/MS spectra are searched against a relevant protein database. Nevertheless, this type of experiment does not give any information about the quantity, and the criteria for match relevance (number of peptides and their scores) are the researcher’s choice. Thus, this type of experiment often serves as a preliminary test for the presence of biological agents in the case of unknown samples, and the results must be further supported by targeted analysis. However, targeted (data independent) experiments are relevant for quantitative and confirmative cases. Here, the method targets previously selected peptides of interest and only scans and fragments these peptides. Targeted (data independent) MS/MS experiments are based on two principles, each taking advantage of a different type of mass spectrometer. First, selected reaction monitoring (SRM) [36] is performed on triple quadrupole (QqQ) mass spectrometers. In this experiment, the first quadrupole serves as a mass filter that targets a particular peptide and sends it further to the second quadrupole, which operates as a collision cell to fragment the peptide. The specific product ion of the fragmentation reaction is then filtered by the third quadrupole. The specific pair of m/z values associated with the precursor peptide and its selected fragment ion is referred to as a “transition” and is usually reported as the parent m/z > fragment m/z. Unlike the classical data-dependent MS experiment, full mass spectra are not recorded in a SRM analysis. Instead, the detector acts as a counting device for the chosen specific ions. Some of their fragments return an intensity value over time [36]. The principle of the second method, parallel reaction monitoring (PRM), is similar to that of SRM but is performed on hybrid quadrupole-Orbitrap mass spectrometers. The targeted precursor ion is filtered and fragmented as described above, but the whole MS/MS spectrum is recorded using the Orbitrap mass analyzer. This allows the quantification of tens to hundreds (and thousands in wide-screen analyses) of targeted proteins in the same run [37]. SRM- and PRM-based methods rely heavily on instrumentation as well as software capable of targeted proteomics method creation and quantitative data analysis. Open-source Skyline software (Version 4.1, the MacCoss Group at the University of Washington, Seattle, WA, USA, released on 11 January 2018) was explicitly developed to aid in this targeted assay development [38]. Quantification in SRM and PRM experiments is performed by extracting the product ion chromatograms and calculating the area under the curve. Absolute quantification of protein (also called AQUA) was described in the early 1980s [39]. The AQUA approach is based on addition of a known quantity of stable isotope-labeled standard peptides. The signal intensity of the target peptide in the experimental sample is compared to that of the heavy peptide and is then back-calculated to the initial concentration of the standard using a pre-determined standard curve to yield the absolute quantification of the target peptide. The development of a quantification method is completed by its validation, which comprises assessment of the detection and quantification limits [40,41]. These limits are further decisive in forensic cases when the presence of a particular toxin is to be reported. Absolute quantification for several protein toxins in various complex matrices has been described [42,43,44]. There is another important group of detection methods that focuses on protein toxins with intrinsic enzymatic activity (such as Bacillus anthracis and Clostridium botulinum toxins) [45]. These methods are based on the combination of biochemical tests with indirect MS detection of the enzymatic reaction products. Their use was first described in 2005 in botulinum neurotoxins [46] and are discussed below.
Compared to the methods using an antibody-antigen interaction (e.g., Western blotting and ELISA), SRM/PRM have several advantages. First is the quality of the assay, as quantification by western blotting is based on a single reagent (antibody), which may be poorly characterized. In contrast, SRM/PRM assays depend on isotopically labeled reference peptides, the quality of which can be easily verified by a fragment ion spectrum. There is also an economic advantage. Given the primary investment in a mass spectrometer and operating costs for the facility, it is still notably cheaper to develop SRM/PRM assays than to screen antibodies for each targeted protein. Finally, performance characteristics, such as limit of detection, linear dynamic range, ability to multiplex, and reproducibility, are also superior in MS-based methods.
3. Detection of Selected Protein Toxins
3.1. Staphylococcal Toxins
Staphylococcus is a genus of Gram-positive bacteria that includes approximately 40 species. Most are harmless and reside on the skin and mucous membranes of humans and other organisms. Nevertheless, some are important as human pathogens. Due to the diversity of this genus, Staphylococci cause a great variety of infections, including skin infections, pneumonia, food poisoning, toxic shock syndrome, and blood poisoning (bacteremia) [47]. The most important species from a toxicological point of view is Staphylococcus aureus.
Staphylococcus aureus is a Gram-positive, round, facultative anaerobe bacterium frequently found in the nose and the respiratory tract and on the skin. S. aureus is not always pathogenic but is a common cause of skin infections such as abscesses, respiratory disease and food poisoning. Pathogenic strains induce infections by producing various virulence factors such as potent protein toxins and expression of a cell-surface protein that binds and inactivates antibodies [48]. For the purpose of this review, the detection and identification of S. aureus enterotoxins (SEs) will be summarized.
Staphylococcal enterotoxins are secreted proteins of approximately 30 kDa that interact with intestinal mucosa and cause emesis and diarrhea. Currently, 23 enterotoxins have been identified as distinct serological individuals [49]. The most common SEs are S. aureus enterotoxin A (SEA) and B (SEB). SEA plays an important role in food poisoning caused by Staphylococcus [50]. SEB is one of the most potent bacterial superantigens, and their toxic effects are based on the activation of cytokine release, ultimately causing cell death by apoptosis. Currently, no treatment or vaccine is available [51]. SEB had been considered and produced as an offensive biologic warfare agent and is identified as a restricted agent by the CDC (Centers for Disease Control) [52].
• Detection of Staphylococcus aureus enterotoxins
The first methods for the detection of SEs were conventional methods such as animal [53,54] and serological tests [55]. The development of molecular biology methods such as PCR brought more sophisticated and faster approaches to detect SEs [12]. Furthermore, there are well-established and sensitive immunoaffinity-based methods available for SEs in various matrices, many of which have detection limits in the range of 1–10 ng/g. Three are three main types of sensors for SE identification using these methods: optical, electrochemical and mass detection techniques. Furthermore, optical detection methods based on colorimetric [56,57,58,59,60,61,62], fluorescence [63,64,65] and chemiluminescence [66] principles as well as highly sophisticated methods, such as electrochemiluminescence [67] and the surface plasmon resonance (SPR) [68] immunoassay, have been developed. ELISA is a fundamental and widely used colorimetric method and is generally the most common method of SE detection. Electrochemical immunoassays [69] present the newest method for simple, sensitive, portable, cheap, and reproducible SEs detection and has outstanding compatibility with the latest technologies. Nevertheless, commercial kits are only available for the detection of five enterotoxins (SEA to SEE) and suffer from serious limitations in terms of sensitivity, specificity and suitability for complex food matrix analysis. A list of methods of SE detection are summarized in Table 1.
Mass spectrometry-based methods, on the other hand, provide abundant specific information. MS analysis of intact SEB was reported and published in the early 1990s [78]. Rapid isolation and identification of staphylococcal exoproteins by LC/MS—followed by the determination of N-terminal amino-acid sequences of separated peaks—was developed [79]. Applications involving the detection of SEs in food matrices have become widely used. Biomolecular interaction analysis mass spectrometry was applied to detect bacterial toxins in food samples [80]. The SEB concentration in milk and mushrooms was determined using a combination of antibody extraction and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). Quantitative MS analysis of SEB in food matrices using labeled peptides and a label-free approach was performed [81]. The objective of this study was to demonstrate that a proteomics-based strategy can be effectively used to detect and quantitate SEB in food matrices within the accepted criteria for bioassays. Another protein standard absolute quantification (PSAQ) strategy represents the ideal alternative methodology, allows the evaluation of the digestive pathogenicity of poorly characterized SEs and helps investigate staphylococcal food poisoning outbreaks that cannot be solved with existing immunological tools [42]. Reference [82] shows how proteomics-based methods can be effectively used to detect, confirm and quantitate SEB in food matrices. Amine-modifying labeling reagents were used to quantitate the protein in complex food matrices using differential isotopic tags, reference internal standards and LC-MS/MS analysis. In this study, the authors show an alternative labeling strategy using acetylation with acetic anhydride (Ac2O/2H6-Ac2O) that is reasonably priced, reliable and provides adequate information about SEB in food matrices. In addition, this method does not require SEB to be manipulated during routine analysis and therefore represents a significant advantage for laboratories controlled by regulatory agencies. Another report presents a label- and antibody-free alternative method based on a bottom-up proteomics approach for the targeted measurement of SEA and SEB in milk and shrimp with UPLC-ESI-MS/MS. This analysis of toxins uses proteotypic amino-acid sequences within the toxin. The use of corresponding 13C-labeled internal standard sequences and tandem mass spectrometry provides high specificity during simultaneous identification and aids the quantification of SEA and SEB toxins in highly complex food extracts at low ppb levels. As the method can be applied without the need for specific reagents or antibodies, it may be extended to other food matrices and enterotoxins for which antibodies have not been developed [83]. The most up to date methods for the detection and identification of SEs in food matrices are shown in refs. [84,85]. In the proteomics-based bottom-up approach [84], the LC technique combined with MS/MS in multiple reaction monitoring (MRM) mode was used to detect and quantify two types of SE (A and B) spiked into a milk and buffer solution. In the proteomic assay developed for simultaneous detection of several CBRN-relevant toxins in food, the PSAQ approach for highly specific detection and absolute quantification strategy was used [85]. Here, CBRN stands for chemical, biological, radiological and nuclear protection. The assay was based on an antibody-free sample preparation followed by bottom-up LC-MS/MS analysis operated in targeted mode. An overview of the newest identification methods of SEs by mass spectrometry is shown in Table 2.
3.2. Bacillus anthracis Toxins
B. anthracis is a Gram-positive, pore-forming, rod-shaped bacterium and the only obligate pathogen from the genus Bacillus. It is one of only a few bacteria known to synthesize a protein capsule (poly-d-gamma-glutamic acid). It forms a calmodulin-dependent adenylate cyclase exotoxin known as anthrax edema factor, along with anthrax lethal factor. Genotypic and phenotypic similarity to B. cereus and B. thuringiensis has been shown, and all form oval endospores. B. anthracis endospores are highly resilient, surviving extremes of temperature, low-nutrient environments, and harsh chemical treatment over decades or even centuries. Because of these features, B. anthracis endospores are particularly well-suited for use (in powdered and aerosol form) as a biological weapon [86]. The U.S. CDC classifies B. anthracis as a category A biological agent [87]. Thus, there are many reasons to study the mechanism of action and methods for detection of the microorganism and its protein toxins. The virulence of B. anthracis species is primarily caused by different components: The poly-d-glutamic acid capsule and the anthrax toxin. The genes for capsule biosynthesis and anthrax toxin are coded by double-stranded DNA situated on plasmids, called pX01 and pX02 [35]. The anthrax toxin is composed of three distinct proteins: protective antigen (PA), lethal factor (LF), and edema factor (EF). Lethal (LeTx) and edema toxin (EdTx) each contain 2 protein types. LeTx is composed of oligomers of PA and LF, and EdTx is made up of PA and EF. These proteins must act together [88]. PA is an 83 kDa protein, and it provides the toxin entrance into the cell. After administration of toxin to the body, PA binds to receptors on the cell surfaces (most often through macrophages) and is cleaved by protease to a 63 kDa fragment, which is attached to the receptor. PA63 forms an oligomer at the cell surface that binds three molecules of LF and EF. EF is a calmodulin-dependent adenylate cyclase that converts ATP to cyclic AMP (cAMP). Decreasing levels of ATP leads to a decrease in chloride ions and water from the affected cells, which leads to the massive edema observed in cutaneous cases of anthrax. EF also inhibits the phagocytic and oxidative burst activity of neutrophils, which increases the vulnerability of the host to spread the infection. LF is a zinc-dependent metalloprotease that cleaves several members of the mitogen-activated protein kinase (MAPK) and thus inhibits the signal transduction pathway. At this point, increasing levels of the cytokines TNF-α and IL-1β in macrophages have been reported. LF activates the oxidative burst pathway, leading to the production of reactive oxygen intermediates [89]. Both toxins act in concert to cause immune deregulation, endothelial dysfunction, advanced septicemia, hemorrhage, and shock, which often leads to death [35].
• Detection of Bacillus anthracis toxins
Detection of B. anthracis microorganism and its toxins have been described in many studies. Some genetic similarities to other members of the genus Bacillus (B. cereus, B. thuringiensis and B. mycoides) make the detection of B. anthracis and its released toxins challenging [90]. Conventional microbiological methods based on morphological examination of the growth culture have long been a gold standard for its identification [35]. Other methods based on the biochemical properties of the organism have been described and usually act as confirmation methods on isolated colonies that are already suspected of being B. anthracis based on their morphological characteristics. Most of the assays are based on detection of the whole organism, bacterial antigens, and/or the nucleic acid. A summary of relevant detection methods of B. anthracis is shown in Table 3.
Enzyme immunoassays detect B. anthracis toxin proteins such as PA, EF, LF or the poly-d-glutamic acid capsule [112,113]. Many immunoassay formats are currently commercially available for a wide variety of detection needs and are outlined in Table 2. The mechanism of action of B. anthracis toxin has been described before, and it is clear that its enzymatic activity can be used to benefit detection. The zinc metalloprotease activity of the anthrax lethal factor was studied for these purposes. Notably, the detection of toxin-specific reaction products may often be easier than the detection of the toxin itself [35,45]. One technique termed Endopep-MS was first mentioned in 2005 [111]. In this study, the toxin activity quantification by MS is based on three levels of measurement. First, the toxin is purified/enriched using monoclonal antibodies (mAbs) covalently bound to magnetic beads that are specific for the toxin. The second step includes toxin-specific enzyme activity directed against a specific substrate, and the last step is MS detection of toxin-generated reaction products [35,45]. Total LeTx and EdTx are captured from the clinical sample using mABs. The captured LF is then added to a reaction buffer containing an optimized peptide substrate with an amino acid sequence, similar to the portion of MAPK cleaved by LF, to give two products with a specific mass. The remaining substrate and two LF-specific products are detected by MALDI-TOF. The EF is caught by mAbs and incubated with ATP and the adenylyl cyclase cofactor calmodulin. The ATP and adenylyl cyclase reaction product cAMP are detected by LC-ESI-MS/MS. Furthermore, the use of isotopically labeled cleavage product allows the use of the method for quantification of LF [111]. The main advantage of monitoring anthrax toxin by MS is early detection of LF in less than 12 h. The LF can be detected before positive bacteremia status, before detection by PCR, and before detection by antigen capture immunoassay [35]. Clinical applications of Endopep-MS using MALDI-TOF-MS were published [114,115].
