
Attaching Onto or Inserting Into an Intramolecular Hydrogen Bond: Exploring and Controlling a Chirality-Dependent Dilemma for Alcohols



Institut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammannstr. 6, 37077 Gottingen, Germany
*
Author to whom correspondence should be addressed.
Symmetry 2022, 14(2), 357; https://doi.org/10.3390/sym14020357
Submission received: 17 December 2021
/
Revised: 8 January 2022
/
Accepted: 31 January 2022
/
Published: 11 February 2022
(This article belongs to the Special Issue Asymmetric Molecules and Chirality Recognition)
Abstract
:Prereactive complexes in noncovalent organocatalysis are sensitive to the relative chirality of the binding partners and to hydrogen bond isomerism. Both effects are present when a transiently chiral alcohol docks on a chiral -hydroxy ester, turning such 1:1 complexes into elementary, non-reactive model systems for chirality induction in the gas phase. With the help of linear infrared and Raman spectroscopy in supersonic jet expansions, conformational preferences are investigated for benzyl alcohol in combination with methyl lactate, also exploring p-chlorination of the alcohol and the achiral homolog methyl glycolate to identify potential London dispersion and chirality effects on the energy sequence. Three of the four combinations prefer barrierless complexation via the hydroxy group of the ester (association). In contrast, the lightest complex predominantly shows insertion into the intramolecular hydrogen bond, such as the analogous lactate and glycolate complexes of methanol. The experimental findings are rationalized with computations, and a uniform helicality induction in the alcohol by the lactate is predicted, independent of insertion into or association with the internal lactate hydrogen bond. p-chlorination of benzyl alcohol has a stabilizing effect on association because the insertion motif prevents a close contact between the chlorine and the hydroxy ester. After simple anharmonicity and substitution corrections, the B3LYP-D3 approach offers a fairly systematic description of the known spectroscopic data on alcohol complexes with -hydroxy esters.
1. Introduction
Hydrogen bonds between oxygen atoms involve strong polarization of the participating chemical bonds, and therefore it matters how the :OH group of an added alcohol interacts with such a preformed :OH:O: unit. Solvation of the donor :OH (:OH:OH:O:) is typically more favorable than additional solvation of the bivalent acceptor :O: from the other side (:OH:O:HO:) because the former can be more cooperative [1,2]. In this sense, cooperativity wins over symmetric solvation of the two lone electron pairs of the acceptor oxygen. Insertion into the :OH:O: unit (:OH:OH:O:) is equivalent to donor solvation, if the two :OH groups are chemically identical and independent of each other. If, however, the preformed :OH:O: hydrogen bond is intramolecular and strained, insertion can be more attractive than donor solvation due to the release of the intramolecular strain in the enlarged hydrogen bonded cycle. On the other hand, insertion of the alcohol may be kinetically hindered because it involves a hydrogen bond-opening barrier, in contrast to more or less barrierless attachment to the :OH:O: unit on its free ends, which has been termed addition [3] or association [4]. Therefore, kinetic and thermodynamic control of the insertion/association ratio in hydrogen-bonded complexes is of considerable interest. The preference is not only controlled by the hydrogen bond energetics at the core [5] but also by remote interactions of the organic groups attached to the three oxygen atoms. Hence, it may serve as a sensitive probe for London dispersion [6] and other interactions between these groups [7,8]. The latter can tip the balance between insertion and association in favorable cases. If the preformed unit is chiral, it may induce a preferred chirality in the docking alcohol, even if that alcohol is on average achiral. This asymmetric or chirality induction is of key importance in organic reactions, and prereactive complexes may play a relevant mechanistic role. Hence, a study of complexes between chiral molecules with intramolecular hydrogen bonds and achiral alcohols in supersonic jet expansion addresses several questions. Can the energy gap between insertion and association be tuned by chemical substitution? Does thermodynamic preference win over kinetic control? Is there an effect of chirality? These questions are schematically summarized in Figure 1.
In the present study, we focus on all three aspects by combining benzyl alcohol [9] and 4-chlorobenzyl alcohol [10] with achiral methyl glycolate [11,12] and chiral methyl lactate [13,14] as hydroxy esters. The monofunctionality of the alcohol minimizes other aggregation topologies [15] than insertion and association. We compare the results to previous studies of other alcohols [3,16] and phenol [17] in complexes with these two hydroxy esters. Where the donating OH group is attached to an aromatic ring, size- and conformationally selective vibrational techniques are possible [16,17]. In the present work, like in the methanol study [3], the main tool is FTIR jet spectroscopy, but important additional insights are obtained from Raman jet spectroscopy because cooperative hydrogen bonds may exhibit different intensity patterns for the hydride stretching fundamentals in these complementary techniques. Although conformationally selective stimulated Raman spectroscopy of selected (typically aromatic) non-volatile molecules is starting to become accessible [18,19], we explore the more general spontaneous Raman scattering variant [20], which also allows for limited volatility of the molecular ingredients.
After describing the experimental and computational methods, we present the experimental spectra obtained for binary supersonic expansions in combination with uniformly scaled harmonic DFT predictions for the most stable cold 1:1 complexes between the alcohol and the hydroxy ester. This leads to largely unambiguous assignments of the topologies realized in the 1:1 complexes and to a rough quantification of their abundance. From the latter, we draw conclusions about the insertion/association preference as a function of chemical substitution and we analyze the possible effect of chirality induction in the lactate complexes by comparison to the achiral glycolate counterparts. The results might also help to rationalize chirality discrimination involving permanently chiral alcohols [16] and they fill a gap in the vibrational characterization of chirality recognition phenomena in the gas phase [21], with implications for solution phenomena [22,23].
2. Materials and Methods
2.1. Experimental Methods
Gas mixtures of the volatile hydroxy esters (methyl (S)-(−)-lactate (abbreviated L, TCI, Eschborn, Germany, >98% and Sigma-Aldrich, Taufkirchen, Germany, 98%); methyl glycolate (G, Alfa Aesar, Kandel, Germany, 98% and Sigma-Aldrich, 98%)) and the less volatile aromatic alcohols (benzyl alcohol (B, Alfa Aesar, 99% and Sigma-Aldrich, 99%); 4-chlorobenzyl alcohol (C, Sigma-Aldrich, 98%)) in a large excess of helium (Linde, Pullach, Germany, 99.996%) were prepared by first mixing in the ester and then flowing the gas through a heated bed of the aromatic alcohol. This was done differently for subsequent FTIR spectroscopy and Raman spectroscopy, but in both cases the gas mixture was expanded through a slit nozzle into a vacuum chamber, where the expansion was crossed by photons and their absorption (Bruker IFS 66v/S FTIR, ceramic glower) or inelastic scattering (Raman, 25 W cw 532 nm Millenia laser) was detected (L-N2 cooled Judson InSb detector in the IR and L-N2-cooled Pylon CCD camera for Raman).
In the FTIR case, helium was guided through a coolable saturator, where it picked up hydroxy ester molecules, into a 69 L reservoir at 1.5 bar. From there, the mixture was led through a channel, in which the aromatic alcohol, deposited on molecular sieve, was placed between two poppet valves. From there, it was expanded through a heatable V-shaped 60 mm × 0.2 mm slit nozzle. To keep the background pressure low, the expansion chamber was connected to a buffer volume (3.6 m3), which was continuously evacuated by three vacuum pumps in sequence (500 m3/h total pumping speed). The FTIR spectrum was recorded by a Bruker IFS 66 v/S spectrometer with KBr beamsplitter and CaF2 optics, the above-mentioned glower as a light source and a resolution of 2 cm−1. One spectrum consists of 350–500 co-averaged pulses. Further details of the experimental setup may be found in references [24,25].
In the Raman case, helium was mixed with the hydroxy ester in a coolable saturator, from where the mixture flowed through a heatable saturator, which contained the aromatic alcohol, into a reservoir at 1.0 bar. The subsequent continuous expansion took place through a heatable 4 mm × 0.2 mm slit nozzle that was located at a distance of 1 mm (BG, CL, CG) or 1.5 mm (BL) from the laser. Perpendicular to the laser and to the gas flow, scattered photons were collected by a monochromator and detected with the CCD camera. A resolution of 1 cm−1 was achieved. The expansion was exposed for 4 min (BL, CL, CG) or 10 min (BG), and the data were co-averaged over 4 (CL), 6 (BL, BG) or 11 (CG) exposures. Further details of the experimental setup are described in reference [20].
2.2. Computational Methods and Nomenclature
To find the most stable mixed dimer structures and to assign the experimental spectra, DFT studies were carried out with ORCA version 4.2.1 [26]. An initial structure search was done both manually and with Grimme’s CREST tool [27]. The structures found were pre-optimized with the B97-3c functional [28] and then optimized using the B3LYP functional [29,30,31] in combination with D3 dispersion correction [32], Becke-Johnson damping [33,34,35,36], a three-body term [37] and the def2-TZVP basis set [38]. Double-harmonic frequency and intensity calculations were added, including Raman intensities. The latter were converted into scattering cross sections. The search for transition states and reaction paths was done with Turbomole version 7.3 [39,40]. To evaluate the role of London dispersion interactions within this study, local energy decomposition analyses (LED) [41,42] were also performed. Computational details can be found in the supporting information (Table S1).
Instead of providing each compound with its own (e.g., monomer-based) scaling factor, all predicted harmonic OH stretching frequencies were scaled with a uniform factor of 0.97 to allow for less biased intersystem comparisons and to cover modes with mixed alcohol and hydroxyester character. This factor includes both anharmonicity effects and DFT errors. As we will see, two significant digits cover all individual assignments, and, more importantly, also the OH:O stretching vibrations for the B dimer (homOg and hetOg [43]) and the analogous C counterparts, to an adequate degree. When switching to methanol or phenol, deviations are expected due to differing DFT performance and different anharmonicity, but the expectation is that these deviations are systematic rather than erratic, at least in a narrow spectral range. This would be less the case if OH:O vibrations were compared to OH: vibrations [43].
In the following, mixed dimers will be labelled by the acronym for the alcohol (B or C), followed by the ester (L or G). An added i (inserted) or a (associated) denotes the topology of the two hydrogen bonds, and for the lactate complexes, hom and het are used to indicate the relative chirality of the alcohol conformation and the permanently chiral ester, as discussed in more detail later on.
3. Results
3.1. Vibrational Spectra
The four binary complexes of interest, BG, BL, CB and CL appear in the OH-stretching infrared and Raman spectra of supersonic expansions involving their components in the He carrier gas. They compete with single component dimers (homodimers) BB, CC, GG and LL, and because all species are hydrogen bond donors, the corresponding signals are shifted from the free OH of B and C, and to a lesser extent from the internally hydrogen-bonded OH signals of G and L. Furthermore, despite the high dilution employed in the expansions, there may be small contributions from trimeric aggregates. Therefore, the spectral congestion can be significant.