3.3. Clostridium Toxins
Clostridium is a genus of Gram-positive bacteria that contains significant human pathogens, including the causative agent of botulism, and is an important cause of severe necrotizing disease in the large intestine. The Clostridium genus comprises approximately a hundred species. The most interesting species related to human and animal diseases are: C. botulinum, C. difficile, C. perfringens and C. tetani.
Members of the C. botulinum strain produce botulinum neurotoxins (BoNT). The diverse group of C. botulinum strains is sorted into four distinct groups according to their ability to produce toxins. Human pathogenic neurotoxins of types A, B, E, and F are produced by the C. botulinum groups I and II, C. butyricum and C. baratii [14]. BoNT are the most lethal bacterial toxins known to mankind and are produced by both natural and synthetic routes, with a lethal dose of approximately 2 ng/kg [116]. BoNT are categorized into seven serotypes from A to G based on their response to antisera. All seven serotypes are synthetized by the bacterium as single-chain polypeptides. The different serotypes are approximately 40% similar at the amino acid level [45]. BoNT are composed of an enzymatically active light chain (LC, 50 kDa) connected by a disulfide bond with a receptor binding heavy chain (HC, 100 kDa). The HC is comprised of two regions: the amino terminal 50 kDa domain with a translocation function and a 50 kDa carboxyl terminal domain. LC domains with zinc-dependent endoprotease activity are responsible for the inhibition of neurotransmitter release. There are three membrane proteins on the presynaptic cells (secretory vesicle) that serve as a target for this endoprotease [35]. The BoNT serotypes have different functional properties. BoNT A, C and E hydrolyze synaptosomal associated protein 25 (SNAP-25), and BoNT B, D, F and G hydrolyze isoforms of synaptobrevin, also known as vesicle associated membrane protein (VAMP-2). BoNT C is unique within the group, as it hydrolyzes a second substrate, syntaxin, in the vesicle docking complex [117,118]. Due to its extreme toxicity, speed of the initiation of symptoms and lack of treatment, a sensitive and rapid BoNT detection method is needed to diagnose botulism in suspected cases before paralysis occurs. Methods of BoNT detection have been reviewed in many publications [14,15,16].
• Detection of BoNT
While the mouse lethality assay has remained the standard test for the detection of botulinum neurotoxins, there has been great progress in the development of alternative in vitro tests in recent years. The mouse lethality test is based on intraperitoneal injection of mice with dilutions of BoNT-suspected samples and sequential observation of these mice for symptoms of botulism and ultimately death, which requires a fully functional toxin capable of binding and entering neurons before cleaving its SNARE (an acronym derived from “SNAP (Soluble NSF Attachment Protein) receptor substrate. It is necessary to find both the maximum sample dilution that kills mice and the minimum dilution that does not kill to estimate the quantity of BoNT in the sample. If the dilution that does not kill is not found and all injected animals die, the sample must be diluted further and the procedure repeated [119]. The quantity of toxin in the sample is then estimated by relating the maximum dilution that kills to the known mouse-lethal dose (MLD50). The toxin serotype is then determined by neutralization of the toxin with serotype-specific antitoxin, which is usually administered prior to toxin injection. Mice are observed for signs of botulism for another 48 h to deduce which specific antitoxin is protective. The mouse bioassay is applicable to all BoNT serotypes, with an LOD of 5 to 10 pg of BoNT/A, but a minimum of four to six days is needed. Variations in the mouse lethality assay have been developed to reduce the number of laboratory mice required [120,121,122,123]. All of these methods can estimate toxin quantity using significantly fewer animals than the mouse lethality assay, but the toxin serotype must be known in advance to correlate symptoms and survival times to dose. If the serotype is unknown, a toxin neutralization assay or alternative assay must be performed.
PCR techniques to identify the presence of C. botulinum DNA were originally used to detect the presence of bacterial spores. The method can detect the presence of as few as 102 spores per reaction mixture for serotypes A, E and F and only 10 spores per reaction mixture for BoNT/B. To enhance the sensitivity, Lindström and colleagues developed an enrichment method that could detect as few as 10−2 spores/gram of sample for the serotypes A, B and F and 10−1 spores/gram of sample for BoNT/E [124]. One critical shortcoming of this method is that enrichment often requires 5 days. Furthermore, the applicability of the assay for the detection of food contamination was diminished by the observation that food matrices could interfere with the sensitivity of the assay. Additionally, if contamination were to occur with the actual toxin and not cells, this traditional PCR method would not be useful. Multiplex technology is conceptually simple for PCR-based assays. Different sets of PCR primers can easily be generated, each highly specific for a gene of interest, allowing for the amplification of multiple targets in one reaction tube. Many multiplex technologies for the detection of BoNT genes have been described [125,126,127,128]. The main disadvantage of the molecular detection of BoNT is the impossibility to detect gene or toxin activity.
Numerous immunological tests have been developed, although many earlier assays, such as radioimmunoassays [129,130], passive hemagglutination [131,132] or the gel diffusion assay [133], have poor sensitivities or specificities. After all, these assays were overcome by more sensitive ELISA. After over four decades of use for BoNT detection [134,135], ELISA is the most commonly applied immunological test. Several variations of ELISA protocol were developed to enhance assay sensitivity [136,137,138,139]. ELISA methodologies have been successfully employed in the detection and quantification of purified botulinum toxin, in C. botulinum cultures that produce the toxin [140], in an extensive variety of food samples [141] (both contaminated food and food artificially spiked with the toxin), and in clinical samples such as serum [142] and feces [143]. Some foods tend to interfere with ELISAs and decrease their sensitivity; therefore, the results should be confirmed by a mouse lethality assay.
The serotype specificity of the ELISA depends on the specificity and cross-reactivity of the antibodies used. All BoNT serotypes are immunogenic and can elicit production of antitoxin antibodies. The seven serotypes differ by up to 70% at the amino acid level, enabling the selection of antibodies with little or no cross-reactivity [144]. Both monoclonal and polyclonal antibodies have been used in ELISA experiments to detect and serotype BoNT, with polyclonal more common due to reduced procurement costs and easier accessibility. Polyclonal antibodies with high specificity against serotypes A, B, E, and F (serotypes generally causing human disease) were employed to identify these BoNT serotypes by amplified-ELISA and ELISA using digoxigenin-labeled antibodies [136] with high sensitivity and no cross-reactivity. Sensitive ELISAs relying on mABs with high serotype specificity have also been developed [145]. Genetic variation within the different serotypes of the toxin may result in decreased affinity for mABs, causing false-negative results [144].
One widely used technology for the detection of peptide bond cleavage is fluorescence resonance energy transfer (FRET) [146]. Multiple variations of fluorophore/quencher modified substrates derived from SNAP-25 have been developed and used for the detection of BoNT/A [147,148].
BoNT has enzymatic activities as well as the previously mentioned B. anthracis toxin. BoNT are Zn2+-dependent endopeptidases that inhibit neurotransmitter release via the specific cleavage of synaptic SNARE complex proteins. Exploitation of this endopeptidase role of BoNT has led to numerous detection methods. Unlike the immunological in vitro methods described above, which are unable to discriminate between an active and inactive form of the toxin, endopeptidase assays detect the active form only. For example, if food that was heated was positive for the presence of BoNT by immunoassay, it may be negative by the mouse lethality assay and endopeptidase assays, as the toxin may be inactive. In this sense, endopeptidase assays are more similar to the mouse lethality bioassay than immunoassays. The first assay based on the endopeptidases activities of BoNT was developed in the early 1990s. In this assay, synthetic peptides with a specific cleavage site were added to the sample, and cleavage products were detected by ELISA [149]. The method was specific to BoNT/B, showing no cross-reactivity with other clostridia neurotoxins, and had sensitivity from 0.6 to 4.5 ng/mL. A rapid endopeptidase-MS to detect and differentiate active BoNT A, B, E, and F was developed [150]. This method has found utilization in various matrices [151]. In the study, in which all conventional tests were used, botulinum toxin was successfully detected in patient serum using the endopeptidase-MS assay, although all conventional tests gave negative results [152].
In recent years, MS analysis has become a promising choice for the characterization and detection of BoNT. A method previously developed for the MS identification of tetanus toxin was first applied to BoNT/A and B [153] as well as BoNT C, D, E, and F to complete the characterization of botulinum toxins [154]. In this study [155], nanoLC-MS/MS with peptic sample pretreatment and neurotoxin database identification was used to characterize the protein composition of botulinum progenitor toxins and assign botulinum progenitor toxins to their proper serotype and strain using currently available sequence information. In contrast to other investigations in this study, crude botulinum toxins directly from proteolytic C. botulinum strains were used rather than standard materials and demonstrated accurate identification of highly toxic botulinum toxins based on peptide sequences. The results demonstrated that the combination of peptic pretreatment and multidimensional nano-LC–MS/MS with an ion trap detector represents substantial progress in botulinum toxin detection [116]. A summary of methods relevant to the characterization of BoNT is in Table 4.
In summary, the detection of BoNT at relevant concentrations is challenging because BoNT are extremely lethal, and therefore the test must be correspondingly sensitive. The currently accepted test for functional detection of BoNT is still the standard mouse lethality bioassay, although it is time consuming, costly, impractical for screening large numbers of samples and cannot be used in the field. A fully functional, highly sensitive replacement assay is the mouse phrenic nerve hemidiaphragm assay, which is currently being validated for pharmaceutical products [156,157]. Assays that detect BoNT proteolytic activity employ naturally occurring or synthetic analogs of SNARE proteins combined with methods for detecting the cleaved products such as FRET or MS.
Clostridium perfringens is a Gram-positive anaerobic spore-forming bacterium that is widely distributed in nature, especially in environmental matrices such as soil or water and the intestinal tracts of humans and animals. Under natural conditions, this bacterium is responsible for local outbreaks of food poisoning associated with the consumption of contaminated food and is frequently connected with inappropriate storage or treatment of food. The bacterium is also a major cause of gas gangrene. The absence of early treatment leads to the spread of toxins in the body, causing shock, coma and death. C. perfringens has been shown to produce 18 toxins: alpha (CPA), beta (CPB), epsilon (ETX), iota (CPI), enterotoxin (CPE), theta/perfringolysin O (PFO), beta-2 (CPB2), TpeL, NetB, NetF, BecA/B, NanI, NanJ, kappa, mu, lambda, clostripain, and delta toxin. C. perfringens strains can be divided into five types (A to E) based on the production of four major extracellular toxins (α, β, ε, and ιA) [158]. C. perfringens types B and D produce the epsilon toxin, which is considered the third most powerful clostridial toxin. Due to the possibility to disperse the toxin as an aerosol and a lack of methods for the treatment and prevention of poisoning, C. perfringens spores and toxins are considered biological warfare agents. See Table 5 for the diversity of C. perfringens toxinotypes and associated diseases [159].
Alpha toxin is a 45 kDa necrotizing toxin, which in purified form has been shown to be a zinc-containing phospholipase C enzyme. Its encoding gene (cpa) is present in all C. perfringens strains. Studies have shown that alpha toxin is the major virulence factor in cases of gas gangrene [160]. Beta toxin (38 kDa) is produced by the B and C types and it is the primary lethal factor in type C. Beta toxins have 28% structural similarity to pore-forming S. aureus alpha toxins, and it is expected that beta toxin acts in a similar way [161]. Epsilon toxin (32 kDa) is produced as a single-chain prototoxin by the B and D types. Epsilon toxin is a pore-forming protein that causes potassium and fluid leakage from cells and is considered a potential biological warfare agent in category B [162]. Iota toxin is known as an AB toxin produced by E strain and is composed of two different proteins: Ia (52 kDa), which has enzymatic activity and Ib (98 kDa). Ib binds to receptors on targeted cells and translocates Ia into the cytosol. Ia causes ADP-ribosylation of actin, resulting in cell rounding and death [163]. The C. perfringens toxins related to human and animal disease are enterotoxins and beta-2 toxin. Enterotoxin is the major toxin responsible for human food poisoning, and beta-2 toxin is a recently described toxin responsible for porcine necrotic enteritis [164].
• Detection of Clostridium perfringens toxins
The bacterium and especially its toxins are usually targets of interest for clinical laboratories, the food industry and biodefense. Immunological [165], molecular biology [166] and MS analysis through application of MALDI-TOF identification [167,168,169,170] are applied in these specialized laboratories. Due to the wide diversity of toxins of C. perfringens, many studies on their detection and identification have been published. In general, the two main molecular approaches for detection of Clostridia toxins are EIA [171] and PCR [172,173,174]. EIA methods are rapid and sensitive and are typically used to target toxins [175,176]. However, cross reactivity is a common problem leading to a high false positive rate, which can misguide the public health response; additionally, the toxins of C. perfringens are unstable and can degrade quickly, which can lead to false negatives if stool or food samples are not analyzed soon after collection or are subjected to improper laboratory conditions. Real-time PCR methods are typically employed to amplify toxin genes. Difficulties with this method can arise, since many of the toxin genes in Clostridium spp. reside on extrachromosomal elements (e.g., plasmids or phages) and can be horizontally transferred to other types of bacteria or even within Clostridium species. The transfer of genetic material between them could be problematic for methods targeting only one or a few genes from a single species. Those drawbacks could be overcome by characterization of Clostridium species utilizing LC-MS for intact proteins [177]. Biodefense laboratories focus on the detection of epsilon toxin as a potential biological warfare agent. The C. perfringens epsilon toxin is the third most potent clostridial toxin in nature following BoNT and tetanus toxins [162]. Since introducing non-animal alternatives, the classic mouse assay involving toxin neutralization with C. perfringens type-specific antisera has been replaced. ELISA technology for specifically detecting ETX in intestinal contents is sensitive and quantitative, giving excellent agreement with the mouse protection test, and thus it is one of the best ways to confirm poisoning [178,179]. Molecular biology methods can identify the ETX gene if it is present [180]. Detection and quantification of the ETX protein is also possible using a novel MS technique [168,181]. MS avoids cross-reactivity issues that are intrinsic to any antibody-based assay, but both immunoassays and MS do not determine the presence of the biological activity of toxins. For this purpose, a latex agglutination test has been developed and published as a cytotoxicity assay using Madin-Darby canine kidney cells [182]. A summary of methods relevant to the characterization of C. perfringens and its toxins is shown in Table 6.
3.4. Selected Plant Protein Toxins
Plants cannot move to escape their predators, so they must have other means of protecting themselves from herbivorous animals. Some plants produce highly toxic protein toxins, several of which are considered biological warfare agents by the CDC. Two significant plant protein toxins from a biodefense and toxicology point of view, namely, ricin and abrin, are produced by Ricinus communis and Abrus precatorius, respectively. Both belong to the family of ribosome-inactivating proteins (RIP). Proteins from the RIP group inactivate the 60S ribosomal subunits of eukaryotic cells by N-glycosidic cleavage, which releases a specific adenine base from the sugar-phosphate backbone of 28S rRNA. They are both naturally occurring lectins with a similar dimeric structure consisting of two protein subunits connected by disulfide bridges. The A-chain is responsible for inhibiting protein synthesis, and the B-chain is a galactose-specific lectin responsible for binding of the toxin to the cell membrane, which precedes its endocytosis. Abrin (58 kDa) is soluble in water and naturally occurs in the seeds of the Abrus precatorius (rosary pea), plant, which is common to tropical regions. Ricin occurs in the seeds of the castor oil plant Ricinus communis. The A-chain and B-chain are of similar molecular weights, approximately 32 kDa and 34 kDa, respectively. In addition to these best-characterized RIP toxins, the class of plant toxins includes lesser known potent chem-bio threat agents, such as viscumin (Viscum album), modeccin (Adenia digitata), and volkensin (Adenia volkensii) [183]. Immunoassays and functional tests are available for the determination of RIP toxins. In functional assays, such as the luciferase luminescence test [184], the enzymatic activity of the A-chain is determined using a cell lysate. Other functional tests assess the enzymatic activity of the A-chain against RNA, using MS to quantify the hydrolysis of adenine in a substrate molecule that is added to the sample [185]. Cell-based cytotoxicity tests that assess the toxin’s full functionality in complex matrices are available [186]. These tests are highly sensitive and can detect toxin activity at concentrations of less than 1 ng/mL. Immunoassays, such as ELISA, using specific antibodies have demonstrated similar sensitivity [187,188].
Ricin from Ricinus communis. Ricin is a naturally occurring toxin found in the seeds of the castor plant (Ricinus communis), which is globally cultivated and processed in large quantities. Ricin has been used as a biothreat agent in the past and has gained national attention due to its remarkable toxicity. The toxicity associated with ricin has long been established, with over 700 human intoxications reported dating as far back as the late 1800s [189]. A summary of notable accounts is summarized in reference [190], including a recent case report of a fatality due to the ingestion of an herbal product containing a lethal level of castor bean powder [191]. In many other recent examples, antigovernment and terrorist groups were involved in the attempted use of ricin as a bioweapon. Its history as a weapon has led to the categorization of ricin by the CDC as a category B biothreat agent [190].
• Detection of ricin
In the case of an intentional release of ricin into the environment, the discrimination of functionally active and denatured ricin is important, especially with regard to emergency operating schedules, forensic analysis and therapy. This information can only be obtained from functional assays, which can be principally differentiated into assays detecting A-chain activity, the B-chain activity, or both. However, the detection of the activity of the isolated subchains provides no information about the activity of the intact 64 kDa ricin molecule. Therefore, the detection of active ricin requires in vivo [192] or in vitro [185] assays for both subchains. To detect functionally active ricin, two methodologies have been used: monitoring of the ribosome inactivating A-chain-induced inhibition or measuring of the release of adenine or depurinated product from the RTA cleaved substrate; the latter method is more convenient. In functional assays where the hydrolysis product is monitored, ricin is usually captured or enriched from environmental samples using an immunoaffinity technique and then reacted with a substrate [193]. The release of labeled or unlabeled free adenine from a ribosome or a synthetic oligonucleotide substrate can be detected by various detection techniques, such as chemiluminescence [194] and MS [195]. Traditionally, immunological methods have been used to detect ricin [196]. Castor seeds contain ricin but also a second lectin, Ricinus communis agglutinin (RCA), which is structurally related to ricin, with over 93% similarity between A-chains and 84% similarity between the B-chains. However, RCA is less toxic [197], which is relevant when choosing an appropriate analytical method. ELISAs can be very sensitive but inclined to non-specific reactions and may give positive responses for RCA, even for denatured ricin lacking toxic activity [198]. In vitro toxicity assays and molecular methods such as PCR provide information on the type of toxic action but do not provide absolute evidence of the substance present in the sample. In some cases of ingestion of ricinus material, the low molecular weight alkaloid ricinin has been used as a marker, as it is easier to detect this compound than the protein ricin [199]. The first MS-based activity test of ricin was presented in 2000 by Fabris, who detected ricin-induced depurination of RNA by direct infusion of ESI. Before this time, MS-based analysis had not produced any direct information about the activity of the toxin. Many other publications have followed [185,195,200,201,202,203]. In reference [193], the sensitivity for the qualitative and quantitative analysis of active ricin by MALDI-TOF MS has been improved. In reference [190], ricin detection methods that can and cannot detect biological activity are reported. Many detection methods provide robust, sensitive, and quantitative detection of ricin, and detection assays that can distinguish between biologically active and inactive ricin are essential for evaluating the lethality of a bioterrorism threat and for monitoring site decontamination procedures. However, many of the biological assays discussed here that can detect toxicity and A-chain activity have limitations in selectivity and cannot distinguish ricin from other harmful toxins. For these reasons, it is necessary to utilize an integrated approach in the development of an ideal ricin detection method. The optimal assay design would have a rapid and efficient enrichment step, an A-chain activity checkpoint, and a selectivity step that can distinguish ricin from other bioactive toxins. An example is given in the study focused on the simultaneous detection of ricin and abrin DNA by qPCR. Ricin and abrin DNA can be distinguished using differently labeled probes and fluorescence signals for ricin and abrin (Alx532) without crosstalk [204]. Other studies for the parallel detection of RIP plant and other protein toxins have been published [66,85,183,205,206].
Abrin from Abrus precatorius. Abrin is a protein toxin obtained from the seeds of Abrus precatorius (jequirity bean), which is similar in structure and properties to ricin. The A- and B-chains have molecular masses of approximately 35.5 kDa and 30 kDa, respectively. Multiple isoforms of these proteins have been isolated, and it is now generally accepted that three isoforms are routinely isolated; the relative molecular weights for the entire molecules were determined as 64 kDa for abrin I and 63 kDa for abrin II and III [207]. Isoforms can occur due to post-translational modifications of proteins or individual peptide chains being encoded by multigene families. Variations within the A- and B-chains, as determined by cDNA sequencing [208] and amino acid sequencing [209], are indicative of multiple-gene control. Abrus agglutinins are also extracted from Abrus precatorius. These are tetramers of approximately 130 kDa. The agglutinins have high amino-acid-sequence similarity to abrin [210], which has significance for detection methods, in particular assays for utilizing polyclonal antibodies against abrin protein, which show cross-reactivity to agglutinins. The presence of agglutinins is not a factor for toxicity, as they are considered to have minimal toxicological significance [211].
• Detection of abrin
Analytical methodologies directed at determining abrin are relatively new. Direct testing of abrin protein has been performed by ELISAs [212,213,214], immunochromatographic strips [215,216,217], multiplex detection immunoassay [206] and electrochemiluminescence assays [213]. Indirect means of testing for abrin include real-time PCR to test for the genes encoding the protein [204] and determination of the small molecule abrine by LC-MS [183,218].
Viscumin from Viscum album. The poisonous properties of mistletoe (Viscum album) have been known since ancient times. The toxic lectin viscumin isolated from Viscum album [219] is a cytotoxic 61 kDa large protein that binds to the galactose residues of cell surface glycoproteins and may be internalized by endocytosis. Viscumin strongly inhibits protein synthesis by inactivating the 60S ribosomal subunit. The structure of this protein is very similar to other RIPs mentioned above (abrin, ricin). Methods for the simultaneous detection of viscumin toxin with other toxins were published. One example is the quantitative immunoassay of biotoxins on hydrogel-based protein microchips, where three-dimensional gel-based microchips with immobilized proteins were used for quantitative immunoassay of a series of plant (ricin and viscumin) and bacterial (staphylococcal enterotoxin B, tetanus and diphtheria toxins, and lethal factor of anthrax) toxins [220]. The chips were incubated with solutions containing one of the toxins and developed with a mixture of Cy3-labeled antibodies against all six toxins. Bright fluorescent signals were observed in gel elements containing immobilized antibodies against the applied toxin. The main factor that determined the sensitivity of parallel analysis was the selection of antibody pairs. In this study, both aspects of a good detection method were demonstrated—high sensitivity in the sandwich analysis of the corresponding toxins and no cross-reaction with other toxins and antibodies. The detection limit for viscumin calculated in parallel analysis using a mixture of the six labeled antibodies was 2 ng/mL, similar to single-antibody tests. Another study for quantitative determination of lectin using the enzyme-linked lectin assay in mistletoe preparation was published with a detection limit of 5 ng/mL [221]. Summary of methods relevant to characterization of selected plant protein toxins is shown in Table 7.
3.5. Additional Underestimated Protein Toxins
Conotoxins are group of neurotoxic peptides isolated from the venom of the marine cone snail of the genus Conus. Conotoxins are peptides consisting of 10 to 30 amino acid residues and typically have one or more disulfide bonds. These patterns of disulfide bridges help to define the number of structural classes of conotoxins, of which μ-conotoxins, ω-conotoxins, and α-conotoxins constitute the major classes. They are soluble in water and acidic acetonitrile and have a variety of mechanisms of actions, most of which have not yet been determined [222]. However, it appears that many of these peptides modulate the activity of ion channels, particularly by blocking potassium and sodium channels in neurons [223]. In recent decades, conotoxins have been the subject of pharmacological interest. Research is being conducted to determine their potential therapeutic value in chronic pain control, Parkinson’s disease, and neuromuscular disorders [224]. Conotoxins appear on selected Agents and Toxins Lists from the CDC [87]. At first, the discovery of each new conotoxin required biochemical purification from venom. Unusual post-translationally modified amino acids were identified in the first group of conotoxins characterized. As more cDNA clones encoding conotoxins were clarified, a molecular cloning/PCR approach for identifying novel conotoxins became possible. The first venom peptide characterized only on the basis of the predicted sequence from a cDNA clone was ω-conotoxin MVIIC. However, one problem with this approach is that if the conotoxin is post-translationally modified, knowledge of the encoding DNA sequence does not necessarily permit an accurate prediction of which amino acids are modified [225].
The method developed for purified α-conotoxin GI in phosphate buffer was published [226]. In this method, a biologically active fluorescein derivative of Conus geographus α-conotoxin (FGI) was used in solution-phase-binding assays with two purified mAbs to detect the toxin in laboratory samples. For competitive ligand-displacement spin-column assays, FGI was premixed with various dilutions of unlabeled ligands and then incubated with the two mAbs (5A1 and 8D2) at room temperature. Competitive displacement assays showed that both mAbs specifically bound α-conotoxin GI with high avidity. Cross-reactivity with α-conotoxins M1 and S1 was not observed for either mAb in a direct ELISA. Another method [227] developed for Conus anemone venom (α-Conotoxins AnIA, AnIB, and AnIC) in buffer was published. An LC/MS analysis with diagnostic screening for the detection of peptides with posttranslational modifications revealed the presence of novel sulfated peptides within the R-conotoxin molecular mass range in Conus anemone crude venom. Three sulfated α-conotoxins (AnIA, AnIB and AnIC) were identified and are within the molecular mass range of other α-conotoxins (i.e., 1400–2200 Da).
Bungarotoxins are a group of closely related neurotoxic proteins of the three-finger toxin superfamily found in the venom of kraits, including Bungarus multicinctus. The α-bungarotoxin inhibits the binding of acetylcholine to nicotinic acetylcholine receptors; β- and γ-bungarotoxins act pre-synaptically, causing excessive acetylcholine release and subsequent depletion. Both α and β forms have been characterized, and the α form is similar to the long or Type II neurotoxins from other elapid venoms [228].
The identification of bungarotoxins is tightly connected with the study of venomics [229,230]. There have been several reports on the diagnosis of snake envenomation, among which ELISA appears to be the most practical and commonly used. A literature review of the various immunoassays developed for the detection of toxins and venoms indicated that the test must not only be specific and sensitive but also rapid, easy to handle and able to withstand wide variation in the environment and field [231]. In reference [232], a highly sensitive, specific, and simple avidin–biotin optical immunoassay (AB-OIA) for the detection of β-BuTx in whole blood, plasma, urine, and tissue homogenates was developed. The assay can detect toxin in whole blood without manipulation, and the entire assay can be performed at room temperature without sophisticated equipment. Affinity purified rabbit IgG anti-β-BuTx antibody was immobilized on an optically active silicon surface (SILIAS™ wafer). The test sample was incubated, and the antigen–antibody reaction was monitored by the addition of a biotinylated monoclonal antibody specific to the toxin, avidin–horseradish peroxidase (HRP) and tetramethylbenzidine substrate. The assay detected β-BuTx levels as low as 16 pg/mL in sample buffer and 100 pg/mL in whole blood or plasma. The AB-OIA was also used to quantitate the postmortem level of β-BuTx in various organs such as brain, liver, and kidney, as well as the tissue at the site of injection. An analytical method based on biosensors was used to detect β-BuTx [233]. In this report, an ISFET-based immunosensor for the detection/quantitation of β-BuTx using a murine monoclonal antibody (mAb15) against β-BuTx and urease conjugated rabbit antibodies specific to the toxin was developed. Antibody immobilization protocols were optimized using SEM and chip ELISA. The ISFET immunosensor detected toxin levels of approximately 15.6 ng/mL.