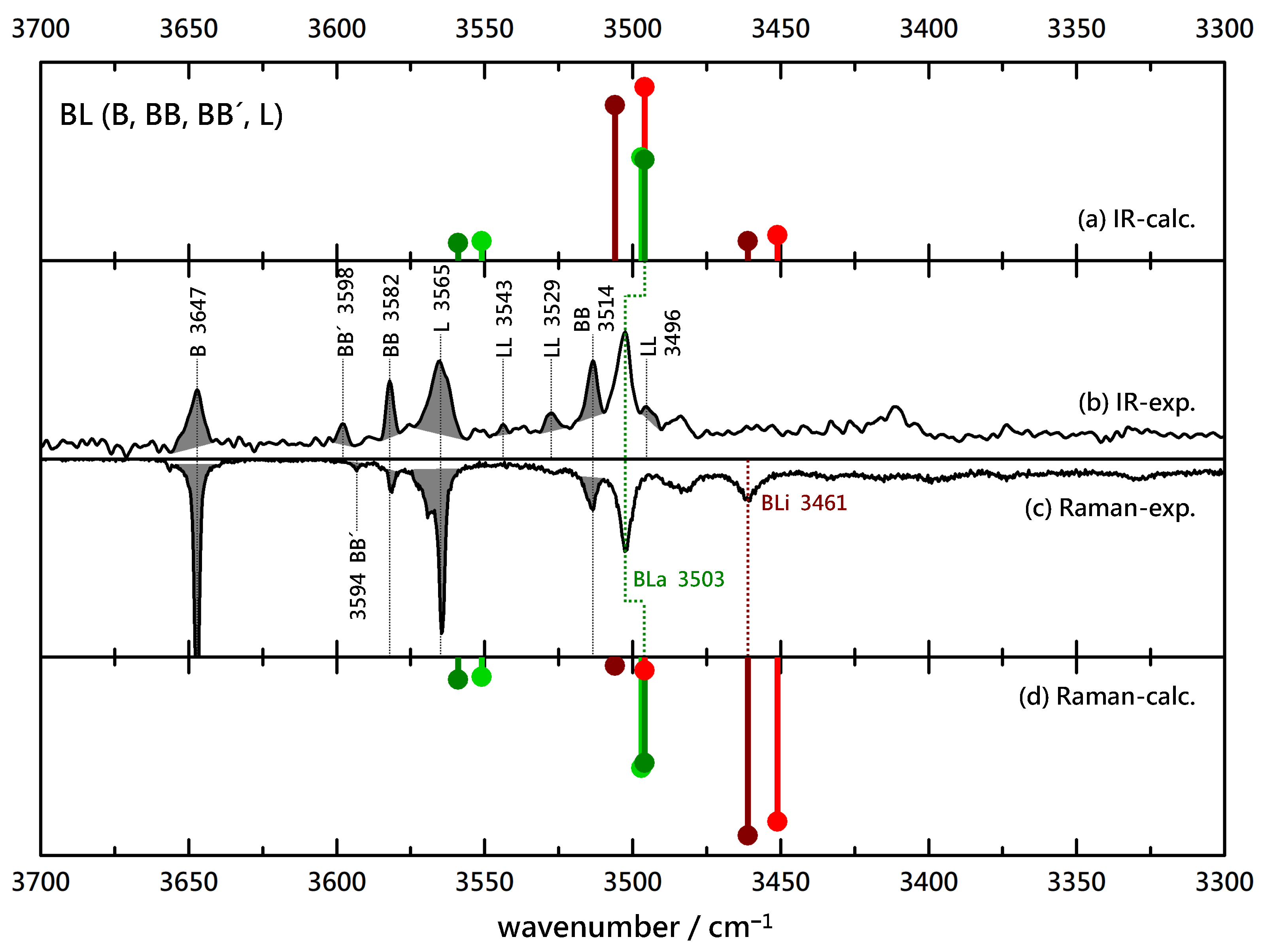
Based on previously published spectra of the homodimers BB, GG and LL [3,43] and the homodimer spectra of CC (Figure S4), as well as estimated relative abundances of these species based on computed intensities, the known contributions can be marked and allow to focus on the new, binary dimer signals. This is shown in Figure 2 for the (FT)IR and Raman (upside down) spectra obtained from relatively concentrated B/L coexpansions. Shaded monomer contributions match in terms of wavenumber in the IR and Raman traces, as do most homodimers, except for the metastable inversion-symmetric BB’, where the strong coupling between vibrations (accentuated by the rule of mutual exclusion) leads to displaced transitions. There is a single strong peak in both spectra at 3503 cm that must be due to a BL complex. If it were dominated by an insertion complex, there would be an even more intense, further downshifted Raman signal from the in-phase stretching motion of the two OH groups. However, there is only a less intense peak at 3461 cm that immediately tells that associated BL complexes dominate inserted complexes. This qualitative assignment can be further supported by harmonic wavenumber predictions for the most stable structures, which are uniformly scaled by 0.97 to best match the known homodimer signals. These suggest that the dominant BL peak in the IR spectrum could indeed have overlapping contributions from strongly IR-active OH stretching vibrations in hom and het variants of BLi and BLa, whereas the insertion/association-discriminating vibrations (upshifted for association, downshifted for insertion) are predicted and found to be too weak to be identified. The single, further downshifted Raman signal suggests that there is some insertion happening but clearly less than association. There is no evidence for a het/hom splitting (predicted to be small but detectable). This is all one can conclude from the BL spectra: more association than insertion, and no evidence for the presence of hom and het complexes at the same time.
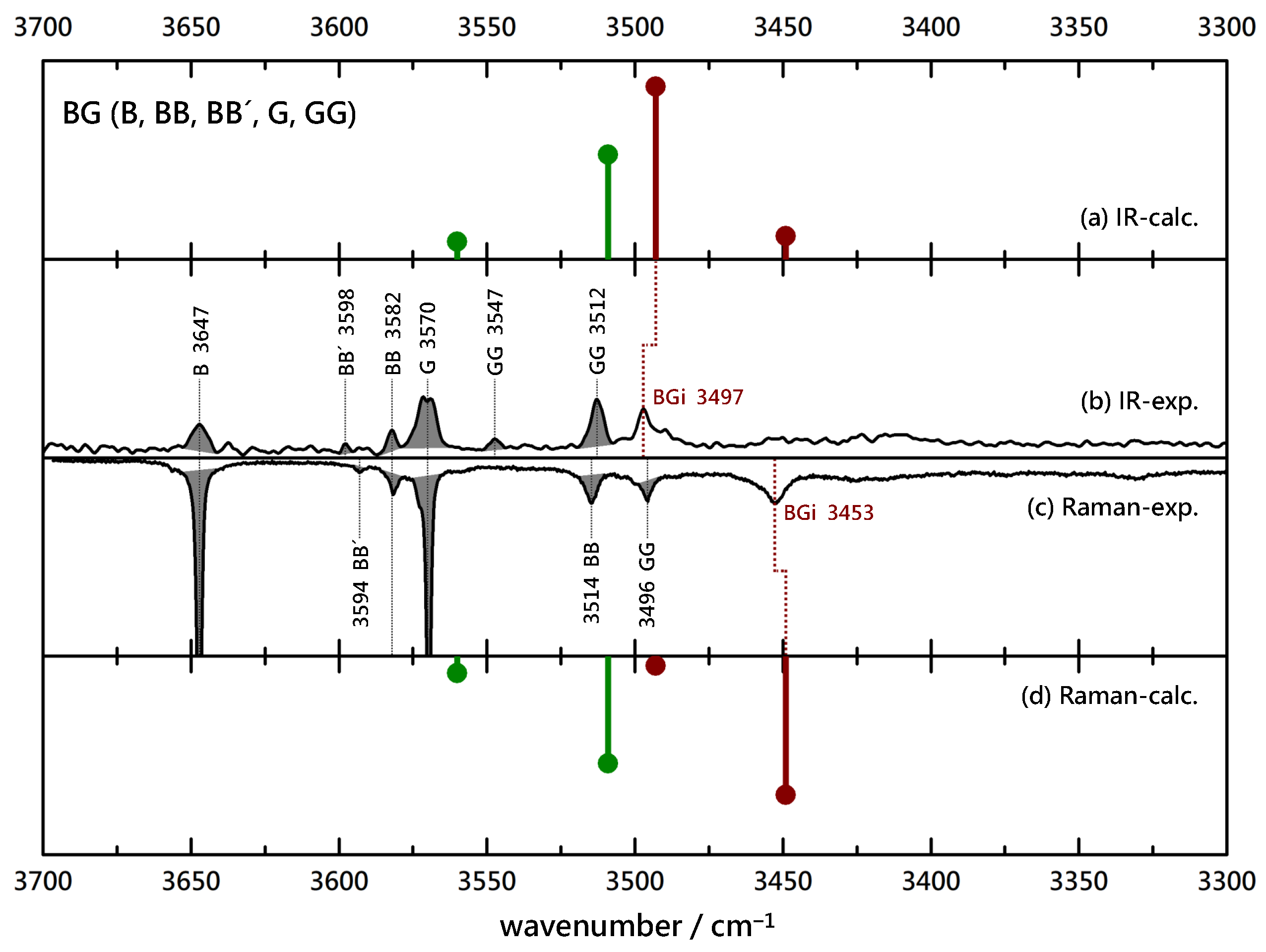
To verify the interpretation, it is instructive to remove the chirality center at L by switching to G. Superficially, Figure 3 shows a rather similar situation. Again, the known B, G, BB and GG contributions are shaded to focus on the binary complexes. Now, the dominant binary peaks do not match in the IR and Raman spectra. This is somewhat obscured by a coincidence with the GG signal in the Raman spectrum, but there cannot be a significant contribution by BG under that peak. Instead, a strongly downshifted BG signal in the Raman spectrum (3453 cm) has no significant IR counterpart and must be due to an insertion complex. The scaled harmonic predictions confirm that and any small BGa contribution would be well hidden behind several homodimer signals. Therefore, the removal of the methyl group at the chiral center of L has the qualitative consequence of switching from association to insertion, which is quite a remarkable effect. Its thermodynamic and kinetic aspects will be analyzed below.
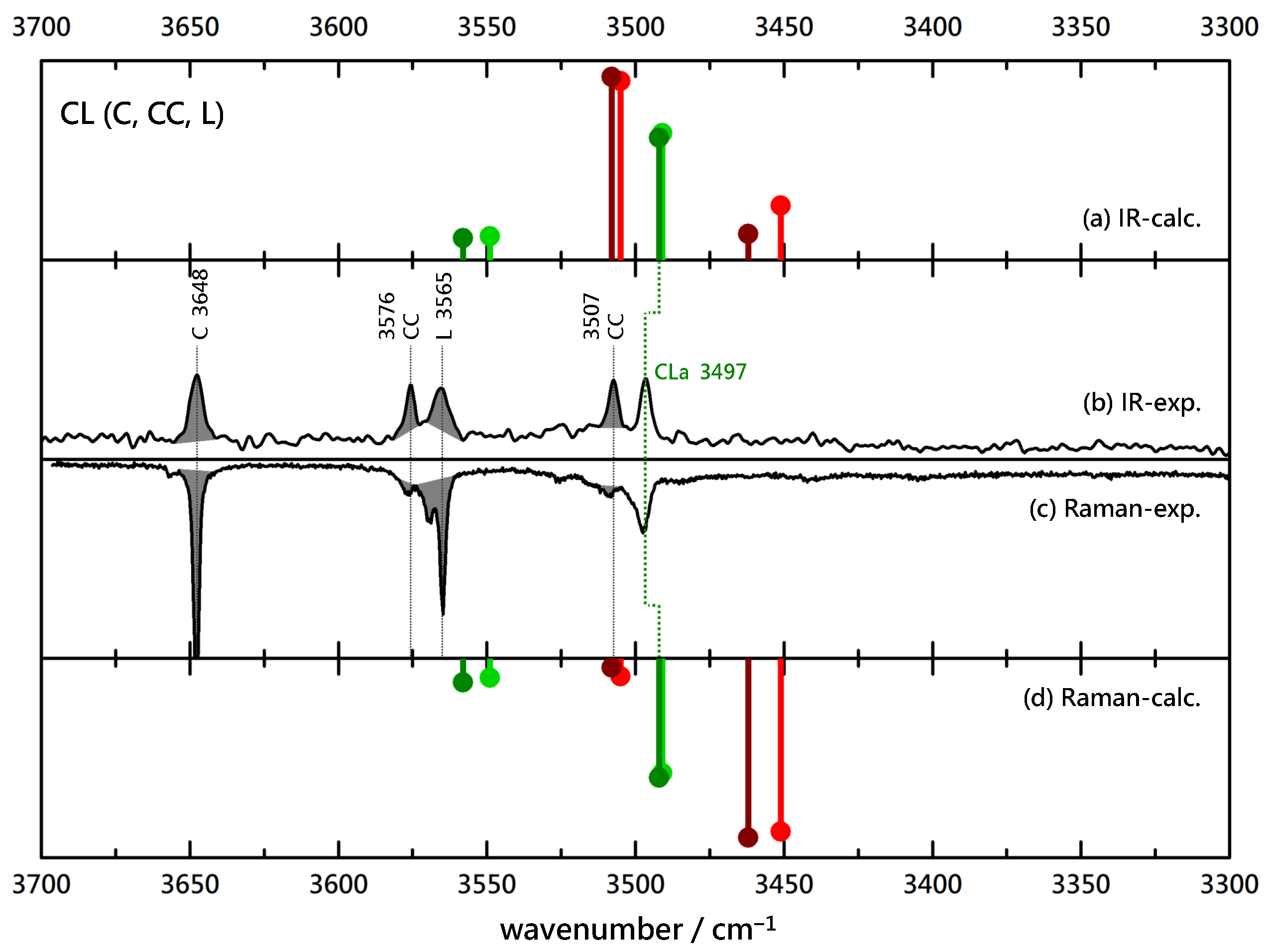
The qualitative consequences of such a small chemical change require further empirical support. Therefore, the benzyl alcohol B is replaced by its para-chloro derivative C in Figure 4 in combination with L. The situation closely resembles the BL case, with a single, coincident, dominant IR and Raman signal for the binary complex. No evidence for a Raman trace due to an inserted complex may be seen, so we conclude exclusive association of C to L. The absence of any splitting of the main peak should not be interpreted as complete relaxation between the hom and het variants towards the more stable one (effective chirality induction from L into C) because theory predicts no significant splitting in this case.
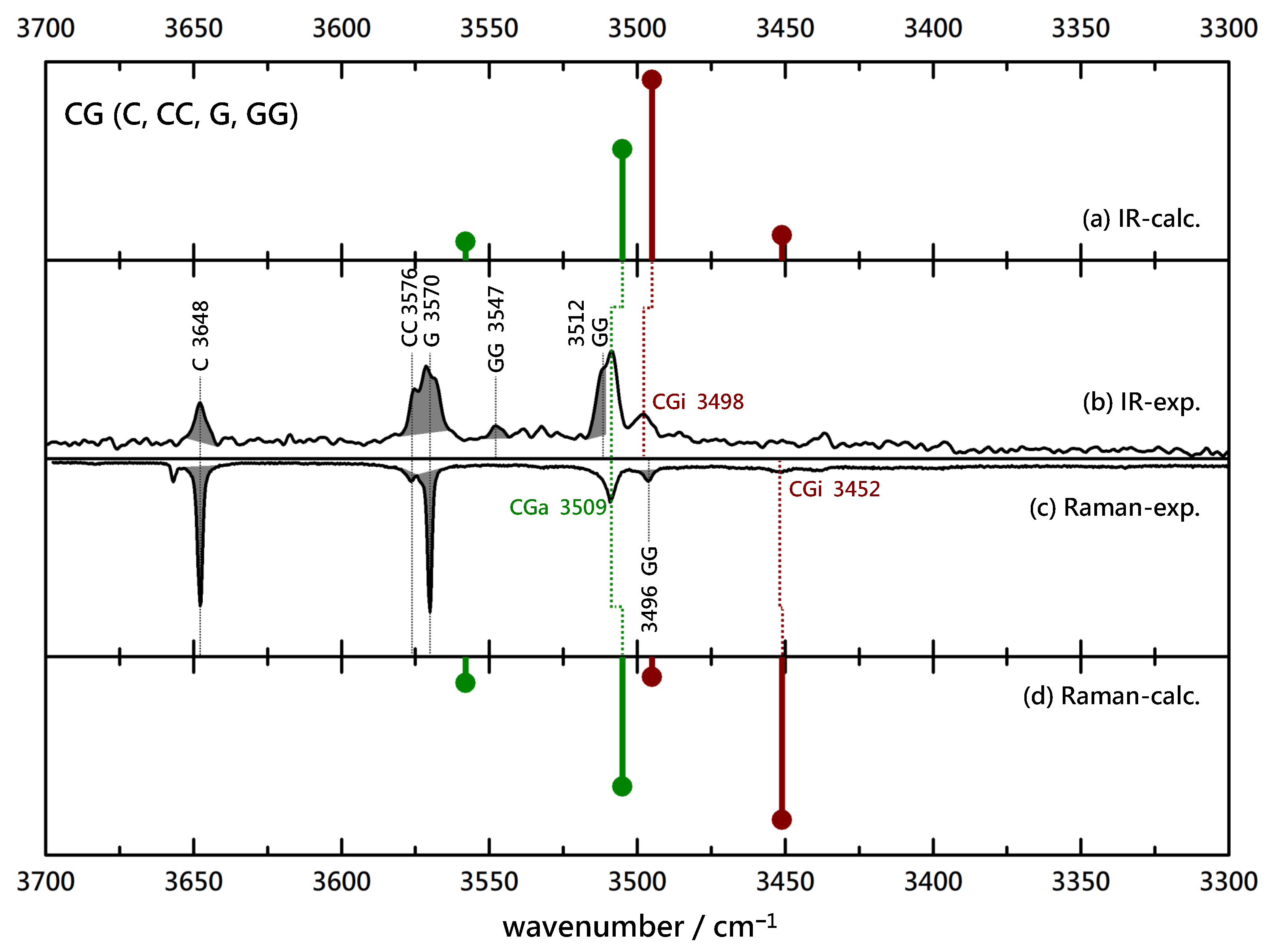
Finally, the achiral pairing of C with G is investigated in Figure 5. Despite some homodimer overlap, one can now identify two IR contributions. The higher wavenumber contribution is matched in the Raman spectrum, indicative of association, whereas the lower and somewhat weaker contribution is not adequately matched when considering the expected contribution of a coinciding GG signal. Instead, a downshifted, very weak contribution may be spotted and indicates a minor insertion signal. Comparison to the harmonic predictions of wavenumber and intensity suggests that association is favored by at least a factor of two, likely more. Again, as in the BL/BG pair, removal of the methyl group at the chiral center appears to promote insertion but to a lesser degree.
Somewhat more quantitative conclusions on relative abundances of monomer and dimer species can be drawn from a comparison of the experimental signal strengths with theoretical intensity predictions in the double-harmonic approximation. These are provided in Tables S3–S6 in the supplementary information. Despite considerable uncertainties due to overlap of bands, the intensity error of the harmonic DFT calculations and competitive aggregation, the resulting rough abundance estimates fit well with the qualitative conclusions drawn based on the inspection of the spectra.
In summary, the experimental spectra reveal three preferences for association and one for insertion of the (substituted) benzyl alcohol onto/into the hydroxy ester. In two instances (BL and CG), there is minor evidence for insertion, and in the other two cases (BG and CL), only one pattern is observed for insertion and association, respectively, such that the four systems nicely explore the balance between these two hydrogen bond topologies. This may be compared with methanol [3], where insertion was predominantly observed, and with more complex alcohols, where association was preferred in the case of a naphthylethanol [16]. No evidence for the simultaneous occurrence of hom/het complexes was observed in the present systems, which may be due to pronounced relaxation into the more stable variant but might also be explained by insufficient spectral resolution in this range.
This diversity of experimental findings calls for a systematic analysis by quantum chemical methods. As the scaled harmonic DFT predictions of the vibrational spectra performed remarkably well with only minor shifts to the observed bands (Figure 2, Figure 3, Figure 4 and Figure 5), one may hope that the employed computational level also gives reliable insights into the relative energetics of the isomers and the interconversion barriers between them. However, this can only be expected if the computational level does not miss an important interaction ingredient, such as dispersion interaction [16].
3.2. Predicted Substitution Trends
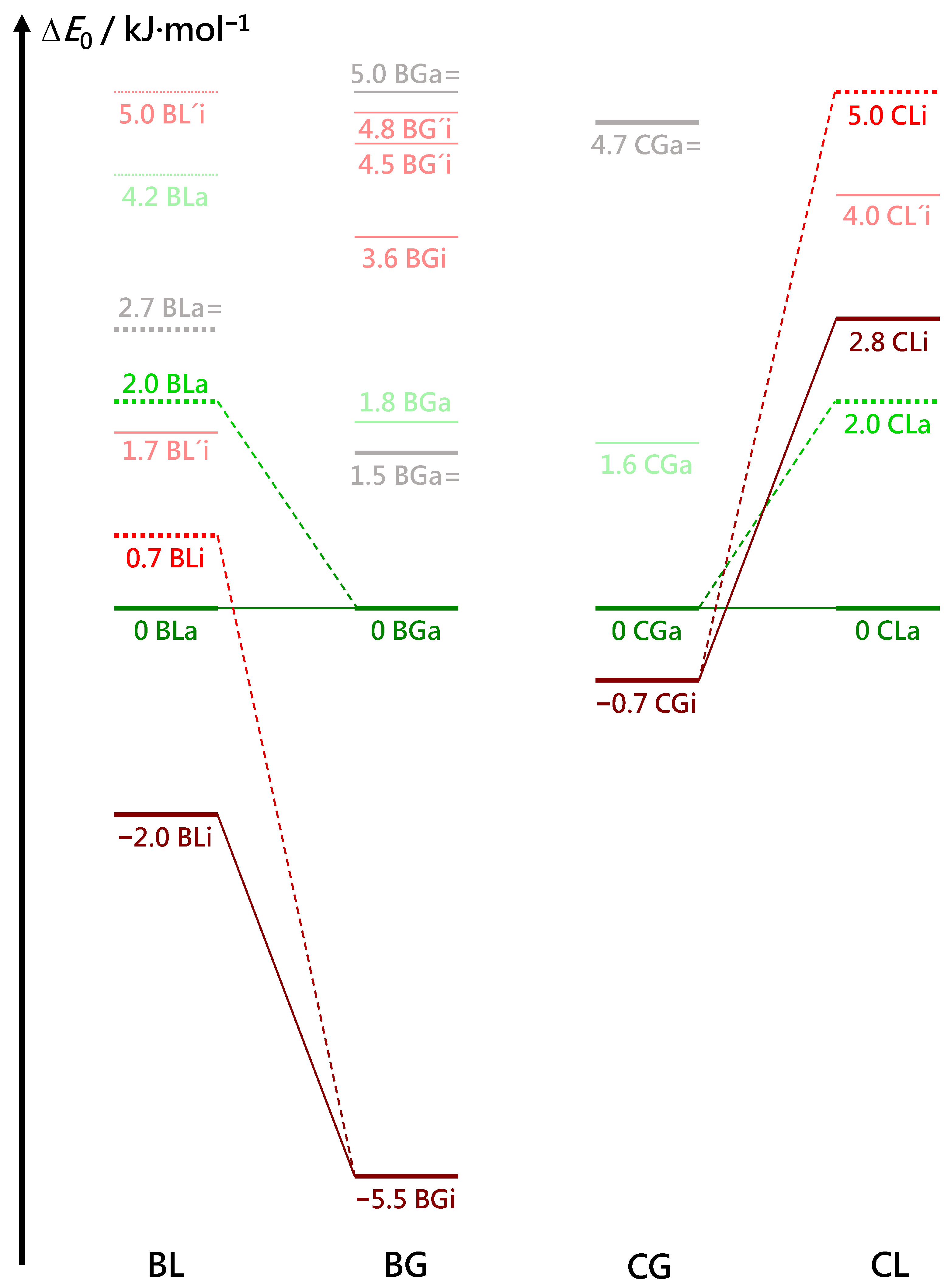
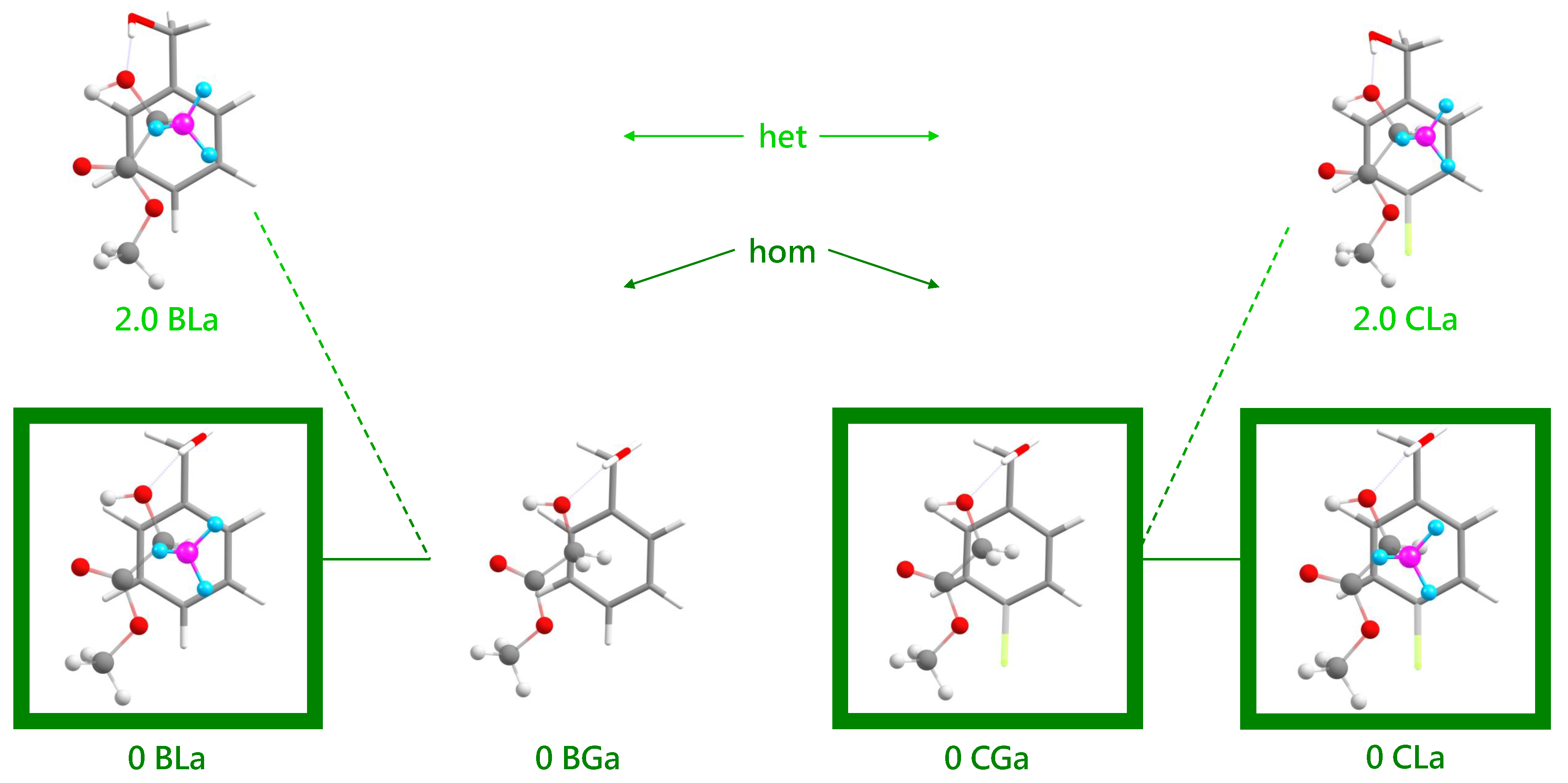
Several energy trends can be extracted from the comparison of BL, BG, CG and CL conformations in Figure 6 (for a more complete representation, see Figure S1 in the supplementary information). All DFT energies are corrected for zero-point vibrational contributions and aligned at zero for the most stable associated complex. This complex has a similar structure in all four cases (see Figure 7), with the alcoholic OH group more or less in line with the two hydroxy ester oxygen atoms connected by the intramolecular hydrogen bond. For the chiral methyl lactate, the arrangement is such that the extra methyl group points away from the aromatic ring, thus minimizing the distance between the two molecular backbone planes. This is best realized for the combination of a g− conformation of benzyl alcohol with the S-configured (−) lactate, which we will arbitrarily call hom(ochiral) in the following. It is equivalent to g+ in combination with methyl R(+)-lactate. In all hom structures, the alcohol OH runs parallel to the CC bond fixing the intramolecular hydrogen bond. As a consequence, the two oxygen atoms of the ester group are relatively far away from the phenyl ring atoms. The het(erochiral) pairings g+/S or g−/R are about 2 kJ·mol−1 higher in energy for BL and for CL in the associated complexes. They also avoid the extra methyl group at the chiral center pointing to the phenyl ring. Instead, the relative chirality of the benzyl