4. Conclusions and Future Perspectives
Based on previous texts, it is clear that a variety of methods are available for each of the reviewed toxins based on different detection principles (e.g., immunochemical assays, MS-based methods, functional assays, and chromatographic methods). In general, to improve the state of detection for a protein toxins, the following is required: (I) Upgrade the preanalytical phase, sample preparation procedures (decrease unfavorable matrix effects, avoid false negative, positive results); (II) and achieve the highest selectivity, robustness, and specificity possible and (III) acquire the most suitable reagents (e.g., purified protein standards, convenient antibodies) to develop the methods. Immunological assays that can detect an antigenic property of a protein are sensitive but inclined to cross-reactions and false-positive results. Molecular biology techniques such as PCR give only indirect evidence of the presence of a certain protein as it reacts to the encoding nucleic acid. Activity tests show a certain type of toxin reaction but not the chemical identity of the toxins. MS-based techniques give direct proof of the molecular structure by measuring the molecular mass, the amino acid sequence, and posttranslational modifications. In addition, modern bioinformatics and proteomics MS approaches, which were developed for the detection and identification of protein-based toxins in different kinds of matrices, have been successfully demonstrated in biodefense applications. However, none of the mentioned methods alone provided complete pictures of the protein-based sample. A combination of techniques for unambiguous identification and tests to measure the biological activity of a particular toxin is the best way to obtain the most information for complex samples.
Acknowledgments
This work was supported by funding from projects of Ministry of Interior, Czech Republic, (project No. VH20172020012: preparation of the collection of biologically significant toxins with the support of European biological European biodefence laboratory network); Ministry of Education, Youth and Sports (project No. SV/FVZ201502) and Ministry of Defense of the Czech Republic—long-term organization development plan Medical Aspects of Weapons of Mass Destruction of the Faculty of Military Health Sciences, University of Defense.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pal, M.; Tsegaye, M.; Girzaw, F.; Bedada, H.; Godishala, V.; Kandi, V. An Overview on Biological Weapons and Bioterrorism. Am. J. Biomed. Res. 2017, 5, 24–34. [Google Scholar] [CrossRef]
- Jansen, H.J.; Breeveld, F.J.; Stijnis, C.; Grobusch, M.P. Biological warfare, bioterrorism, and biocrime. Clin. Microbiol. Infect. 2014, 20, 488–496. [Google Scholar] [CrossRef] [PubMed]
- NIAID Emerging Infectious Diseases/Pathogens | NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (accessed on 12 June 2017).
- COMMISSION DELEGATED REGULATION (EU) 2016/1969 of 12 September 2016 Amending Council Regulation (EC) No 428/2009 Setting up a Community Regime for the Control of Exports, Transfer, Brokering and Transit of Dual-Use Items. Available online: https://danishbusinessauthority.dk/sites/default/files/media/reg._2016-1969_new_control_list.pdf (accessed on 1 November 2017).
- Lubran, M.M. Bacterial toxins. Ann. Clin. Lab. Sci. 1988, 18, 58–71. [Google Scholar] [PubMed]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.-G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010, 584, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Duriez, E.; Armengaud, J.; Fenaille, F.; Ezan, E. Mass spectrometry for the detection of bioterrorism agents: From environmental to clinical applications. J. Mass Spectrom. JMS 2016, 51, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Dorner, B.G.; Zeleny, R.; Harju, K.; Hennekinne, J.-A.; Vanninen, P.; Schimmel, H.; Rummel, A. Biological toxins of potential bioterrorism risk: Current status of detection and identification technology. TrAC Trends Anal. Chem. 2016, 85, 89–102. [Google Scholar] [CrossRef]
- Demirev, P.A.; Fenselau, C. Mass spectrometry in biodefense. J. Mass Spectrom. JMS 2008, 43, 1441–1457. [Google Scholar] [CrossRef] [PubMed]
- OPCW Hosts Series of Science and Technology Meetings. Available online: https://www.opcw.org/news/article/opcw-hosts-series-of-science-and-technology-meetings/ (accessed on 6 November 2017).
- Wilson, I.G.; Cooper, J.E.; Gilmour, A. Detection of enterotoxigenic Staphylococcus aureus in dried skimmed milk: Use of the polymerase chain reaction for amplification and detection of staphylococcal enterotoxin genes entB and entC1 and the thermonuclease gene nuc. Appl. Environ. Microbiol. 1991, 57, 1793–1798. [Google Scholar] [PubMed]
- Jay, M.J.; Loessner, J.M.; Golden, A.D. Bioassay and Related Methods. In Modern Food Microbiology; Food Science Text Series; Springer: New York, NY, USA, 2005; pp. 285–298. ISBN 978-0-387-23180-8. [Google Scholar]
- Lindström, M.; Korkeala, H. Laboratory diagnostics of botulism. Clin. Microbiol. Rev. 2006, 19, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Singh, B.R.; Sharma, S. Botulism diagnostics: From clinical symptoms to in vitro assays. Crit. Rev. Microbiol. 2007, 33, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Whiting, R.C. Methods for detection of Clostridium botulinum toxin in foods. J. Food Prot. 2005, 68, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Solberg, M.; Post, L.S.; Furgang, D.; Graham, C. Bovine serum eliminates rapid nonspecific toxic reactions during bioassay of stored fish for Clostridium botulinum toxin. Appl. Environ. Microbiol. 1985, 49, 644–649. [Google Scholar] [PubMed]
- Dezfulian, M.; Bartlett, J.G. Detection of Clostridium botulinum type B toxin in the presence of a lethal substance interfering with toxin neutralization. Diagn. Microbiol. Infect. Dis. 1985, 3, 105–112. [Google Scholar] [CrossRef]
- Aydin, S. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides 2015, 72, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.E.; Kulinski, S.S.; Reichard, D.W.; Metzger, J.F. Detection of Clostridium botulinum type G toxin by enzyme-linked immunosorbent assay. Appl. Environ. Microbiol. 1981, 42, 1018–1022. [Google Scholar] [PubMed]
- Park, C.E.; Akhtar, M.; Rayman, M.K. Nonspecific reactions of a commercial enzyme-linked immunosorbent assay kit (TECRA) for detection of staphylococcal enterotoxins in foods. Appl. Environ. Microbiol. 1992, 58, 2509–2512. [Google Scholar] [PubMed]
- Wieneke, A.A. Comparison of four kits for the detection of staphylococcal enterotoxin in foods from outbreaks of food poisoning. Int. J. Food Microbiol. 1991, 14, 305–312. [Google Scholar] [CrossRef]
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Investig. 1960, 39, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, S.J. Radioimmunoassay: Review of basic principles. Semin. Nucl. Med. 1975, 5, 125–152. [Google Scholar] [CrossRef]
- Gan, S.D.; Patel, K.R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. J. Investig. Dermatol. 2013, 133, e12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dietrich, R.; Didier, A.; Doyscher, D.; Märtlbauer, E. Recent Developments in Antibody-Based Assays for the Detection of Bacterial Toxins. Toxins 2014, 6, 1325–1348. [Google Scholar] [CrossRef] [PubMed]
- Hejtmancik, K.E.; Peterson, J.W.; Markel, D.E.; Kurosky, A. Radioimmunoassay for the antigenic determinants of cholera toxin and its components. Infect. Immun. 1977, 17, 621–628. [Google Scholar] [PubMed]
- Harris, C.C.; Yolken, R.H.; Krokan, H.; Hsu, I.C. Ultrasensitive enzymatic radioimmunoassay: Application to detection of cholera toxin and rotavirus. Proc. Natl. Acad. Sci. USA 1979, 76, 5336–5339. [Google Scholar] [CrossRef] [PubMed]
- Godal, A.; Olsnes, S.; Pihl, A. Radioimmunoassays of abrin and ricin in blood. J. Toxicol. Environ. Health 1981, 8, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Eagle, M.R.; Houston, L.L. Radioimmunoassay of ricin A- and B-chains applied to samples of ricin A-chain prepared by chromatofocusing and by DEAE Bio-Gel A chromatography. Biochim. Biophys. Acta 1982, 719, 341–348. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ladhani, S.; Robbie, S.; Garratt, R.C.; Chapple, D.S.; Joannou, C.L.; Evans, R.W. Development and Evaluation of Detection Systems for Staphylococcal Exfoliative Toxin A Responsible for Scalded-Skin Syndrome. J. Clin. Microbiol. 2001, 39, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Wu, D.; Lim, D.V.; Jin, S. Sensitive detection of multiplex toxins using antibody microarray. Anal. Biochem. 2010, 401, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Skinner, C.; Patfield, S.; Stanker, L.H.; Fratamico, P.; He, X. New High-Affinity Monoclonal Antibodies against Shiga Toxin 1 Facilitate the Detection of Hybrid Stx1/Stx2 In Vivo. PLoS ONE 2014, 9, e99854. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Gallegos-Candela, M.; Lins, R.C.; Kuklenyik, Z.; Woolfitt, A.; Moura, H.; Kalb, S.; Quinn, C.P.; Barr, J.R. Quantitative Mass Spectrometry for Bacterial Protein Toxins—A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis. Molecules 2011, 16, 2391–2413. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinform. Oxf. Engl. 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, D.M.; Kai, M. Preparation of stable isotope-incorporated peptide internal standards for field desorption mass spectrometry quantification of peptides in biologic tissue. Biomed. Mass Spectrom. 1983, 10, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. MCP 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; MacLean, B.; Tomazela, D.M.; Liebler, D.C.; MacCoss, M.J. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 2012, 12, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Hennekinne, J.-A.; Garin, J.; Brun, V. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Adrait, A.; Lebert, D.; Trauchessec, M.; Dupuis, A.; Louwagie, M.; Masselon, C.; Jaquinod, M.; Chevalier, B.; Vandenesch, F.; Garin, J.; et al. Development of a Protein Standard Absolute Quantification (PSAQ™) assay for the quantification of Staphylococcus aureus enterotoxin A in serum. J. Proteom. 2012, 75, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Dupré, M.; Gilquin, B.; Fenaille, F.; Feraudet-Tarisse, C.; Dano, J.; Ferro, M.; Simon, S.; Junot, C.; Brun, V.; Becher, F. Multiplex quantification of protein toxins in human biofluids and food matrices using immunoextraction and high-resolution targeted mass spectrometry. Anal. Chem. 2015, 87, 8473–8480. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Boyer, A.E.; Barr, J.R. Mass Spectrometric Detection of Bacterial Protein Toxins and Their Enzymatic Activity. Toxins 2015, 7, 3497–3511. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Moura, H.; Woolfitt, A.R.; Kalb, S.R.; McWilliams, L.G.; Pavlopoulos, A.; Schmidt, J.G.; Ashley, D.L.; Barr, J.R. From the mouse to the mass spectrometer: Detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal. Chem. 2005, 77, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Masalha, M.; Borovok, I.; Schreiber, R.; Aharonowitz, Y.; Cohen, G. Analysis of Transcription of the Staphylococcus aureus Aerobic Class Ib and Anaerobic Class III Ribonucleotide Reductase Genes in Response to Oxygen. J. Bacteriol. 2001, 183, 7260–7272. [Google Scholar] [CrossRef] [PubMed]
- Schlievert, P.M.; Case, L.C. Molecular analysis of staphylococcal superantigens. Methods Mol. Biol. 2007, 391, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal Enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- Fries, B.C.; Varshney, A.K. Bacterial Toxins-Staphylococcal Enterotoxin B. Microbiol. Spectr. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Zapor, M.; Fishbain, J.T. Aerosolized biologic toxins as agents of warfare and terrorism. Respir. Care Clin. N. Am. 2004, 10, 111–122. [Google Scholar] [CrossRef]
- Fulton, F. Staphylococcal Enterotoxin—With Special Reference to the Kitten Test. Br. J. Exp. Pathol. 1943, 24, 65–72. [Google Scholar]
- Scheuber, P.H.; Mossmann, H.; Beck, G.; Hammer, D.K. Direct skin test in highly sensitized guinea pigs for rapid and sensitive determination of staphylococcal enterotoxin B. Appl. Environ. Microbiol. 1983, 46, 1351–1356. [Google Scholar] [PubMed]
- Wu, S.; Duan, N.; Gu, H.; Hao, L.; Ye, H.; Gong, W.; Wang, Z. A Review of the Methods for Detection of Staphylococcus aureus Enterotoxins. Toxins 2016, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Saunders, G.C.; Bartlett, M.L. Double-antibody solid-phase enzyme immunoassay for the detection of staphylococcal enterotoxin A. Appl. Environ. Microbiol. 1977, 34, 518–522. [Google Scholar] [PubMed]
- Notermans, S.; Verjans, H.L.; Bol, J.; van Schothorst, M. Enzyme linked immunosorbent assay (ELISA) for determination of Staphylococcus aureus enterotoxin type B. Health Lab. Sci. 1978, 15, 28–31. [Google Scholar] [PubMed]
- Stiffler-Rosenberg, G.; Fey, H. Simple assay for staphylococcal enterotoxins A, B, and C: Modification of enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1978, 8, 473–479. [Google Scholar] [PubMed]
- Fey, H.; Pfister, H.; Rüegg, O. Comparative evaluation of different enzyme-linked immunosorbent assay systems for the detection of staphylococcal enterotoxins A, B, C, and D. J. Clin. Microbiol. 1984, 19, 34–38. [Google Scholar] [PubMed]
- Wieneke, A.A.; Gilbert, R.J. The use of a sandwich ELISA for the detection of staphylococcal enterotoxin A in foods from outbreaks of food poisoning. Epidemiol. Infect. 1985, 95, 131–138. [Google Scholar] [CrossRef]