alcohol switches and the alcoholic OH as well as the ester backbone run more parallel to the para axis of the benzyl group. It is not so obvious why this comes with an energy penalty, based on the structures alone. This will be discussed below. However, Figure S1 in the supplementary information shows that this energy penalty is already present in the achiral G ester (realized in the BGa and CGa isomers 1.8 and 1.6 kJ·mol−1 above the most stable structures, respectively, see Figure 6). It is thus due to the different interactions of the enantiotopic faces of G with a given alcohol conformation. When moving to L by adding a methyl group, only one of these faces remains sufficiently flat to favorably interact with the alcohol. The alternative complex structure, which accepts the methyl group pointing to the phenyl ring, is even higher in energy and thus gives way to the better alignment of the molecular planes.
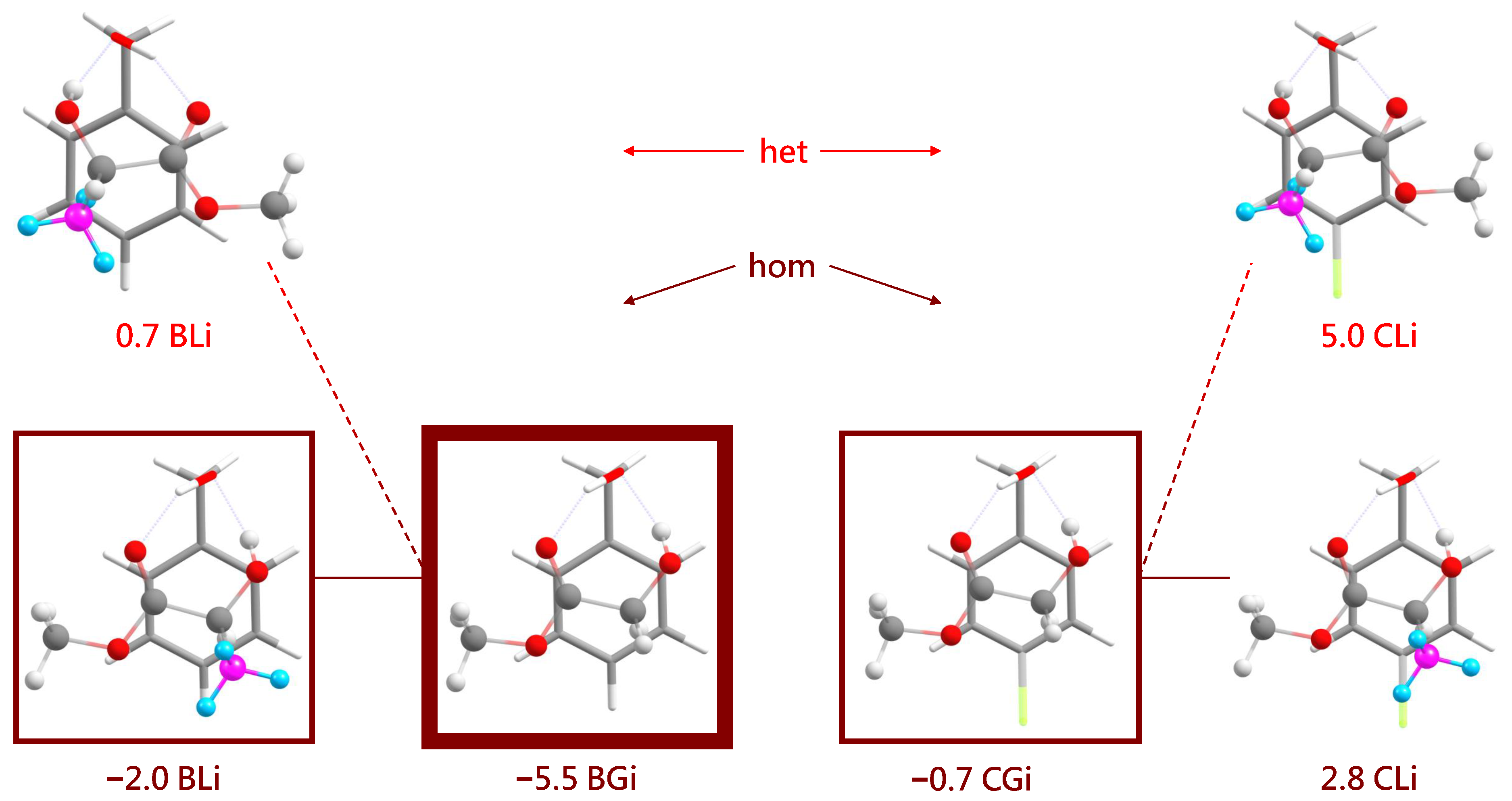
The most stable inserted complexes (Figure 8) involve a triangular arrangement of the alcohol O and the two ester oxygens that sacrifice their intramolecular hydrogen bond. They also prefer a homochiral pairing for L, again by about 2–3 kJ·mol−1 but now clearly due to the interference of the chirality-generating methyl group with an alignment of the ester and phenyl planes in the het case. The intramolecular hydrogen bond arrangement does not allow for het stacking while having the extra methyl group pointing away from the phenyl ring. This is nicely reflected in the angle between the aromatic plane and the ester plane. It is only 3–6 for all associated complexes, illustrating their ability to accomodate for stacking. For the inserted (hom) complexes, it amounts to 14–17 because the insertion reduces intermolecular flexibility. For the two inserted het complexes (BL and CL), the angle between the two planes increases to 46 to accomodate the chirality-generating methyl group. It appears plausible that this comes with an energy penalty for heterochiral insertion, and that is what Figure 8 confirms.
Relative to the associated complexes, the most stable inserted complexes show a large variation in energy (Figure 6). For benzyl alcohol, there is a 3.5 kJ·mol−1 penalty when moving from glycolate to lactate, although the extra methyl group in L points away from the aromatic plane. A possible structural explanation is that the extra methyl group can undergo less dispersion interaction with the ring in the inserted geometry, compared to the associated reference structure. In addition, and perhaps more importantly, there could be inductive effects of the methyl group on the acceptor strength of the ester hydroxy group that give the associated structure an advantage. Interestingly, the same insertion penalty is found for C instead of B despite the vicinity of the Cl atom and the new methyl group, when moving from G to L, and the structures are indeed rather similar to those for B. This supports the through-bond inductive interpretation. Remarkably, chlorination of B in the para position has a large (≈5 kJ·mol−1) destabilising effect on the relative energy of the most stable insertion structures, independent of whether the ester is chiral or not. Insertion is perhaps disfavored due to an unfavorable interaction of the polar C-H group in the ester with the polar C-Cl bond in the alcohol, or association is favored by attraction between the ester group and the C-Cl bond. The interplay of these three energy trends among the most stable conformations leads to a large energy advantage of BGi over BGa, which is attenuated in BLi and CGi. For CLi, where the two insertion penalties are added, the association complex becomes even more stable than the insertion complex, although it still contains a strained intramolecular hydrogen bond.
Together, these three 2–5 kJ·mol−1-trends nicely explain why BG is the only system where the inserted complex dominates in the experimental spectra. For BL and CG, the insertion advantage is much smaller, closer to or even within the accuracy of the hybrid functional, and the barrier associated with it prevents a dominance of the inserted conformers in the jet expansion. They are only observed as minor constituents. For CL, insertion is so unattractive due to the unfavorable interaction of C-H with C-Cl and the less favorable CH–ring interaction that only the associated complex is observed.