- Hahn, I.F.; Pickenhahn, P.; Lenz, W.; Brandis, H. An avidin-biotin ELISA for the detection of staphylococcal enterotoxins A and B. J. Immunol. Methods 1986, 92, 25–29. [Google Scholar] [CrossRef]
- Nia, Y.; Rodriguez, M.; Zeleny, R.; Herbin, S.; Auvray, F.; Fiebig, U.; Avondet, M.-A.; Munoz, A.; Hennekinne, J.-A. Organization and ELISA-Based Results of the First Proficiency Testing to Evaluate the Ability of European Union Laboratories to Detect Staphylococcal Enterotoxin Type B (SEB) in Buffer and Milk. Toxins 2016, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Cao, C.J.; Thompson, R.G.; Valdes, J.J. A simple and rapid fluorescence-based immunoassay for the detection of staphylococcal enterotoxin B. Mol. Cell. Probes 2003, 17, 125–126. [Google Scholar] [CrossRef]
- Hun, X.; Zhang, Z. A novel sensitive staphylococcal enterotoxin C1 fluoroimmunoassay based on functionalized fluorescent core-shell nanoparticle labels. Food Chem. 2007, 105, 1623–1629. [Google Scholar] [CrossRef]
- Vinayaka, A.C.; Thakur, M.S. An immunoreactor-based competitive fluoroimmunoassay for monitoring staphylococcal enterotoxin B using bioconjugated quantum dots. Analyst 2012, 137, 4343–4348. [Google Scholar] [CrossRef] [PubMed]
- Szkola, A.; Linares, E.M.; Worbs, S.; Dorner, B.G.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Seidel, M. Rapid and simultaneous detection of ricin, staphylococcal enterotoxin B and saxitoxin by chemiluminescence-based microarray immunoassay. Analyst 2014, 139, 5885–5892. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, M.; Kostov, Y.; Rasooly, A. ELISA-LOC: Lab-on-a-chip for enzyme-linked immunodetection. Lab Chip 2010, 10, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Sd, S.; Rc, S.; Ap, L.; Ce, F. Surface plasmon resonance detection using antibody-linked magnetic nanoparticles for analyte capture, purification, concentration, and signal amplification., Surface Plasmon Resonance (SPR) Detection Using Antibody-Linked Magnetic Nanoparticles for Analyte Capture, Purification, Concentration and Signal Amplification. Anal. Chem. 2009, 81, 2357–2363. [Google Scholar] [CrossRef]
- Tang, D.; Tang, J.; Su, B.; Chen, G. Ultrasensitive electrochemical immunoassay of staphylococcal enterotoxin B in food using enzyme-nanosilica-doped carbon nanotubes for signal amplification. J. Agric. Food Chem. 2010, 58, 10824–10830. [Google Scholar] [CrossRef] [PubMed]
- Park, C.E.; Szabo, R. Evaluation of the reversed passive latex agglutination (RPLA) test kits for detection of staphylococcal enterotoxins A, B, C, and D in foods. Can. J. Microbiol. 1986, 32, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, H.; Igarashi, H. Rapid latex agglutination test for detection of staphylococcal enterotoxins A to E that uses high-density latex particles. Appl. Environ. Microbiol. 1988, 54, 2345–2348. [Google Scholar] [PubMed]
- Rose, S.A.; Bankes, P.; Stringer, M.F. Detection of staphylococcal enterotoxins in dairy products by the reversed passive latex agglutination (SET-RPLA) kit. Int. J. Food Microbiol. 1989, 8, 65–72. [Google Scholar] [CrossRef]
- Pereira, M.L.; Heneine, L.G.; Santos, E.J.; Carmo, L.S.; Pereira, J.L.; Bergdoll, M.S. Prevention of nonspecific reactions on reversed passive latex agglutination assay (RPLA) for detecting low amounts of staphylococcal enterotoxins. Rev. Latinoam. Microbiol. 1997, 39, 57–63. [Google Scholar] [PubMed]
- Hall, H.E.; Angelotti, R.; Lewis, K.H. Quantitative detection of staphylococcal Enterotoxin B in food by gel-diffusion methods. Public Health Rep. 1963, 78, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Salomon, L.L.; Tew, R.W. Assay of staphylococcal enterotoxin B by latex agglutination. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1968, 129, 539–542. [Google Scholar] [CrossRef]
- Lee, C.L.; Lin, C.C. Detection of staphylococcal enterotoxin by latex agglutination inhibition test. Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za Zhi 1984, 17, 77–80. [Google Scholar] [PubMed]
- Read, R.B.; Bradshaw, J.; Pritchard, W.L.; Black, L.A. Assay of Staphylococcal Enterotoxin from Cheese. J. Dairy Sci. 1965, 48, 420–424. [Google Scholar] [CrossRef]
- Kientz, C.E.; Hulst, A.G.; Wils, E.R. Determination of staphylococcal enterotoxin B by on-line (micro) liquid chromatography-electrospray mass spectrometry. J. Chromatogr. A 1997, 757, 51–64. [Google Scholar] [CrossRef]
- Kawano, Y.; Ito, Y.; Yamakawa, Y.; Yamashino, T.; Horii, T.; Hasegawa, T.; Ohta, M. Rapid isolation and identification of staphylococcal exoproteins by reverse phase capillary high performance liquid chromatography-electrospray ionization mass spectrometry. FEMS Microbiol. Lett. 2000, 189, 103–108. [Google Scholar] [CrossRef]
- Nedelkov, D.; Rasooly, A.; Nelson, R.W. Multitoxin biosensor-mass spectrometry analysis: A new approach for rapid, real-time, sensitive analysis of staphylococcal toxins in food. Int. J. Food Microbiol. 2000, 60, 1–13. [Google Scholar] [CrossRef]
- Callahan, J.H.; Shefcheck, K.J.; Williams, T.L.; Musser, S.M. Detection, confirmation, and quantification of staphylococcal enterotoxin B in food matrixes using liquid chromatography–mass spectrometry. Anal. Chem. 2006, 78, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.D.; Letellier, A.; Beaudry, F. Analysis of Staphylococcus enterotoxin B using differential isotopic tags and liquid chromatography quadrupole ion trap mass spectrometry. Biomed. Chromatogr. BMC 2012, 26, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Zuberovic Muratovic, A.; Hagström, T.; Rosén, J.; Granelli, K.; Hellenäs, K.-E. Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins 2015, 7, 3637–3656. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Tsilia, V.; Rajkovic, A.; De Cremer, K.; Van Loco, J. Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk. Toxins 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Gilquin, B.; Jaquinod, M.; Louwagie, M.; Kieffer-Jaquinod, S.; Kraut, A.; Ferro, M.; Becher, F.; Brun, V. A proteomics assay to detect eight CBRN-relevant toxins in food. Proteomics 2017, 17, 1600357. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Clancy, H.A.; Baeumner, A.J. Bacillus anthracis: Toxicology, epidemiology and current rapid-detection methods. Anal. Bioanal. Chem. 2006, 384, 73–84. [Google Scholar] [CrossRef] [PubMed]
- CDC—Biosafety Home. Available online: https://www.cdc.gov/biosafety/ (accessed on 4 July 2017).
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Asp. Med. 2009, 30, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Halverson, K.M.; Ribot, W.; Lane, D.; Kenny, T.; Abshire, T.G.; Ezzell, J.W.; Hoover, T.A.; Powell, B.; Little, S.; et al. Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J. Biol. Chem. 2005, 280, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Peng, D.; Song, X.; Ruan, L.; Mahillon, J.; Sun, M. Differentiation of Bacillus anthracis, B. cereus, and B. thuringiensis on the Basis of the csaB Gene Reflects Host Source. Appl. Environ. Microbiol. 2013, 79, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- Hurtle, W.; Bode, E.; Kulesh, D.A.; Kaplan, R.S.; Garrison, J.; Bridge, D.; House, M.; Frye, M.S.; Loveless, B.; Norwood, D. Detection of the Bacillus anthracis gyrA gene by using a minor groove binder probe. J. Clin. Microbiol. 2004, 42, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Acharya, G.; Doorneweerd, D.D.; Chang, C.-L.; Henne, W.A.; Low, P.S.; Savran, C.A. Label-free optical detection of anthrax-causing spores. J. Am. Chem. Soc. 2007, 129, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Zahavy, E.; Heleg-Shabtai, V.; Zafrani, Y.; Marciano, D.; Yitzhaki, S. Application of fluorescent nanocrystals (q-dots) for the detection of pathogenic bacteria by flow-cytometry. J. Fluoresc. 2010, 20, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.D.; Mansfield, B.; Yip, P.; Cohen, S.J.; Sonenshein, A.L.; Hitt, B.A.; Davis, C.E. Novel technology for rapid species-specific detection of Bacillus spores. Biomol. Eng. 2006, 23, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Young, M.A.; Lyandres, O.; Van Duyne, R.P. Rapid detection of an anthrax biomarker by surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 2005, 127, 4484–4489. [Google Scholar] [CrossRef] [PubMed]
- Farrell, S.; Halsall, H.B.; Heineman, W.R. Immunoassay for B. globigii spores as a model for detecting B. anthracis spores in finished water. Analyst 2005, 130, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Zahavy, E.; Fisher, M.; Bromberg, A.; Olshevsky, U. Detection of frequency resonance energy transfer pair on double-labeled microsphere and Bacillus anthracis spores by flow cytometry. Appl. Environ. Microbiol. 2003, 69, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.I.; Cheun, H.I.; Watarai, M.; Uchida, I.; Takeshi, K. Detection of anthrax spores from the air by real-time PCR. Lett. Appl. Microbiol. 2001, 33, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.L.; Heroux, K.; Kearney, J.; Arasteh, A.; Gostomski, M.; Emanuel, P.A. Bacillus Spore Inactivation Methods Affect Detection Assays. Appl. Environ. Microbiol. 2001, 67, 3665–3670. [Google Scholar] [CrossRef] [PubMed]
- Stopa, P.J. The flow cytometry of Bacillus anthracis spores revisited. Cytometry 2000, 41, 237–244. [Google Scholar] [CrossRef]
- Gatto-Menking, D.L.; Yu, H.; Bruno, J.G.; Goode, M.T.; Miller, M.; Zulich, A.W. Sensitive detection of biotoxoids and bacterial spores using an immunomagnetic electrochemiluminescence sensor. Biosens. Bioelectron. 1995, 10, 501–507. [Google Scholar] [CrossRef]
- Phillips, A.P.; Martin, K.L. Comparison of direct and indirect immunoradiometric assays (IRMA) for Bacillus anthracis spores immobilised on multispot microscope slides. J. Appl. Bacteriol. 1983, 55, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Moayeri, M.; Chen, Z.; Harma, H.; Zhao, J.; Hu, H.; Purcell, R.H.; Leppla, S.H.; Hewlett, I.K. Detection of Anthrax Toxin by an Ultrasensitive Immunoassay Using Europium Nanoparticles. Clin. Vaccine Immunol. CVI 2009, 16, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, Z.P.; Sirisena, M. Development of automated amperometric detection of antibodies against Bacillus anthracis protective antigen. Anal. Bioanal. Chem. 2007, 389, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Zhang, J.-B.; Zhang, Z.-P.; Zhou, Y.-F.; Yang, R.-F.; Chen, J.; Guo, Y.-C.; You, F.; Zhang, X.-E. Construction of single chain variable fragment (ScFv) and BiscFv-alkaline phosphatase fusion protein for detection of Bacillus anthracis. Anal. Chem. 2006, 78, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Rucker, V.C.; Havenstrite, K.L.; Herr, A.E. Antibody microarrays for native toxin detection. Anal. Biochem. 2005, 339, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sastry, K.S.R.; Tuteja, U.; Batra, H.V. Generation and characterization of monoclonal antibodies to protective antigen of Bacillus anthracis. Indian J. Exp. Biol. 2003, 41, 123–128. [Google Scholar] [PubMed]
- Bell, C.A.; Uhl, J.R.; Hadfield, T.L.; David, J.C.; Meyer, R.F.; Smith, T.F.; Cockerill, F.R. Detection of Bacillus anthracis DNA by LightCycler PCR. J. Clin. Microbiol. 2002, 40, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.J.; Erler, A.M.; Nasarabadi, S.L.; Skowronski, E.W.; Imbro, P.M. A multiplexed PCR-coupled liquid bead array for the simultaneous detection of four biothreat agents. Mol. Cell. Probes 2005, 19, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Okinaka, R.T. Ultrasensitive, direct detection of a specific DNAsequence of Bacillus anthracis in solution. Analyst 2000, 125, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Quinn, C.P.; Woolfitt, A.R.; Pirkle, J.L.; McWilliams, L.G.; Stamey, K.L.; Bagarozzi, D.A.; Hart, J.C.; Barr, J.R. Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal. Chem. 2007, 79, 8463–8470. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.H.; Ezzell, J.W.; Abshire, T.G.; Kidd, S.; Kaufmann, A.F. Evaluation of serologic tests for diagnosis of anthrax after an outbreak of cutaneous anthrax in Paraguay. J. Infect. Dis. 1989, 160, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.P.; Semenova, V.A.; Elie, C.M.; Romero-Steiner, S.; Greene, C.; Li, H.; Stamey, K.; Steward-Clark, E.; Schmidt, D.S.; Mothershed, E.; et al. Specific, sensitive, and quantitative enzyme-linked immunosorbent assay for human immunoglobulin G antibodies to anthrax toxin protective antigen. Emerg. Infect. Dis. 2002, 8, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Kuklenyik, Z.; Boyer, A.E.; Lins, R.; Quinn, C.P.; Gallegos-Candela, M.; Woolfitt, A.; Pirkle, J.L.; Barr, J.R. Comparison of MALDI-TOF-MS and HPLC-ESI-MS/MS for endopeptidase activity-based quantification of Anthrax lethal factor in serum. Anal. Chem. 2011, 83, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Gallegos-Candela, M.; Quinn, C.P.; Woolfitt, A.R.; Brumlow, J.O.; Isbell, K.; Hoffmaster, A.R.; Lins, R.C.; Barr, J.R. High-sensitivity MALDI-TOF MS quantification of anthrax lethal toxin for diagnostics and evaluation of medical countermeasures. Anal. Bioanal. Chem. 2015, 407, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Klaubert, B.; Vujtovic-Ockenga, N.; Wermter, R.; Schad, K.; von Meyer, L. Determination of botulinum toxins after peptic sample pre-treatment by multidimensional nanoscale liquid chromatography and nano-electrospray ion-trap mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Pellizzari, R.; Rossetto, O.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: Mechanism of action and therapeutic uses. Philos. Trans. R. Soc. B Biol. Sci. 1999, 354, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; et al. Genetic Diversity among Botulinum Neurotoxin-Producing Clostridial Strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.B.; Borodic, G.E.; First, E.R.; Maccallum, R.D. Measurement of Botulinum Toxin Activity: Evaluation of the Lethality Assay. Toxicol. Appl. Pharmacol. 1994, 128, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kameyama, S.; Sakaguchi, G. Assay in mice for low levels of Clostridium botulinum toxin. Int. J. Food Microbiol. 1990, 11, 271–277. [Google Scholar] [CrossRef]