It is rewarding that these conclusions from dispersion-corrected B3LYP calculations do not change qualitatively when single-point energies at DLPNO-CCSD(T) level are added (Table 1 and Table 2). The relative energies typically change by less than 0.5 kJ·mol−1. The only exceptions are the BG and CG complexes, where the stability advantage of the inserted complex is reduced by 1–2 kJ·mol−1. For BG, this does not change the experimental and DFT evidence for the BGi global minimum, whereas for CG, it may indicate a switch from CGi to CGa in terms of the global minimum. This would actually match the experimental evidence for more CGa species, without having to invoke a kinetic argument. In summary, there is a high degree of consistency between the experimental findings and the theoretical energy sequences, and a significant kinetic hindrance for insertion only has to be invoked for BL, where a 1–2 kJ·mol−1 energy advantage of BLi is not obvious from the spectra.
3.3. Interconversion Paths
The competition between insertion and association complexes not only requires an analysis of their relative energies but also of the interconversion paths between them. This interconversion is illustrated for all four complexes in the supplementary material (Figure S2). It involves a transition state with hydrogen bond bifurcation. Other than in conventional bifurcation, where one hydrogen is shared between two oxygens [44], the carbonyl oxygen is shared between the two OH groups. The barrier height, as viewed from the associated complex, is typically larger than 10 kJ·mol−1, except for BG, where it is predicted below 7 kJ·mol−1 and thus relatively easy to overcome even in a supersonic jet expansion. This further supports the insertion preference for BG, in addition to the energetic driving force. The experimental findings are schematically summarized in Figure 9.
4. Discussion
4.1. Energy Decomposition Analysis
To explain some of the theoretical energy rankings, it is instructive to attempt some energy decomposition. For the insertion-association preference, this requires particular caution. If one looks at the dispersion contribution to the total interaction energy between the alcohol and hydroxy ester fragment, as provided by a LED analysis of the DLPNO-CCSD(T) data or similar decomposition schemes, one finds a clear dispersion preference for association. Namely, the dispersion contribution (strong and weak pairs together) amounts to 76–78% of the total electronic interaction energy for all six association complexes, whereas it varies between 63 and 71% for the six insertion complexes (Table 1 and Table 2). In contrast, leaving away the D3 dispersion correction as a very rough qualitative approach leads to unsystematic results. It suggests that dispersion favors insertion for BG, association for CG and CL, and remains inconclusive for BL. The absolute size of the LED dispersion contribution provides yet another picture, clearly being larger for insertion in all four combinations (39–41 kJ·mol−1 for the most stable insertion complexes) than for association (32–38 kJ·mol−1 for the most stable associated complexes). The explanation for these contradictory trends lies in the deformation energy to prepare, in particular the hydroxy ester for insertion of the alcohol. This is on the order of 10 kJ·mol−1 and renders the interaction energy comparison between insertion and association complexes less useful than a comparison of total energies. Overall, it appears that the influence of dispersion on association vs. insertion is mixed. The large difference between BG and CG can be clarified by energy decomposition. The interaction energy of the inserted complex differs by less than 1 kJ·mol−1, and so does the dispersion contribution. In contrast, the associated CG complex interacts 5 kJ·mol−1 more strongly than BG, and more than 4 kJ·mol−1 are due to dispersion interaction. Therefore, the exceptional stability of BGi is actually a lack of stability of BGa, which is compensated for in CGa by dispersion interaction between the Cl atom and the methoxy group of the ester. This stabilization of associated complexes by chlorine substitution in para position is also active in the CL complexes and contributes to the absence of insertion in experiment.
More robust conclusions can be drawn for the role of dispersion in chirality induction because the deformation energies should be similar for hom and het pairings, if one does not mix insertion and assocation complexes. For BL and CL, the CCSD(T) interaction energy is always larger for hom complexes than for the corresponding het complexes, by 1–3 kJ·mol−1. This is also true for the total energy, both at CCSD(T) level (2–3 kJ·mol−1) and at DFT level (2–3 kJ·mol−1). The contribution of dispersion energy to this systematic hom preference is 1–2 kJ·mol−1 for associated complexes, which means that dispersion slightly supports the homochirality of the two binding partners, but other interactions do so as well. This fits the somewhat unclear structural situation discussed before, where the energetic hom preference was not so obvious. For inserted complexes, the situation is different. Here, the dispersion preference for hom is 4–5 kJ·mol−1 and thus larger than the total hom preference. The explanation is quite straightforward because in the inserted geometry, het complexes are less coplanar due to the methyl group placed between the planes. The hom preference of London dispersion is so pronounced that without its action, insertion complexes would likely prefer a heterochiral arrangement, whereas for association complexes the dispersion influence is quite subtle and het complexes would also be disfavored in the absence of London dispersion forces.
4.2. Experimental Evidence for Metastable Complexes
After understanding the reasons for heterochiral metastability in these alcohol-hydroxy ester complexes, we should briefly come back to the observed spectra. Experimentally, it is difficult to rule out such het complexes, because their spectra do not differ much from the hom variants. At least there are no prominent spectral doublets, even in cases where the hom/het spectral splitting is predicted to be significant. A resolution of this uncertainty might come from microwave spectroscopy, which should be able to unambiguously identify the preferred relative chirality of the alcohol conformation and the ester.
It is also difficult to experimentally rule out anticooperative associated complexes, where the alcohol docks on the carbonyl oxygen and competes with the intramolecular hydrogen bond to the other carbonyl lone pair (:OH:O:HO:). Small fractions of such isomers might be hidden behind homodimer and monomer signals. However, these complexes (indicated by = in Figure 6) are always predicted at least 5 kJ·mol−1 higher in energy than the most stable complexes and are always less stable than the most stable associated complex of a given alcohol-ester combination. This is also the case for complexes in which the ester conformation or coordination deviates from the most stable structures, denoted with a prime. Furthermore, hydrogen bond coordination of the methoxy group was not predicted to be competitive.
As discussed before, the computed barriers for interconversion between associated and inserted complexes are typically larger than 10 kJ·mol−1 and thus help to explain why only small amounts of inserted complexes (BL and CG) or no inserted complexes (CL) are observed experimentally. Only for BG, the barrier is less than 10 kJ·mol−1 (see Figure S2 in the supplementary information for the interconversion path), and the driving force for insertion is so large that only the inserted complex is observed and the metastable associated isomer is elusive.
In summary, the predicted energy sequence of the alcohol-ester conformations and of the activation barriers for insertion is fully consistent with the experimental observation, but experiment cannot strictly rule out small contributions of competing isomers.
4.3. Comparison with Other Alcohols and Phenol
For methanol (M) in combination with L and G, it was experimentally verified for the 1:1 complexes that insertion wins over association, despite the insertion barrier and its potential kinetic hindrance [3]. In the present work, we find the same preference for benzyl alcohol (B), at least in combination with G. Evidently, the insertion barrier is still small enough and the driving force persists. The combination of phenol with L also revealed both kinds of aggregation topologies [17]. This is apparently no more the case for a larger aromatic, naphthyl-based alcohol [16] (N), where exclusively associated complexes are assigned and inserted topologies are expected to be less stable due to the less flexible hinge between the monomer units. The chlorinated variant of benzyl alcohol C is a borderline case between plain phenyl- and naphthyl-substituted methanols, where the associated complex already dominates but the inserted complex may still be detected (at least in combination with G). Perhaps, dispersion interaction of the methoxy group in -hydroxy esters with distant (para) ring substituents of the benzyl alcohol (including an annealed ring for N) represents a generic stabilization motif for association complexes. Insertion topologies do not profit from that kind of interaction because the hydroxy ester backbone runs orthogonal to the para-axis.
If all these topological assignments from experiment are correct, one may expect that the uniformly scaled B3LYP-D3 method makes reasonably consistent predictions for the observed OH fundamental band positions in the most stable 1:1 complexes of methanol, phenol, (chlorinated) benzyl alcohol and 2-naphthyl-1-ethanol. This is explored in Table 3, with the underlying raw data being listed in Table S2 in the supplementary information. In the left half, one can see that the eight assignments from the present work indeed match the scaled prediction very systematically, with negative deviations of less than 10 cm. However, no ester-based OH stretching fundamental could be assigned for associated complexes due to their weak IR and Raman intensity. Using the same scaling factor, the four known inserted complex transitions for methanol [3] are also predicted uniformly but systematically underestimated by 17–30 cm. This is actually expected due to a systematic substitution dependence of B3LYP calculations for alcohol monomers [45], amounting to about 15 cm between methanol and benzyl alcohol and adding to the deviation found in the benzyl alcohol based complexes. Therefore, all 12 FTIR/Raman band positions for BL, BG, CL, CG, ML and MG are consistent with each other at the B3LYP-D3 level. This is also the case for the alcoholic OH stretching fundamental of the most stable associated (S)-2-naphthyl-1-ethanol (N) 1:1 complexes with L and G [16], whereas there is a considerable mismatch for their ester OH stretch fundamentals. Because there is no independent FTIR verification of weakly IR-active ester-localized OH vibrations in either B, G, M or N complexes, this unsystematic scaling has to remain open. We note that the hydroxyester monomer OH transitions for L and G are off by +25 cm−1 and +21 cm−1, respectively, using our uniform theoretical scaling approach. This may offer an explanation in the same spirit as for the methanol complexes, although it remains unclear why this offset is not found for inserted complexes. Furthermore, it should be noted that a uniform scaling factor of 0.97 will necessarily fail for either much stronger or much weaker hydrogen bonds, and in particular for isolated alcohol monomers [45].
It is bound to a relatively narrow range of transition wavenumbers, due to hybrid DFT deficiencies and the harmonic approximation, and even there it can fail for different types of hydrogen bond acceptors [43].
One may finally compare our results to a recent study of inserted and associated complexes of phenol (P) with L [17]. We do not compare the structures Ia and IIa assigned by Hong et al. (also based on electronic spectroscopy), but instead the most stable inserted and associated complex found with the theoretical method used in this work, for consistency. These are the inserted structure IIb from reference [17] (marked i in Table 3) and an associated structure (a) that has not been discussed in that reference and is shown in Figure S3 in the supplementary information. Deviations between theory and experiment are again quite systematic for the inserted structure and the alcohol-based OH stretching fundamental in the associated complex with differences between –3 cm−1 and +9 cm−1. For the monomer-like ester-based OH vibration in the association complex, theory again deviates significantly. This pattern occurs for PL, NL and NG and is an indication that the simple uniform harmonic scaling approach is skewed for those cases in which the intramolecular hydrogen bond of the ester is not broken, in addition to the well known methanol case.
5. Conclusions
When the transiently chiral benzyl alcohol forms a complex with methyl lactate, it has two decisions to make. Will it insert into the intramolecular lactate hydrogen bond in an activated process or will it attach to the hydroxy group without significant reaction barrier? Will it adjust to the chirality of the lactate or will this be irrelevant? Despite the subtlety of the interaction, different levels of theory suggest that benzyl alcohol uniformly adjusts to the lactate chirality, independent of whether the ring is para-substituted by chlorine and whether the alcohol inserts into or associates onto the lactate. For insertion, this preference is strongly dispersion-driven, for association less so, because the hydrogen bond pattern can avoid a conflict with the chirality-generating methyl group.
The question of insertion vs. association finds less uniform answers across methyl lactate and its achiral counterpart methyl glycolate. Experimentally, it is clear that in three out of the four combinations, association is preferred. Only the combination of benzyl alcohol with methyl glycolate prefers insertion, despite the existence of an insertion barrier. It is remarkable that a modification as subtle as hydrogen/chlorine substitution six bonds away from the interacting OH group of the alcohol changes this preference, whereas, for example, the replacement of OH by SH in benzyl alcohol has much more subtle effects on its self-aggregation via hydrogen bonding [50]. The hydrogen bond topology switches with chlorination and methylation are in good agreement with the theoretical predictions. Dispersion-corrected B3LYP calculations appear to capture all essential interactions and only differ in detail from DLPNO-CCSD(T) electronic energy checks, beyond the conclusiveness of the experimental spectra due to kinetic hindrance issues. Furthermore, a simple, uniformly scaled harmonic prediction of transition wavenumbers is remarkably consistent except for a known deficiency for methanol and a less well understood deficiency for the intramolecularly hydrogen-bonded OH stretch in association complexes. Others are invited to check their favorite DFT approaches for the performance in this chirality induction and hydrogen bond topology test set. The microwave spectroscopy technique [47,51] promises to provide finer details of the chirality induction processes that these hydrogen-bonded model systems exhibit, which are at the heart of some cases of enantioselective organocatalysis [52,53,54]. Finally, it should be pointed out that chirality induction is just one way to subtly modulate intermolecular interactions, whereas the insights from our study, including halogenation effects, can also be useful for experiment–theory comparisons in achiral noncovalent catalysis [55,56].