- Adler, M.; Scovill, J.; Parker, G.; Lebeda, F.J.; Piotrowski, J.; Deshpande, S.S. Antagonism of botulinum toxin-induced muscle weakness by 3,4-diaminopyridine in rat phrenic nerve-hemidiaphragm preparations. Toxicon 1995, 33, 527–537. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Sheridan, R.E.; Adler, M. A study of zinc-dependent metalloendopeptidase inhibitors as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1995, 33, 551–557. [Google Scholar] [CrossRef]
- Torii, Y.; Goto, Y.; Takahashi, M.; Ishida, S.; Harakawa, T.; Sakamoto, T.; Kaji, R.; Kozaki, S.; Ginnaga, A. Quantitative determination of biological activity of botulinum toxins utilizing compound muscle action potentials (CMAP), and comparison of neuromuscular transmission blockage and muscle flaccidity among toxins. Toxicon 2010, 55, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.; Keto, R.; Markkula, A.; Nevas, M.; Hielm, S.; Korkeala, H. Multiplex PCR assay for detection and identification of Clostridium botulinum types A, B, E, and F in food and fecal material. Appl. Environ. Microbiol. 2001, 67, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Fach, P.; Gibert, M.; Griffais, R.; Guillou, J.P.; Popoff, M.R. PCR and gene probe identification of botulinum neurotoxin A-, B-, E-, F-, and G-producing Clostridium spp. and evaluation in food samples. Appl. Environ. Microbiol. 1995, 61, 389–392. [Google Scholar] [PubMed]
- Takeshi, K.; Fujinaga, Y.; Inoue, K.; Nakajima, H.; Oguma, K.; Ueno, T.; Sunagawa, H.; Ohyama, T. Simple method for detection of Clostridium botulinum type A to F neurotoxin genes by ploymerase chain reaction. Microbiol. Immunol. 1996, 40, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Fach, P.; Micheau, P.; Mazuet, C.; Perelle, S.; Popoff, M. Development of real-time PCR tests for detecting botulinum neurotoxins A, B, E, F producing Clostridium botulinum, Clostridium baratii and Clostridium butyricum. J. Appl. Microbiol. 2009, 107, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Krämer, K.M.; Schulze, M.; Pauly, D.; Jacob, D.; Gessler, F.; Nitsche, A.; Dorner, B.G.; Dorner, M.B. Pentaplexed quantitative real-time PCR assay for the simultaneous detection and quantification of botulinum neurotoxin-producing clostridia in food and clinical samples. Appl. Environ. Microbiol. 2010, 76, 4387–4395. [Google Scholar] [CrossRef] [PubMed]
- Boroff, D.A.; Shu-Chen, G. Radioimmunoassay for Type A Toxin of Clostridium botulinum. Appl. Microbiol. 1973, 25, 545–549. [Google Scholar] [PubMed]
- Ashton, A.C.; Crowther, J.S.; Dolly, J.O. A sensitive and useful radioimmunoassay for neurotoxin and its haemagglutinin complex from Clostridium botulinum. Toxicon 1985, 23, 235–246. [Google Scholar] [CrossRef]
- Sonnenschein, B. Use of the reversed passive hemagglutination in detection of Clostridium botulinum type A, B, and E toxin (author’s transl). Zentralbl. Bakteriol. Orig. A 1978, 240, 221–234. [Google Scholar]
- Johnson, H.M.; Brenner, K.; Angelotti, R.; Hall, H.E. Serological Studies of Types A, B, and E Botulinal Toxins by Passive Hemagglutination and Bentonite Flocculation. J. Bacteriol. 1966, 91, 967–974. [Google Scholar] [PubMed]
- Mestrandrea, L.W. Rapid Detection of Clostridium botulinum Toxin by Capillary Tube Diffusion. Appl. Microbiol. 1974, 27, 1017–1022. [Google Scholar] [PubMed]
- Kozaki, S.; Dufrenne, J.; Hagenaars, A.M.; Notermans, S. Enzyme linked immunosorbent assay (ELISA) for detection of Clostridium botulinum type B toxin. Jpn. J. Med. Sci. Biol. 1979, 32, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Notermans, S.; Dufrenne, J.; Kozaki, S. Enzyme-linked immunosorbent assay for detection of Clostridium botulinum type E toxin. Appl. Environ. Microbiol. 1979, 37, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.L. Comparison of amplified ELISA and mouse bioassay procedures for determination of botulinal toxins A, B, E, and F. J. AOAC Int. 2001, 84, 85–88. [Google Scholar] [PubMed]
- Ferreira, J.L.; Eliasberg, S.J.; Harrison, M.A.; Edmonds, P. Detection of preformed type A botulinal toxin in hash brown potatoes by using the mouse bioasssay and a modified ELISA test. J. AOAC Int. 2001, 84, 1460–1464. [Google Scholar] [PubMed]
- Ferreira, J.L.; Maslanka, S.; Johnson, E.; Goodnough, M. Detection of botulinal neurotoxins A, B, E, and F by amplified enzyme-linked immunosorbent assay: Collaborative study. J. AOAC Int. 2003, 86, 314–331. [Google Scholar] [PubMed]
- Ferreira, J.L.; Eliasberg, S.J.; Edmonds, P.; Harrison, M.A. Comparison of the mouse bioassay and enzyme-linked immunosorbent assay procedures for the detection of type A botulinal toxin in food. J. Food Prot. 2004, 67, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Dezfulian, M.; Bartlett, J.G. Selective isolation and rapid identification of Clostridium botulinum types A and B by toxin detection. J. Clin. Microbiol. 1985, 21, 231–233. [Google Scholar] [PubMed]
- Sharma, S.K.; Ferreira, J.L.; Eblen, B.S.; Whiting, R.C. Detection of type A, B, E, and F Clostridium botulinum neurotoxins in foods by using an amplified enzyme-linked immunosorbent assay with digoxigenin-labeled antibodies. Appl. Environ. Microbiol. 2006, 72, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Rivera, V.R.; Neal, D. Development of sensitive colorimetric capture ELISAs for Clostridium botulinum neurotoxin serotypes E and F. Toxicon 2002, 40, 797–802. [Google Scholar] [CrossRef]
- Dezfulian, M.; Hatheway, C.L.; Yolken, R.H.; Bartlett, J.G. Enzyme-linked immunosorbent assay for detection of Clostridium botulinum type A and type B toxins in stool samples of infants with botulism. J. Clin. Microbiol. 1984, 20, 379–383. [Google Scholar] [PubMed]
- Smith, T.J.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Tsai, R.; LaPorte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; et al. Sequence Variation within Botulinum Neurotoxin Serotypes Impacts Antibody Binding and Neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar] [CrossRef] [PubMed]
- Stanker, L.H.; Merrill, P.; Scotcher, M.C.; Cheng, L.W. Development and partial characterization of high-affinity monoclonal antibodies for botulinum toxin type A and their use in analysis of milk by sandwich ELISA. J. Immunol. Methods 2008, 336, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, C.; Li, X.; Chen, S. A Simple, Rapid and Sensitive FRET Assay for Botulinum Neurotoxin Serotype B Detection. PLoS ONE 2014, 9, e114124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poras, H.; Ouimet, T.; Orng, S.-V.; Fournié-Zaluski, M.-C.; Popoff, M.R.; Roques, B.P. Detection and Quantification of Botulinum Neurotoxin Type A by a Novel Rapid In Vitro Fluorimetric Assay. Appl. Environ. Microbiol. 2009, 75, 4382–4390. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Stanker, L.H.; Carter, J.M.; Do, P.M.; Cheng, L.W.; He, X.; Brandon, D.L. Detection of botulinum neurotoxin-A activity in food by peptide cleavage assay. Int. J. Food Microbiol. 2008, 126, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Hallis, B.; James, B.A.; Shone, C.C. Development of novel assays for botulinum type A and B neurotoxins based on their endopeptidase activities. J. Clin. Microbiol. 1996, 34, 1934–1938. [Google Scholar] [PubMed]
- Barr, J.R.; Moura, H.; Boyer, A.E.; Woolfitt, A.R.; Kalb, S.R.; Pavlopoulos, A.; McWilliams, L.G.; Schmidt, J.G.; Martinez, R.A.; Ashley, D.L. Botulinum Neurotoxin Detection and Differentiation by Mass Spectrometry. Emerg. Infect. Dis. 2005, 11, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Krilich, J.C.; Dykes, J.K.; Lúquez, C.; Maslanka, S.E.; Barr, J.R. Detection of Botulinum Toxins A, B, E, and F in Foods by Endopep-MS. J. Agric. Food Chem. 2015, 63, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Feldberg, L.; Gura, S.; Brosh-Nissimov, T.; Guri, A.; Zimhony, O.; Shapiro, E.; Beth-Din, A.; Stein, D.; Ozeri, E.; et al. Early, Real-Time Medical Diagnosis of Botulism by Endopeptidase-Mass Spectrometry. Clin. Infect. Dis. 2015, 61, e58–e61. [Google Scholar] [CrossRef] [PubMed]
- Van Baar, B.L.M.; Hulst, A.G.; de Jong, A.L.; Wils, E.R.J. Characterisation of botulinum toxins type A and B, by matrix-assisted laser desorption ionisation and electrospray mass spectrometry. J. Chromatogr. A 2002, 970, 95–115. [Google Scholar] [CrossRef]
- Van Baar, B.L.M.; Hulst, A.G.; de Jong, A.L.; Wils, E.R.J. Characterisation of botulinum toxins type C, D, E, and F by matrix-assisted laser desorption ionisation and electrospray mass spectrometry. J. Chromatogr. A 2004, 1035, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Hines, H.B.; Lebeda, F.; Hale, M.; Brueggemann, E.E. Characterization of Botulinum Progenitor Toxins by Mass Spectrometry. Appl. Environ. Microbiol. 2005, 71, 4478–4486. [Google Scholar] [CrossRef] [PubMed]
- Rasetti-Escargueil, C.; Liu, Y.; Rigsby, P.; Jones, R.G.A.; Sesardic, D. Phrenic nerve-hemidiaphragm as a highly sensitive replacement assay for determination of functional botulinum toxin antibodies. Toxicon 2011, 57, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, H.; Rummel, A. Botulinum Neurotoxins: Qualitative and Quantitative Analysis Using the Mouse Phrenic Nerve Hemidiaphragm Assay (MPN). Toxins 2015, 7, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.R.A.; Moreira, G.M.S.G.; da Cunha, C.E.P.; Mendonça, M.; Salvarani, F.M.; Moreira, Â.N.; Conceição, F.R. Recombinant Alpha, Beta, and Epsilon Toxins of Clostridium perfringens: Production Strategies and Applications as Veterinary Vaccines. Toxins 2016, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Popoff, M.R. Clostridium perfringens: Toxinotype and genotype. Trends Microbiol. 1999, 7, 104–110. [Google Scholar] [CrossRef]
- Sakurai, J.; Nagahama, M.; Oda, M. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Halldórsson, H.; Andrésson, O.S. Clostridium perfringens beta-toxin forms multimeric transmembrane pores in human endothelial cells. Microb. Pathog. 2000, 28, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.G.; Barth, G.; Barth, H.; Popoff, M.R. Clostridium perfringens Epsilon Toxin: A Malevolent Molecule for Animals and Man? Toxins 2013, 5, 2138–2160. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Oda, M.; Tsuge, H.; Kobayashi, K. Clostridium perfringens Iota-Toxin: Structure and Function. Toxins 2009, 1, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Farzan, A.; Kircanski, J.; DeLay, J.; Soltes, G.; Songer, J.G.; Friendship, R.; Prescott, J.F. An investigation into the association between cpb2-encoding Clostridium perfringens type A and diarrhea in neonatal piglets. Can. J. Vet. Res. 2013, 77, 45–53. [Google Scholar] [PubMed]
- Hamad, M.A.; Habra, N.; Allouz, A.K. Biotyping of Clostridium perfringens strains isolated from enterotoxemia cases in sheep using ELISA technique. Iraqi J. Vet. Sci. 2010, 24, 17–22. [Google Scholar]
- Tansuphasiri, U. Development of duplex PCR assay for rapid detection of enterotoxigenic isolates of Clostridium perfringens. Southeast Asian J. Trop. Med. Public Health 2001, 32, 105–113. [Google Scholar] [PubMed]
- Kull, S.; Pauly, D.; Störmann, B.; Kirchner, S.; Stämmler, M.; Dorner, M.B.; Lasch, P.; Naumann, D.; Dorner, B.G. Multiplex detection of microbial and plant toxins by immunoaffinity enrichment and matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2010, 82, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.I.; Kumar, B.; Kamboj, D.V. Multiplex detection of protein toxins using MALDI-TOF-TOF tandem mass spectrometry: Application in unambiguous toxin detection from bioaerosol. Anal. Chem. 2012, 84, 10500–10507. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed]
- Brodzik, C.; Augustynowicz, E.; Korzeniowska-Kowal, A.; Lutyhska, A. Application of the MALDI-TOF for identification of Clostridium perfringens strains. Medycyna Doswiadczalna I Mikrobiologia 2016, 68, 13–21. [Google Scholar] [PubMed]
- Krt, B. Development and evaluation of various enzyme-linked immunosorbent assays for the detection of Clostridium perfringens beta anti-toxins. FEMS Immunol. Med. Microbiol. 1999, 24, 293–297. [Google Scholar] [PubMed]
- Baums, C.G.; Schotte, U.; Amtsberg, G.; Goethe, R. Diagnostic multiplex PCR for toxin genotyping of Clostridium perfringens isolates. Vet. Microbiol. 2004, 100, 11–16. [Google Scholar] [CrossRef]
- Albini, S.; Brodard, I.; Jaussi, A.; Wollschlaeger, N.; Frey, J.; Miserez, R.; Abril, C. Real-time multiplex PCR assays for reliable detection of Clostridium perfringens toxin genes in animal isolates. Vet. Microbiol. 2008, 127, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.; López-Enríquez, L.; Rodríguez-Lázaro, D. Quantitative Detection of Clostridium perfringens by Real-Time PCR in Raw Milk. Food Anal. Methods 2017, 10, 1139–1147. [Google Scholar] [CrossRef]