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/sym14020357/s1, Figure S1: Energy scheme, Figure S2: Interconversion paths, Figure S3: Associated PL dimer, Figure S4: Experimental IR and Raman spectra of C, Table S1: Computational keywords, Table S2: Raw data for the analysis of the uniformly scaled B3LYP-D3 method, Table S3: Relative abundances of species in the BL spectra, Table S4: Relative abundances of species in the BG spectra, Table S5: Relative abundances of species in the CL spectra, and Table S6: Relative abundances of species in the CG spectra.
Author Contributions
Conceptualization, M.A.S.; methodology, M.L.; validation, M.L. and M.A.S.; investigation, M.L. and E.S.; resources, M.A.S.; data curation, M.L.; writing—original draft preparation, M.A.S.; writing—review and editing, M.L., E.S. and M.A.S.; visualization, M.L.; supervision, M.A.S.; funding acquisition, M.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-271107160/SPP1807 and 405832858 (compute cluster). The APC was waived by the journal Symmetry.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The key spectra and structures presented in this study are openly available at Göttingen Research Online under https://doi.org/10.25625/LS2SQ8 (accessed on 8 February 2022) and under https://doi.org/10.25625/NFIKNJ (accessed on 8 February 2022), respectively.
Acknowledgments
We thank Sophie Schweer for valuable support in the Raman measurements and the mechanics and electronics workshops for the construction and maintenance of the jet expansion setups. We further thank Robert Medel for inspiring discussions.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| B | Benzyl alcohol |
| C | 4-Chlorobenzyl alcohol |
| L | Methyl (S)-(−)-lactate |
| G | Methyl glycolate |
| P | Phenol |
| N | (S)-2-Naphthyl-1-ethanol |
| M | Methanol |
| CCD | Charge Coupled Device |
| L-N2 | Liquid Nitrogen |
| DFT | Density Functional Theory |
| CREST | Conformer-Rotamer Ensemble Sampling Tool |
| B3LYP | Becke 3-parameter Lee-Yang-Parr density functional |
| D3 | Third-generation Grimme parametrization of density functional dispersion |
| correction | |
| BJ | Becke and Johnson damping function for small interatomic distances |
| LED | Local energy decomposition |
| DLPNO-CCSD(T) | Domain based local pair-natural orbital singles and doubles coupled cluster |
| with perturbative triples correction |
References
- Maes, G.; Smets, J. Hydrogen bond cooperativity: A quantitative study using matrix-isolation FT-IR spectroscopy. J. Phys. Chem. 1993, 97, 1818–1825. [Google Scholar] [CrossRef]
- López de la Paz, M.; Ellis, G.; Pérez, M.; Perkins, J.; Jiménez-Barbero, J.; Vicent, C. Carbohydrate Hydrogen-Bonding Cooperativity—Intramolecular Hydrogen Bonds and Their Cooperative Effect on Intermolecular Processes—Binding to a Hydrogen-Bond Acceptor Molecule. Eur. J. Org. Chem. 2002, 2002, 840–855. [Google Scholar] [CrossRef]
- Borho, N.; Suhm, M.A.; Le Barbu-Debus, K.; Zehnacker, A. Intra- vs. intermolecular hydrogen bonding: Dimers of alpha-hydroxyesters with methanol. Phys. Chem. Chem. Phys. 2006, 8, 4449–4460. [Google Scholar] [CrossRef] [PubMed]
- Agreiter, J.; Knight, A.; Duncan, M. ZEKE-PFI spectroscopy of the Al–(H2O) and Al–(D2O) complexes. Chem. Phys. Lett. 1999, 313, 162–170. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]
- Melandri, S. “Union is strength”: How weak hydrogen bonds become stronger. Phys. Chem. Chem. Phys. 2011, 13, 13901–13911. [Google Scholar] [CrossRef]
- Scuderi, D.; Le Barbu-Debus, K.; Zehnacker, A. The role of weak hydrogen bonds in chiral recognition. Phys. Chem. Chem. Phys. 2011, 13, 17916–17929. [Google Scholar] [CrossRef] [PubMed]
- Medel, R.; Camiruaga, A.; Saragi, R.T.; Pinacho, P.; Pérez, C.; Schnell, M.; Lesarri, A.; Suhm, M.A.; Fernández, J.A. Rovibronic signatures of molecular aggregation in the gas phase: Subtle homochirality trends in the dimer, trimer and tetramer of benzyl alcohol. Phys. Chem. Chem. Phys. 2021, 23, 23610–23624. [Google Scholar] [CrossRef]
- Hashimoto, M.; Harada, M. Conformational Phase Transitions Associated with Reversal of Hydrogen Bond Direction in 4-Chloro- and 4-Bromobenzyl Alcohols. An X-Ray Study. Z. Naturforsch. A 2003, 58, 63–67. [Google Scholar] [CrossRef]
- Jarmelo, S.; Maria, T.M.R.; Leitão, M.L.P.; Fausto, R. Structural and vibrational characterization of methyl glycolate in the low temperature crystalline and glassy states. Phys. Chem. Chem. Phys. 2000, 2, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.; Caminati, W.; Hollenstein, H. Torsional motions in methyl glycolate. J. Mol. Spectrosc. 1989, 137, 87–103. [Google Scholar] [CrossRef]
- Ottaviani, P.; Velino, B.; Caminati, W. Jet cooled rotational spectrum of methyl lactate. Chem. Phys. Lett. 2006, 428, 236–240. [Google Scholar] [CrossRef]
- Borho, N.; Xu, Y. Rotational spectrum of a chiral α-hydroxyester: Conformation stability and internal rotation barrier heights of methyl lactate. Phys. Chem. Chem. Phys. 2007, 9, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Asselin, P.; Madebène, B.; Soulard, P.; Georges, R.; Goubet, M.; Huet, T.R.; Pirali, O.; Zehnacker-Rentien, A. Competition between inter- and intra-molecular hydrogen bonding: An infrared spectroscopic study of jet-cooled amino-ethanol and its dimer. J. Chem. Phys. 2016, 145, 224313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seurre, N.; Le Barbu-Debus, K.; Lahmani, F.; Zehnacker, A.; Borho, N.; Suhm, M.A. Chiral recognition between lactic acid derivatives and an aromatic alcohol in a supersonic expansion: Electronic and vibrational spectroscopy. Phys. Chem. Chem. Phys. 2006, 8, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Moon, C.J.; Jang, H.; Min, A.; Choi, M.Y.; Heo, J.; Kim, N.J. Isomer-Specific Induced Circular Dichroism Spectroscopy of Jet-Cooled Phenol Complexes with (−)-Methyl L-Lactate. J. Phys. Chem. Lett. 2018, 9, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Maué, D.; Strebert, P.H.; Bernhard, D.; Rösel, S.; Schreiner, P.R.; Gerhards, M. Dispersion-Bound Isolated Dimers in the Gas Phase: Observation of the Shortest Intermolecular CH···H–C Distance via Stimulated Raman Spectroscopy. Angew. Chem. Int. Ed. 2021, 60, 11305–11309. [Google Scholar] [CrossRef]
- Shachar, A.; Kallos, I.; de Vries, M.S.; Bar, I. Revealing the Structure and Noncovalent Interactions of Isolated Molecules by Laser-Desorption/Ionization-Loss Stimulated Raman Spectroscopy and Quantum Calculations. J. Phys. Chem. Lett. 2021, 12, 11273–11279. [Google Scholar] [CrossRef]
- Forsting, T.; Gottschalk, H.C.; Hartwig, B.; Mons, M.; Suhm, M.A. Correcting the record: The dimers and trimers of trans-N-methylacetamide. Phys. Chem. Chem. Phys. 2017, 19, 10727–10737. [Google Scholar] [CrossRef] [Green Version]
- Zehnacker, A.; Suhm, M. Chirality Recognition between Neutral Molecules in the Gas Phase. Angew. Chem. Int. Ed. 2008, 47, 6970–6992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, G.; Losada, M.; Xu, Y. Vibrational absorption, vibrational circular dichroism, and theoretical studies of methyl lactate self-aggregation and methyl lactate-methanol intermolecular interactions. J. Chem. Phys. 2010, 132, 234513. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, S.A.; Spicher, S.; Gerasimova, T.P.; Grimme, S. Revisiting conformations of methyl lactate in water and methanol. J. Chem. Phys. 2021, 155, 024507. [Google Scholar] [CrossRef] [PubMed]
- Altnöder, J.; Lee, J.J.; Otto, K.E.; Suhm, M.A. Molecular Recognition in Glycolaldehyde, the Simplest Sugar: Two Isolated Hydrogen Bonds Win Over One Cooperative Pair. ChemistryOpen 2012, 1, 269–275. [Google Scholar] [CrossRef]
- Hartwig, B.; Lange, M.; Poblotzki, A.; Medel, R.; Zehnacker, A.; Suhm, M.A. The reduced cohesion of homoconfigurational 1,2-diols. Phys. Chem. Chem. Phys. 2020, 22, 1122–1136. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Bannwarth, C.; Hansen, A.; Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 2018, 148, 064104. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree–Fock model of intermolecular interactions. J. Chem. Phys. 2005, 123, 024101. [Google Scholar] [CrossRef]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- TURBOMOLE V7.3 2018, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. 2007. Available online: http://www.turbomole.com (accessed on 16 December 2021).