- McClane, B.A.; Strouse, R.J. Rapid detection of Clostridium perfringens type A enterotoxin by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1984, 19, 112–115. [Google Scholar] [PubMed]
- Nagahama, M.; Kobayashi, K.; Ochi, S.; Sakurai, J. Enzyme-linked immunosorbent assay for rapid detection of toxins from Clostridium perfringens. FEMS Microbiol. Lett. 1991, 84, 41–44. [Google Scholar] [CrossRef]
- Everley, R.A.; Mott, T.M.; Toney, D.M.; Croley, T.R. Characterization of Clostridium species utilizing liquid chromatography/mass spectrometry of intact proteins. J. Microbiol. Methods 2009, 77, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.D.; Martin, P.K.; Sharpe, R.T. Detection of Clostridium perfringens epsilon toxin by ELISA. Res. Vet. Sci. 1987, 42, 255–256. [Google Scholar] [PubMed]
- Layana, J.E.; Fernandez Miyakawa, M.E.; Uzal, F.A. Evaluation of different fluids for detection of Clostridium perfringens type D epsilon toxin in sheep with experimental enterotoxemia. Anaerobe 2006, 12, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Plumb, J.J.; Blackall, L.L.; Kelly, W.R. PCR detection of Clostridium perfringens producing different toxins in faeces of goats. Lett. Appl. Microbiol. 1997, 25, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Seyer, A.; Fenaille, F.; Féraudet-Tarisse, C.; Volland, H.; Popoff, M.R.; Tabet, J.-C.; Junot, C.; Becher, F. Rapid quantification of clostridial epsilon toxin in complex food and biological matrixes by immunopurification and ultraperformance liquid chromatography-tandem mass spectrometry. Anal. Chem. 2012, 84, 5103–5109. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.W.; Williamson, E.D.; Havard, H.; Modi, N.; Brown, J. Evaluation of a new cytotoxicity assay for Clostridium perfringens type D epsilon toxin. FEMS Microbiol. Lett. 1994, 116, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.-Å.; Artursson, E.; Bergström, T.; Östin, A.; Nilsson, C.; Åstot, C. Identification of RIP-II toxins by affinity enrichment, enzymatic digestion and LC-MS. Anal. Chem. 2015, 87, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.L. Microtiter-based assay for evaluating the biological activity of ribosome-inactivating proteins. Pharmacol. Toxicol. 2001, 88, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Becher, F.; Duriez, E.; Volland, H.; Tabet, J.C.; Ezan, E. Detection of Functional Ricin by Immunoaffinity and Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Worbs, S.; Kirchner, S.; Shatohina, O.; Dorner, M.B.; Dorner, B.G. Real-time cytotoxicity assay for rapid and sensitive detection of ricin from complex matrices. PLoS ONE 2012, 7, e35360. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Rivera, V.R.; Hewetson, J.F.; Merrill, G.A. Detection of ricin by colorimetric and chemiluminescence ELISA. Toxicon 1994, 32, 1371–1377. [Google Scholar] [CrossRef]
- Cook, D.L.; David, J.; Griffiths, G.D. Retrospective identification of ricin in animal tissues following administration by pulmonary and oral routes. Toxicology 2006, 223, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Worbs, S.; Köhler, K.; Pauly, D.; Avondet, M.-A.; Schaer, M.; Dorner, M.B.; Dorner, B.G. Ricinus communis Intoxications in Human and Veterinary Medicine-A Summary of Real Cases. Toxins 2011, 3, 1332–1372. [Google Scholar] [CrossRef] [PubMed]
- Bozza, W.P.; Tolleson, W.H.; Rivera Rosado, L.A.; Zhang, B. Ricin detection: Tracking active toxin. Biotechnol. Adv. 2015, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Assiri, A.S. Ricin poisoning causing death after ingestion of herbal medicine. Ann. Saudi Med. 2012, 32, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Beyer, N.H.; Kogutowska, E.; Hansen, J.J.; Engelhart Illigen, K.E.; Heegaard, N.H.H. A mouse model for ricin poisoning and for evaluating protective effects of antiricin antibodies. Clin. Toxicol. 2009, 47, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Baudys, J.; Barr, J.R.; Kalb, S.R. Improved Sensitivity for the Qualitative and Quantitative Analysis of Active Ricin by MALDI-TOF Mass Spectrometry. Anal. Chem. 2016, 88, 6867–6872. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.B.; Schramm, V.L. Detecting Ricin: Sensitive Luminescent Assay for Ricin A-Chain Ribosome Depurination Kinetics. Anal. Chem. 2009, 81, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Barr, J.R. Mass spectrometric detection of ricin and its activity in food and clinical samples. Anal. Chem. 2009, 81, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.D. Understanding Ricin from a Defensive Viewpoint. Toxins 2011, 3, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Lamb, F.I.; Pappin, D.J.; Lord, J.M. The primary sequence of Ricinus communis agglutinin. Comparison with ricin. J. Biol. Chem. 1985, 260, 15682–15686. [Google Scholar] [PubMed]
- Kumar, O.; Pradhan, S.; Sehgal, P.; Singh, Y.; Vijayaraghavan, R. Denatured ricin can be detected as native ricin by immunological methods, but nontoxic in vivo. J. Forensic Sci. 2010, 55, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Mouser, P.; Filigenzi, M.S.; Puschner, B.; Johnson, V.; Miller, M.A.; Hooser, S.B. Fatal ricin toxicosis in a puppy confirmed by liquid chromatography/mass spectrometry when using ricinine as a marker. J. Vet. Diagn. Investig. 2007, 19, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hines, H.B.; Brueggemann, E.E.; Hale, M.L. High-performance liquid chromatography-mass selective detection assay for adenine released from a synthetic RNA substrate by ricin A chain. Anal. Biochem. 2004, 330, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, V.L.H.; Nilles, J.M.; Rice, J.S.; Connell, T.R.; Schenning, A.M.; Reilly, L.M.; Durst, H.D. Ricin activity assay by direct analysis in real time mass spectrometry detection of adenine release. Anal. Chem. 2010, 82, 798–800. [Google Scholar] [CrossRef] [PubMed]
- McGrath, S.C.; Schieltz, D.M.; McWilliams, L.G.; Pirkle, J.L.; Barr, J.R. Detection and Quantification of Ricin in Beverages Using Isotope Dilution Tandem Mass Spectrometry. Anal. Chem. 2011, 83, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Schieltz, D.M.; McGrath, S.C.; McWilliams, L.G.; Rees, J.; Bowen, M.D.; Kools, J.J.; Dauphin, L.A.; Gomez-Saladin, E.; Newton, B.N.; Stang, H.L.; et al. Analysis of active ricin and castor bean proteins in a ricin preparation, castor bean extract, and surface swabs from a public health investigation. Forensic Sci. Int. 2011, 209, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Felder, E.; Mossbrugger, I.; Lange, M.; Wölfel, R. Simultaneous detection of ricin and abrin DNA by real-time PCR (qPCR). Toxins 2012, 4, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Kirchner, S.; Stoermann, B.; Schreiber, T.; Kaulfuss, S.; Schade, R.; Zbinden, R.; Avondet, M.-A.; Dorner, M.B.; Dorner, B.G. Simultaneous quantification of five bacterial and plant toxins from complex matrices using a multiplexed fluorescent magnetic suspension assay. Analyst 2009, 134, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E.; Venkateswaran, K.V.; O’Brien, T.W. Simultaneous multiplex detection and confirmation of the proteinaceous toxins abrin, ricin, botulinum toxins, and Staphylococcus enterotoxins A, B, and C in food. J. Agric. Food Chem. 2010, 58, 6600–6607. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Maiti, T.K.; Podder, S.K. Purification and characterization of three toxins and two agglutinins from Abrus precatorius seed by using lactamyl-Sepharose affinity chromatography. Anal. Biochem. 1991, 194, 101–109. [Google Scholar] [CrossRef]
- Hung, C.H.; Lee, M.C.; Lee, T.C.; Lin, J.Y. Primary structure of three distinct isoabrins determined by cDNA sequencing. Conservation and significance. J. Mol. Biol. 1993, 229, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Podder, S.K. A- and B-subunit variant distribution in the holoprotein variants of protein toxin abrin: Variants of abrins I and III have constant toxic A subunits and variant lectin B subunits. Arch. Biochem. Biophys. 1997, 344, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tsai, C.C.; Lin, S.C.; Wang, L.I.; Hsu, C.I.; Hwang, M.J.; Lin, J.Y. Primary structure and function analysis of the Abrus precatorius agglutinin A chain by site-directed mutagenesis. Pro(199) of amphiphilic alpha-helix H impairs protein synthesis inhibitory activity. J. Biol. Chem. 2000, 275, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Bagaria, A.; Surendranath, K.; Ramagopal, U.A.; Ramakumar, S.; Karande, A.A. Structure-function analysis and insights into the reduced toxicity of Abrus precatorius agglutinin I in relation to abrin. J. Biol. Chem. 2006, 281, 34465–34474. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E. Toxicity and detection of ricin and abrin in beverages. J. Food Prot. 2008, 71, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E.; Walker, J.L.; O’Brien, T.W. Detection of abrin in food using enzyme-linked immunosorbent assay and electrochemiluminescence technologies. J. Food Prot. 2008, 71, 1868–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, X.-L.; Li, Y.-S.; Pan, F.-G.; Zhang, Y.-Y.; Zhang, J.-H.; Wang, X.-R.; Ren, H.-L.; Lu, S.-Y.; Li, Z.-H.; et al. Development of a monoclonal antibody-based sandwich-type enzyme-linked immunosorbent assay (ELISA) for detection of abrin in food samples. Food Chem. 2012, 135, 2661–2665. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Nie, C.; Wang, J.; Wang, J.; Kang, L.; Zhou, Y.; Wang, J.-L. Colloidal gold-based immunochromatographic test strip for rapid detection of abrin in food samples. J. Food Prot. 2012, 75, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, X.; Liu, G.; Zhang, B.; Zhang, Y.; Kong, T.; Tang, J.; Li, D.; Wang, Z. A colloidal gold probe-based silver enhancement immunochromatographic assay for the rapid detection of abrin-a. Biosens. Bioelectron. 2011, 26, 3710–3713. [Google Scholar] [CrossRef] [PubMed]
- Ramage, J.G.; Prentice, K.W.; Morse, S.A.; Carter, A.J.; Datta, S.; Drumgoole, R.; Gargis, S.R.; Griffin-Thomas, L.; Hastings, R.; Masri, H.P.; et al. Comprehensive laboratory evaluation of a specific lateral flow assay for the presumptive identification of abrin in suspicious white powders and environmental samples. Biosecur. Bioterror. Biodef. Strategy Pract. Sci. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.; Koester, C. Quantitation of Abrine, an Indole Alkaloid Marker of the Toxic Glycoproteins Abrin, by Liquid Chromatography/Tandem Mass Spectrometry When Spiked into Various Beverages. J. Agric. Food Chem. 2008, 56, 11139–11143. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Stirpe, F.; Sandvig, K.; Pihl, A. Isolation and characterization of viscumin, a toxic lectin from Viscum album L. (mistletoe). J. Biol. Chem. 1982, 257, 13263–13270. [Google Scholar] [PubMed]
- Rubina, A.Y.; Dyukova, V.I.; Dementieva, E.I.; Stomakhin, A.A.; Nesmeyanov, V.A.; Grishin, E.V.; Zasedatelev, A.S. Quantitative immunoassay of biotoxins on hydrogel-based protein microchips. Anal. Biochem. 2005, 340, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Jäggy, C.; Musielski, H.; Urech, K.; Schaller, G. Quantitative determination of lectins in mistletoe preparations. Arzneimittel-Forschung 1995, 45, 905–909. [Google Scholar] [PubMed]
- Layer, R.T.; McIntosh, J.M. Conotoxins: Therapeutic Potential and Application. Mar. Drugs 2006, 4, 119–142. [Google Scholar] [CrossRef]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Imperial, J.S.; Concepcion, G.P. Snail Peptides. In Handbook of Biologically Active Peptides, 2nd ed.; Kastin, A.J., Ed.; Academic Press: Boston, MA, USA, 2013; Chapter 61; pp. 437–450. ISBN 978-0-12-385095-9. [Google Scholar]
- Ashcom, J.D.; Stiles, B.G. Characterization of alpha-conotoxin interactions with the nicotinic acetylcholine receptor and monoclonal antibodies. Biochem. J. 1997, 328 Pt 1, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated alpha-conotoxins from the cone snail Conus anemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Prasarnpun, S.; Walsh, J.; Awad, S.S.; Harris, J.B. Envenoming bites by kraits: The biological basis of treatment-resistant neuromuscular paralysis. Brain 2005, 128, 2987–2996. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.-L.; Gao, J.-F.; Zhang, Y.-X.; Shen, S.-S.; He, Y.; Wang, J.; Ma, X.-M.; Ji, X. Proteomic characterization and comparison of venoms from two elapid snakes (Bungarus multicinctus and Naja atra) from China. J. Proteom. 2016, 138, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Yang, C.C. Separation and Characterization of the A Chain and B Chain in β1-Bungarotoxin from Bungarus Multicinctus (Taiwan Banded Krait) Venom. J. Protein Chem. 1993, 12, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Selvanayagam, Z.E.; Gopalakrishnakone, P. Tests for detection of snake venoms, toxins and venom antibodies: Review on recent trends (1987–1997). Toxicon 1999, 37, 565–586. [Google Scholar] [CrossRef]
- Dong, L.V.; Selvanayagam, Z.E.; Gopalakrishnakone, P.; Eng, K.H. A new avidin–biotin optical immunoassay for the detection of beta-bungarotoxin and application in diagnosis of experimental snake envenomation. J. Immunol. Methods 2002, 260, 125–136. [Google Scholar] [CrossRef]
- Selvanayagam, Z.E.; Neuzil, P.; Gopalakrishnakone, P.; Sridhar, U.; Singh, M.; Ho, L.C. An ISFET-based immunosensor for the detection of β-Bungarotoxin. Biosens. Bioelectron. 2002, 17, 821–826. [Google Scholar] [CrossRef]
Figure 1.