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Schneider, W.B.; Bistoni, G.; Sparta, M.; Saitow, M.; Riplinger, C.; Auer, A.A.; Neese, F. Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework. J. Chem. Theory Comput. 2016, 12, 4778–4792. [Google Scholar] [CrossRef]
- Altun, A.; Neese, F.; Bistoni, G. Local energy decomposition analysis of hydrogen-bonded dimers within a domain-based pair natural orbital coupled cluster study. Beilstein J. Org. Chem. 2018, 14, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Medel, R.; Suhm, M.A. Understanding benzyl alcohol aggregation by chiral modification: The pairing step. Phys. Chem. Chem. Phys. 2020, 22, 25538–25551. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, C.; Jeffrey, G.; Taylor, R. A survey of O-H···O hydrogen bond geometries determined by neutron diffraction. J. Mol. Struct. 1981, 70, 255–271. [Google Scholar] [CrossRef]
- Medel, R.; Suhm, M.A. Predicting OH stretching fundamental wavenumbers of alcohols for conformational assignment: Different correction patterns for density functional and wave-function-based methods. Phys. Chem. Chem. Phys. 2021, 23, 5629–5643. [Google Scholar] [CrossRef]
- Le Barbu-Debus, K.; Lahmani, F.; Zehnacker-Rentien, A.; Guchhait, N.; Panja, S.S.; Chakraborty, T. Fluorescence spectroscopy of jet-cooled chiral (±)-indan-1-ol and its cluster with (±)-methyl- and ethyl-lactate. J. Chem. Phys. 2006, 125, 174305. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Sukhorukov, O.; Jäger, W.; Xu, Y. Chirped-Pulse and Cavity-Based Fourier Transform Microwave Spectra of the Methyl Lactate···Ammonia Adduct. Angew. Chem. Int. Ed. 2013, 52, 4402–4405. [Google Scholar] [CrossRef]
- Le Barbu-Debus, K.; Broquier, M.; Mahjoub, A.; Zehnacker-Rentien, A. Chiral recognition in jet-cooled complexes of (1R,2S)-(+)-cis-1-amino-2-indanol and methyl lactate: On the importance of the CH⋯π interaction. Phys. Chem. Chem. Phys. 2009, 11, 7589–7598. [Google Scholar] [CrossRef]
- Mahjoub, A.; Le Barbu-Debus, K.; Zehnacker, A. Structural Rearrangement in the Formation of Jet-Cooled Complexes of Chiral (S)-1,2,3,4-Tetrahydro-3-isoquinolinemethanol with Methyl Lactate: Chirality Effect in Conformer Selection. J. Phys. Chem. A 2013, 117, 2952–2960. [Google Scholar] [CrossRef]
- Saragi, R.T.; Juanes, M.; Pinacho, R.; Rubio, J.E.; Fernández, J.A.; Lesarri, A. Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry 2021, 13, 2022. [Google Scholar] [CrossRef]
- Thomas, J.; Sukhorukov, O.; Jäger, W.; Xu, Y. Direct Spectroscopic Detection of the Orientation of Free OH Groups in Methyl Lactate–(Water)1,2 Clusters: Hydration of a Chiral Hydroxy Ester. Angew. Chem. Int. Ed. 2014, 53, 1156–1159. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef]
- Seayad, J.; List, B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis Based on C–I⋯π Halogen Bonds: Electrophilic Activation of 2-Alkenylindoles by Cationic Halogen-Bond Donors for [4 + 2] Cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef] [PubMed]
- Sysoeva, A.A.; Novikov, A.S.; Il’in, M.V.; Suslonov, V.V.; Bolotin, D.S. Predicting the catalytic activity of azolium-based halogen bond donors: An experimentally-verified theoretical study. Org. Biomol. Chem. 2021, 19, 7611–7620. [Google Scholar] [CrossRef]
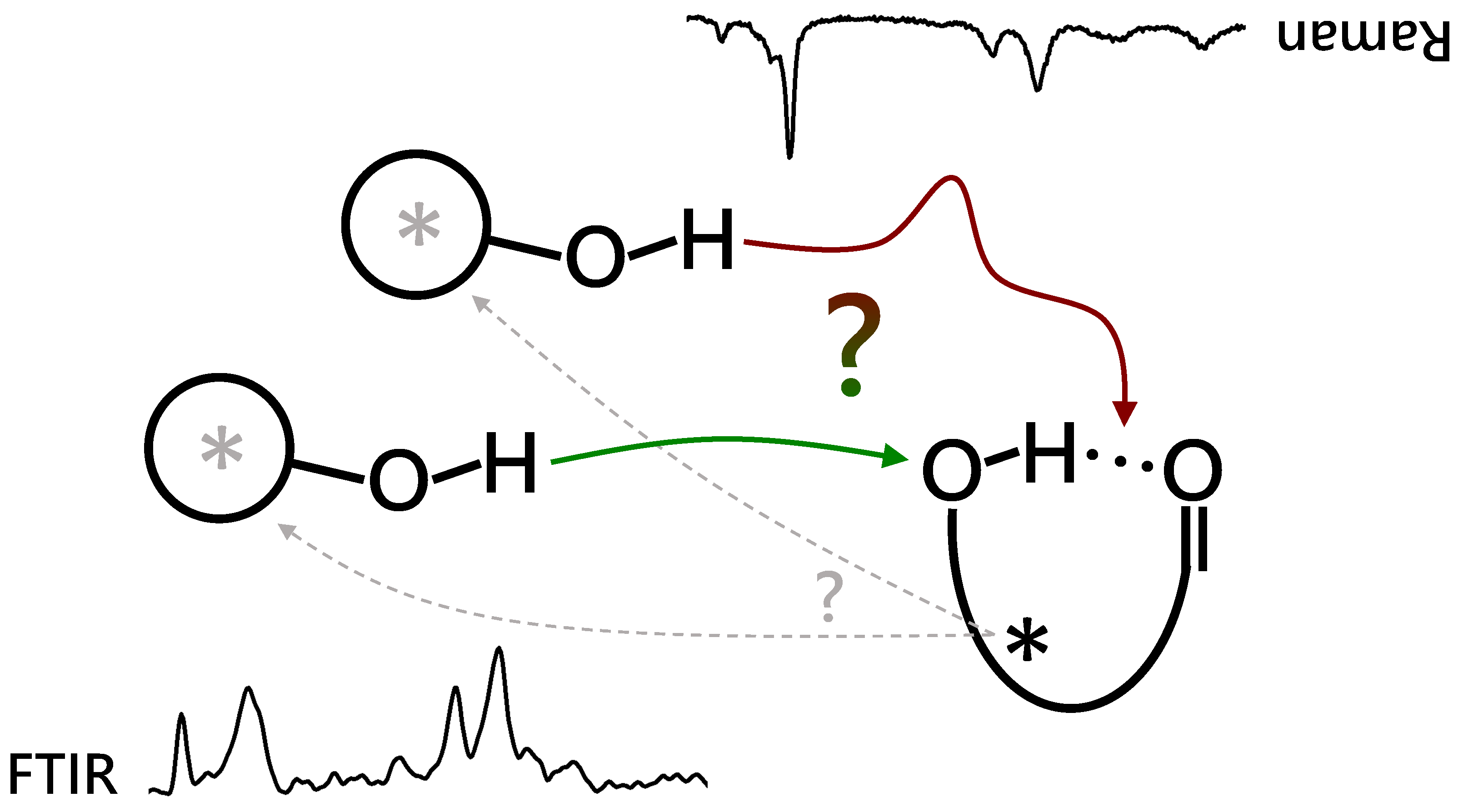
Figure 1.
Different complexation options between an alcoholic OH and an internally OH:O=C hydrogen bonded ester, either inserting (red arrow, activation barrier) the alcohol into or attaching it (green arrow) onto the internal hydrogen bond. FTIR (lower left) and Raman (upper right, inverted) spectra are combined to elucidate the actual preferences under supersonic jet expansion conditions. The permanent (black *) and transient chirality (grey *) of the binding partners can potentially lead to chirality induction processes (dashed arrows).
Figure 1.
Different complexation options between an alcoholic OH and an internally OH:O=C hydrogen bonded ester, either inserting (red arrow, activation barrier) the alcohol into or attaching it (green arrow) onto the internal hydrogen bond. FTIR (lower left) and Raman (upper right, inverted) spectra are combined to elucidate the actual preferences under supersonic jet expansion conditions. The permanent (black *) and transient chirality (grey *) of the binding partners can potentially lead to chirality induction processes (dashed arrows).

Figure 2.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of B(enzyl alcohol)-L(actate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) BL complexes (the darker color corresponds to the more stable isomer). Shaded areas represent known contributions from homodimers BB (and isomeric BB’), LL and monomers B and L, as marked by their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.
Figure 2.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of B(enzyl alcohol)-L(actate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) BL complexes (the darker color corresponds to the more stable isomer). Shaded areas represent known contributions from homodimers BB (and isomeric BB’), LL and monomers B and L, as marked by their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.

Figure 3.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of B(enzyl alcohol)-G(lycolate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) BG complexes. Shaded areas represent known contributions from homodimers BB (and isomeric BB’), GG and monomers B and G, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.
Figure 3.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of B(enzyl alcohol)-G(lycolate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) BG complexes. Shaded areas represent known contributions from homodimers BB (and isomeric BB’), GG and monomers B and G, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.

Figure 4.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of (4-)C(hlorobenzyl alcohol)-L(actate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) CL complexes (the darker color corresponds to the more stable isomer). Shaded areas represent known contributions from homodimer CC and monomers C and L, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.
Figure 4.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of (4-)C(hlorobenzyl alcohol)-L(actate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength and Raman scattering cross section) of inserted (red) and associated (green) CL complexes (the darker color corresponds to the more stable isomer). Shaded areas represent known contributions from homodimer CC and monomers C and L, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.

Figure 5.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of (4-)C(hlorobenzyl alcohol)-G(lycolate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength, and Raman scattering cross section) of inserted (red) and associated (green) CG complexes. Shaded areas represent known contributions from homodimers CC and GG and monomers C and G, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.
Figure 5.
Comparison of experimental IR (upwards, (b)) and Raman (downwards, (c)) spectra of (4-)C(hlorobenzyl alcohol)-G(lycolate) coexpansions with He and simulated stick spectra (uppermost (a) and lowermost (d) trace, B3LYP-D3(BJ, abc)/def2-TZVP, double-harmonic approximation, wavenumber-scaled by 0.97, relative intensity based on IR band strength, and Raman scattering cross section) of inserted (red) and associated (green) CG complexes. Shaded areas represent known contributions from homodimers CC and GG and monomers C and G, as marked with their wavenumbers, leaving unshaded features as the best candidates for mixed complexes.

Figure 6.
Vibrational zero point-corrected energy differences (B3LYP-D3(BJ, abc)/def2-TZVP) between different conformers for the four systems BL, BG, CG and CL. The most stable associated complexes are aligned at zero, and all conformers up to an energy difference of 5 kJ·mol−1 above that complex are shown. Red (green) colors indicate inserted (associated) structures. Complexes in which the alcohol is hydrogen-bonded to the carbonyl oxygen atom of the ester are marked in grey and labeled =. A prime indicates structures where the ester conformation or coordination deviates from the most stable structures. Solid lines are drawn for hom stuctures in which the (S)-(−)-lactate induces a gauche(−) conformation in the alcohol and dashed lines mark het complexes. The lines do not imply structural similarity but rather best-in-class property. The connecting line pattern thus emphasizes the systematic hom preference (chirality induction by L in B and C).
Figure 6.
Vibrational zero point-corrected energy differences (B3LYP-D3(BJ, abc)/def2-TZVP) between different conformers for the four systems BL, BG, CG and CL. The most stable associated complexes are aligned at zero, and all conformers up to an energy difference of 5 kJ·mol−1 above that complex are shown. Red (green) colors indicate inserted (associated) structures. Complexes in which the alcohol is hydrogen-bonded to the carbonyl oxygen atom of the ester are marked in grey and labeled =. A prime indicates structures where the ester conformation or coordination deviates from the most stable structures. Solid lines are drawn for hom stuctures in which the (S)-(−)-lactate induces a gauche(−) conformation in the alcohol and dashed lines mark het complexes. The lines do not imply structural similarity but rather best-in-class property. The connecting line pattern thus emphasizes the systematic hom preference (chirality induction by L in B and C).

Figure 7.
The most stable associated structures for the four systems BL, BG, CG and CL (all homochiral) and the most stable heterochiral complexes for the lactate cases. Structures observed in the experiment are framed. The chirality-generating methyl group is highlighted by different colors. The different hues of green for hom and het complexes match those in Figure 2, Figure 3, Figure 4 and Figure 5 and are chosen to emphasize that it is difficult to discriminate between them experimentally.
Figure 7.
The most stable associated structures for the four systems BL, BG, CG and CL (all homochiral) and the most stable heterochiral complexes for the lactate cases. Structures observed in the experiment are framed. The chirality-generating methyl group is highlighted by different colors. The different hues of green for hom and het complexes match those in Figure 2, Figure 3, Figure 4 and Figure 5 and are chosen to emphasize that it is difficult to discriminate between them experimentally.

Figure 8.
The most stable inserted structures for the four systems BL, BG, CG and CL (all homochiral) and the most stable heterochiral complexes for the lactate cases. The experimentally dominant inserted structure is framed as in Figure 7, whereas minor observed inserted contributions are marked by thinner frames. The different hues of red for hom and het complexes match those in Figure 2, Figure 3, Figure 4 and Figure 5 and are chosen to emphasize that it is difficult to discriminate between them experimentally.
Figure 8.
The most stable inserted structures for the four systems BL, BG, CG and CL (all homochiral) and the most stable heterochiral complexes for the lactate cases. The experimentally dominant inserted structure is framed as in Figure 7, whereas minor observed inserted contributions are marked by thinner frames. The different hues of red for hom and het complexes match those in Figure 2, Figure 3, Figure 4 and Figure 5 and are chosen to emphasize that it is difficult to discriminate between them experimentally.

Figure 9.
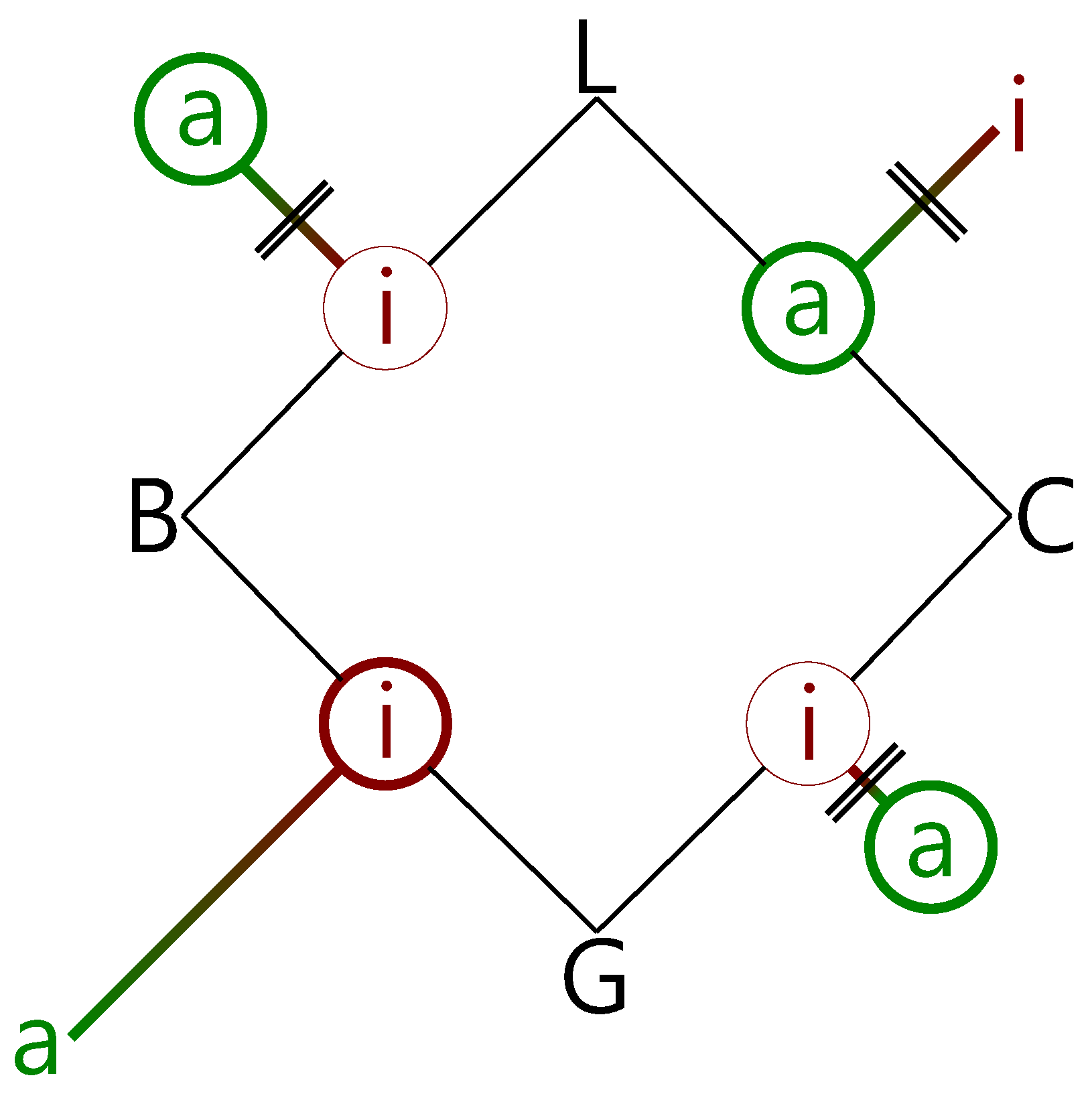
Schematic summary of the experimental findings for the combination of the alcohols B and C with the esters G and L, in terms of insertion (i) and association (a) complexes (circles). The predicted winning complexes are placed on the edges of the square, and the less stable topologies are placed away from the edges, with connecting lines qualitatively proportional in length to their predicted energy penalty. If the barrier exceeds 10 kJ·mol−1, the connecting lines are crossed (||). Observed structures are surrounded by thick circles if dominant and by thin circles if minor. The observations are consistent with insertion dominating for barriers less than 10 kJ·mol−1, if association is energetically disfavored.
Figure 9.
Schematic summary of the experimental findings for the combination of the alcohols B and C with the esters G and L, in terms of insertion (i) and association (a) complexes (circles). The predicted winning complexes are placed on the edges of the square, and the less stable topologies are placed away from the edges, with connecting lines qualitatively proportional in length to their predicted energy penalty. If the barrier exceeds 10 kJ·mol−1, the connecting lines are crossed (||). Observed structures are surrounded by thick circles if dominant and by thin circles if minor. The observations are consistent with insertion dominating for barriers less than 10 kJ·mol−1, if association is energetically disfavored.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Glycolate-based vibrational zero point-corrected B3LYP energy differences (B3LYP), electronic B3LYP and CCSD(T) energy differences ( (B3LYP), (CCSD(T))) and relative contributions of the dispersion correction for the most stable associated and inserted complexes. Also given are absolute CCSD(T) interaction energies (CCSD(T)) and CCSD(T) dispersion contributions (LED) as well as their ratios (LED). All energies are given in kJ·mol−1.
Table 1.
Glycolate-based vibrational zero point-corrected B3LYP energy differences (B3LYP), electronic B3LYP and CCSD(T) energy differences ( (B3LYP), (CCSD(T))) and relative contributions of the dispersion correction for the most stable associated and inserted complexes. Also given are absolute CCSD(T) interaction energies (CCSD(T)) and CCSD(T) dispersion contributions (LED) as well as their ratios (LED). All energies are given in kJ·mol−1.
| (B3LYP) | (B3LYP) | (CCSD(T)) | (CCSD(T)) | (LED) | (LED) | (B3LYP) | |
|---|---|---|---|---|---|---|---|
| BGi | 0 | 0 | 0 | −59.2 | −38.9 | 0.66 | 0 |
| BGa | 5.5 | 7.4 | 5.8 | −42.3 | −32.3 | 0.76 | 2.2 |
| CGi | 0 | 0 | 0 | −58.9 | −39.7 | 0.67 | 0 |
| CGa | 0.7 | 2.1 | 0.9 | −47.2 | −36.7 | 0.78 | −1.2 |
Table 2.
Lactate-based vibrational zero point-corrected B3LYP energy differences (B3LYP), electronic B3LYP and CCSD(T) energy differences ( (B3LYP), (CCSD(T))) and relative contributions of the dispersion correction for the most stable associated and inserted hom complexes and their het counterparts. Also given are absolute CCSD(T) interaction energies (CCSD(T)) and CCSD(T) dispersion contributions (LED) as well as their ratios (LED). All energies are given in kJ·mol−1.
Table 2.
Lactate-based vibrational zero point-corrected B3LYP energy differences (B3LYP), electronic B3LYP and CCSD(T) energy differences ( (B3LYP), (CCSD(T))) and relative contributions of the dispersion correction for the most stable associated and inserted hom complexes and their het counterparts. Also given are absolute CCSD(T) interaction energies (CCSD(T)) and CCSD(T) dispersion contributions (LED) as well as their ratios (LED). All energies are given in kJ·mol−1.
| (B3LYP) | (B3LYP) | (CCSD(T)) | (CCSD(T)) | (LED) | (LED) | (B3LYP) | |
|---|---|---|---|---|---|---|---|
| BLi (hom) | 0 | 0 | 0 | −58.6 | −40.4 | 0.69 | 0 |
| BLa (hom) | 2.0 | 3.4 | 3.0 | −44.2 | −33.8 | 0.76 | 2.6 |
| BLi (het) | 2.7 | 2.7 | 2.6 | −56.5 | −35.6 | 0.63 | 5.5 |
| BLa (het) | 4.0 | 5.8 | 5.5 | −41.7 | −32.7 | 0.78 | 3.3 |
| CLa (hom) | 0 | 0 | 0 | −49.6 | −38.0 | 0.77 | 0 |
| CLa (het) | 2.0 | 2.3 | 2.3 | −47.0 | −36.5 | 0.78 | 1.3 |
| CLi (hom) | 2.8 | 1.8 | 1.9 | −58.5 | −41.4 | 0.71 | 0.5 |
| CLi (het) | 5.0 | 3.8 | 4.0 | −57.5 | −36.4 | 0.63 | 5.8 |
Table 3.
Performance of the uniformly scaled harmonic wavenumber prediction for the experimental bands assigned in this work (left column) and in previous investigations of alcohol-hydroxyester complexes (right column, P = phenol, M = methanol, N = (S)-2-naphthyl-1-ethanol in combination with (R)-lactate). For PL, the most stable inserted (i) and the most stable associated complex (a) are considered. For NL, only the folded OH-addition complex is considered and adopted for NG. The OH stretching vibrations of the inserted complexes explored in this work are strongly coupled, resulting in a symmetric (s) and an antisymmetric (as) mode.
Table 3.
Performance of the uniformly scaled harmonic wavenumber prediction for the experimental bands assigned in this work (left column) and in previous investigations of alcohol-hydroxyester complexes (right column, P = phenol, M = methanol, N = (S)-2-naphthyl-1-ethanol in combination with (R)-lactate). For PL, the most stable inserted (i) and the most stable associated complex (a) are considered. For NL, only the folded OH-addition complex is considered and adopted for NG. The OH stretching vibrations of the inserted complexes explored in this work are strongly coupled, resulting in a symmetric (s) and an antisymmetric (as) mode.
| Label | Mode | () | System | Label | () |
|---|---|---|---|---|---|
| This Work | /cm−1 | /cm−1 | |||
| BLa | 3503 (OHB) | −7 | PL [17] | i (OHP) | −3 |
| BLi | 3461 (OHs) | 0 | PL [17] | i (OHL) | +6 |
| BGi | 3497 (OHas) | −4 | PL [17] | a (OHP) | +9 |
| BGi | 3453 (OHs) | −4 | PL [17] | a (OHL) | +44 |
| CLa | 3497 (OHC) | −5 | NL [16] | SR1,A (OHN) | −1 |
| CGa | 3509 (OHC) | −4 | NL [16] | SR1,A (OHL) | +25 |
| CGi | 3498 (OHas) | −3 | NG [16] | (OHN) | −1 |
| CGi | 3452 (OHs) | −1 | NG [16] | (OHG) | +27 |
| ML [3] | 1-Ia (OHM) | −22 | |||
| ML [3] | 1-Ib (OHL) | −17 | |||
| MG [3] | 0-Ia (OHM) | −25 | |||
| MG [3] | 0-Ib (OHG) | −30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lange, M.; Sennert, E.; Suhm, M.A. Attaching Onto or Inserting Into an Intramolecular Hydrogen Bond: Exploring and Controlling a Chirality-Dependent Dilemma for Alcohols. Symmetry 2022, 14, 357. https://doi.org/10.3390/sym14020357
AMA Style
Lange M, Sennert E, Suhm MA. Attaching Onto or Inserting Into an Intramolecular Hydrogen Bond: Exploring and Controlling a Chirality-Dependent Dilemma for Alcohols. Symmetry. 2022; 14(2):357. https://doi.org/10.3390/sym14020357
Chicago/Turabian StyleLange, Manuel, Elisabeth Sennert, and Martin A. Suhm. 2022. "Attaching Onto or Inserting Into an Intramolecular Hydrogen Bond: Exploring and Controlling a Chirality-Dependent Dilemma for Alcohols" Symmetry 14, no. 2: 357. https://doi.org/10.3390/sym14020357
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.