Summary of liquid chromatography–mass spectrometry (LC-MS)-based methods in protein-based toxins analysis: (a) protein of interest (red) in complex matrix of other proteins and small molecules; (b) SDS-PAGE separation; (c) immunoaffinity purification; (d) enzymatic digest; (e) data-dependent analysis: LC-MS/MS analysis of all peptides in the mixture, all measured MS/MS spectra are searched against protein database, and in the case of a spectral match, the presence of the agent is confirmed, regardless of its quantity. (f) In a targeted (data independent) MS/MS experiment, peptides of interest are fragmented repeatedly during their elution times, and (g) extracted ion chromatograms of selected fragment ions are then used for protein quantification (exported from Skyline, unpublished results).
Figure 1.
Summary of liquid chromatography–mass spectrometry (LC-MS)-based methods in protein-based toxins analysis: (a) protein of interest (red) in complex matrix of other proteins and small molecules; (b) SDS-PAGE separation; (c) immunoaffinity purification; (d) enzymatic digest; (e) data-dependent analysis: LC-MS/MS analysis of all peptides in the mixture, all measured MS/MS spectra are searched against protein database, and in the case of a spectral match, the presence of the agent is confirmed, regardless of its quantity. (f) In a targeted (data independent) MS/MS experiment, peptides of interest are fragmented repeatedly during their elution times, and (g) extracted ion chromatograms of selected fragment ions are then used for protein quantification (exported from Skyline, unpublished results).


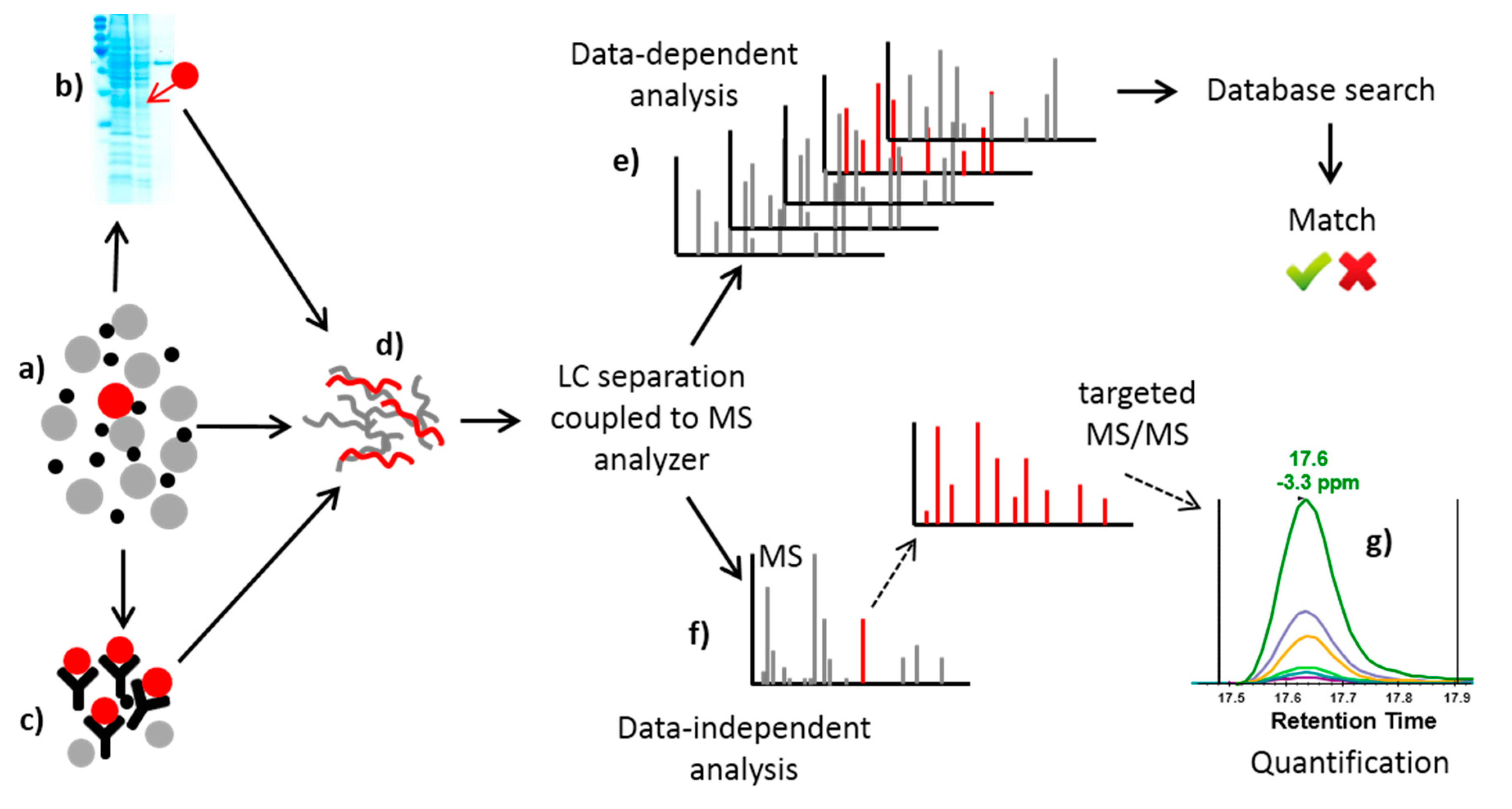{kind=link}
Table 1.
Summary of methods relevant to the detection of SEs.
| Target Protein | Method of Detection | References |
|---|---|---|
| SEs | Kitten emesis test | [53] |
| Reversed passive latex agglutination kit | [70,71,72,73] | |
| Enzyme-linked immunosorbent assay | [58,59] | |
| SEA | Double-antibody solid-phase enzyme immunoassay | [56] |
| Enzyme-linked immunosorbent assay | [60] | |
| SEB | Direct skin test | [54] |
| Single-gel diffusion test | [74] | |
| Latex agglutination test | [75] | |
| Latex agglutination inhibition test | [76] | |
| Enzyme-linked immunosorbent assay | [57,62] | |
| Fluorescence-based immunoassay | [63] | |
| Immunoreactor-based competitive fluoroimmunoassay | [65] | |
| ELISA-Lab-on-a-chip | [67] | |
| Surface plasmon resonance | [68] | |
| Electrochemical immunoassay using enzyme-nanosilica-doped carbon nanotubes for signal amplification | [69] | |
| SEA, SEB | Double-gel diffusion assay | [77] |
| Avidin-biotin ELISA | [61] | |
| SEC | Fluoroimmunoassay based on functionalized fluorescent core-shell nanoparticle labels | [64] |
| Staphylococcal enterotoxin genes | Polymerase chain reaction | [12] |
Table 2.
Mass spectrometry methods for detection of SEs in various matrices.
| Toxin | Matrix | Detection (QqQ) | Standards (PSAQ) | LOD | LOQ * | Reference |
|---|---|---|---|---|---|---|
| SEB | Apple juice | (QTOF) | - | 60 ng/mL | N/A | [81] |
| SEA | Chinese dessert (Coco-pearls) | + | + | N/A | N/A | [42] |
| SEB | Chicken meat | (QIT) | - | N/A | N/A | [82] |
| SEA | Milk | + | + | SEA: 2.5 ng/g | Milk: 2.5 ppb | [83] |
| SEB | Shrimp | SEB: 10 ng/g | Shrimp: 5 ppm | |||
| SEA | Milk | + | + | SEA: 4 ng/g | N/A | [84] |
| SEB | SEB: 8 ng/g | |||||
| SEA | Soup | + | + | SEA: 78 ng/mL | N/A | [85] |
| SEB | SEB: 141 ng/mL | |||||
| SED | SED: 48 ng/mL |
Abbreviation: QIT: Quadrupole ion trap. QTOF: Quadrupole Time-of-Flight. N/A: not available. PSAQ Protein standard absolute quantification (isotopically labeled standard). * LOQ (limit of quantification) determined by applying FDA guidelines for bioanalytical method validation, i.e., accuracy between 80% and 120%, precision 20% and signal-to-noise ratio >5. When accuracy was <80% or >120% or precision was >20%, LLOQ (lower LOQ) was determined based on a signal-to-noise ratio >10. (+): detected, (-): not detected by QqQ or PSAQ.
Table 3.
Summary of methods relevant to detection of B. anthracis.
| Target of Method | Method of Detection | Reference |
|---|---|---|
| Gene | RT-PCR-F | [91] |
| Spores | Light transmission | [92] |
| Fluorescence | [93] | |
| MS | [94] | |
| SERS | [95] | |
| Bead-based sandwich immunoassay | [96] | |
| FRET-flow cytometry | [97] | |
| RT-PCR-F | [98] | |
| ELISA flow cytometry | [99] | |
| Flow cytometry | [100] | |
| ECL | [101] | |
| Immunoradiometric assay | [102] | |
| Protective antigen | Fluorescence | [103] |
| ENIA | [103] | |
| AM | [104] | |
| AFM | [105] | |
| Antibody microarray | [106] | |
| ELISA | [107] | |
| DNA | RT-PCR-F | [108] |
| PCR | [109] | |
| Fluorescence | [110] | |
| Lethal factor | MS | [111] |
| Antibody microarray | [106] |
Abbreviations: amperometry (AM); atomic force microscopy (AFM); enzyme linked immunosorbent assay (ELISA); electrochemiluminescence (ECL); europium nanoparticle-based immunoassay (ENIA); fluorescence resonance energy transfer (FRET); real-time polymerase chain reaction and fluorescence (RT-PCR-F); surface enhanced Raman scattering (SERS).
Table 4.
Summary of methods relevant to characterization of BoNT.
| Target of Method | Technique | Reference |
|---|---|---|
| Botulinum toxin activity | Mouse lethality assay | [119,120] |
| Rat compound muscle action potentials test | [123] | |
| Spores | Polymerase chain reaction | [124] |
| Genes | Polymerase chain reaction | [125,126,127,128] |
| BoNT | Radioimmunoassay | [129,130] |
| Passive hemagglutination | [131,132] | |
| Gel diffusion assay | [133] | |
| Enzyme-linked immune sorbent assay | [136,137,138,139,140,141,142,143,144,145] | |
| Fluorescence resonance energy transfer technology | [146] | |
| In vitro fluorimetric assay | [147] | |
| Endopeptidase assay | [149] | |
| Endopeptidase-MS assay | [150,151,152] | |
| Mass spectrometry | [116,153,154,155] |
Table 5.
Diversity of C. perfringens toxinotypes and associated diseases.
| Toxinotype | Major Toxins | Minor Toxins | Associated Disease | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α | β | ε | ι | CPE | λ | θ | δ | Humans | Animals | |
| A | ++ | − | − | − | + | − | + | − | Gangrene, GI diseases | Diarrhea (foals, pig) NE in fowl |
| B | + | + | + | − | − | + | − | + | NE | Dysentery in newborn lambs Hemorrhagic enteritis in neonatal calves and foals Enterotoxemia in sheep |
| C | + | + | − | − | + | − | − | + | NE in piglets Enterotoxemia in sheep | |
| D | + | − | + | − | + | + | − | − | Enterotoxemia in lambs, sheep, calves and goats | |
| E | + | − | − | + | + | + | − | − | Enterotoxemia in calves | |
Abbreviation: GI: gastrointestinal, NE: necrotic enteritis. –: no production of toxin, +: production of toxins, ++: strong production of toxins.
Table 6.
Summarization of methods relevant to characterization of Clostridium perfringens and its toxins.
Table 6.
Summarization of methods relevant to characterization of Clostridium perfringens and its toxins.
| Target of Method | Technique | Reference |
|---|---|---|
| C. perfringens strains | ELISA | [165] |
| MS | [170,177] | |
| Gene | PCR | [166,170,172,173,174] |
| Toxins | MS | [167,168,181] |
| EIA | [171,175,176] | |
| ELISA | [178,179] | |
| PCR | [180] | |
| Clostridium toxin activity | Cytotoxicity assay | [182] |
Table 7.
Summary of methods relevant to characterization of selected plant protein toxins.
| Target of Method | Technique | Reference |
|---|---|---|
| RIPs | MS | [183] |
| Multiplex fluorescent magnetic suspension assay | [186] | |
| ELISA | [212] | |
| Hydrogen-based protein microchips | [220] | |
| Biological activity of RIP toxins | Microtiter-based assay (luciferase luminescence test) | [184] |
| Ricin | MS | [85,185,193,195,200,201,202,203] |
| ELISA | [187,188,198] | |
| Mouse model | [192] | |
| Luminescent assay | [194] | |
| Chemiluminescence-based microarray immunoassay | [66] | |
| Ricinine | MS | [199] |
| Genes (abrin) | PCR | [204] |
| Abrin | ELISA | [213,214] |
| Immunochromatographic assay | [215,216] | |
| MS | [218] | |
| Viscumin | Enzyme-linked lectin assay | [221] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Duracova, M.; Klimentova, J.; Fucikova, A.; Dresler, J. Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins 2018, 10, 99. https://doi.org/10.3390/toxins10030099
AMA Style
Duracova M, Klimentova J, Fucikova A, Dresler J. Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins. 2018; 10(3):99. https://doi.org/10.3390/toxins10030099
Chicago/Turabian StyleDuracova, Miloslava, Jana Klimentova, Alena Fucikova, and Jiri Dresler. 2018. "Proteomic Methods of Detection and Quantification of Protein Toxins" Toxins 10, no. 3: 99. https://doi.org/10.3390/toxins10030099
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.