
Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond

Department of Chemistry, Johannes Gutenberg University of Mainz, D-55128 Mainz, Germany
*
Author to whom correspondence should be addressed.
Sustain. Chem. 2022, 3(4), 430-454; https://doi.org/10.3390/suschem3040027
Submission received: 26 August 2022
/
Revised: 9 October 2022
/
Accepted: 11 October 2022
/
Published: 13 October 2022
Abstract
:The electrochemical generation of highly reactive and hazardous bromine under controlled conditions as well as the reduction of surplus oxidizers and reagent waste has placed electrochemical synthesis in a highlighted position. In particular, the electrochemical dibromination and bromofunctionalization of alkenes and alkynes have received significant attention, as the forming of synthetically important derivatives can be generated from bench-stable and safe bromide sources under “green” conditions. Readily available and non-corrosive bromide salts have been utilized with a dual role as both a reagent and supporting electrolyte. However, this trend seems to change with the preparation of organobromine species. In this review, the electrochemical dibromination and bromofunctionalization of alkenes and alkynes was addressed in terms of their bromine sources and sustainability.

1. Introduction
With the increasing amount of economic, environmental and political pressure, the chemical industry faces the challenge of providing the public and the scientific community a sustainable and greener alternative to conventional chemical methods [1]. Electro-organic synthesis has become one of the most attractive research topics in recent years, providing highly versatile synthetic methodologies [2,3,4,5,6]. Using electrons as “traceless” redox reagents allows the elimination of the excess use of harmful redox chemicals with a high atom efficiency [7]. The electro-generation of highly reactive species in-situ allows the utilization of inherently safe surrogates and provides a simplified approach [8]. These advantages, in combination with the valorization of renewable feedstock and “green electricity”, makes electrochemical functionalization particularly attractive.
There have been many recent reviews discussing C–C [9,10,11,12,13,14,15], C–N [13,16,17,18], C–O [12,13,17], C–S [19,20] and C–P [12,21] bond formation under electrochemical conditions [22,23]. In parallel to this, the implementation of halogens in electro-organic synthesis has also received significant attention [24,25]. The highly versatile and valuable organohalides that serve as prevalent structural motifs in pharmaceuticals [26,27], natural products [28] and intermediates [29] have encouraged electrochemists to provide more sustainable methodologies for the installation of halo-substituents. The precise control of electron transfer at the electrode via constant electric current or potential without the addition of metal-catalysts or exogeneous oxidants overcomes the conventional halogenation methods that burden conventional synthetic protocols [30,31,32]. Furthermore, the use of inexpensive and bench-stable halides allows the controlled generation of halonium species, halogen radicals or halogens in-situ. In particular, the electrochemical generation of bromine has received significant attention. Bromine can be easily generated under electrochemical conditions and equally holds its place as a direct halogenating reagent as well as a mediator [24,33,34]. A great example of electrochemically generated bromine as a mediator in organic synthesis was demonstrated via the electrooxidation of alcohols [35], as well as via the Clauson–Kaas electro-alkoxylation of furanes in the presence of bromide and methanol [36]. The latter was also demonstrated in the preparation of the natural product hispanolone [37].
In this review, the electrochemical dibromination and bromofunctionalization of alkenes and alkynes will be discussed in terms of their bromine source and sustainability.
2. Bromine and Bromination Agents
Bromine is a dark brown-red, volatile, noxious liquid at room temperature. Due to its high reactivity, it is mainly found as alkali bromides in brines, minerals and the Earth’s crust. One of the richest sources is the Dead Sea, which has a bromide content of ca. 5 g/L [38]. However, these sources are diminishing, and their prices are increasing steadily. Currently, bromine is produced on a global scale at 390,000 t/year, and bromine demand has been increasing over the last 5 years and is predicted to rise dramatically [39].
In particular, vicinal 1,2-dibromides are prevalent motifs used in fine chemicals and flame retardants [29]. Almost half of the globally produced bromine is used to produce the latter, which can easily leach out, posing a persistent environmental problem [40,41]. As well as its environmental toxicity, bromine’s storage and transportation equally pose hazards [42,43].
The most common bromination methods include the direct use of elemental bromine or HBr, which is toxic, corrosive and creates hazardous reagent waste. Alternatively, inorganic or organic bromide salts with an external oxidant allows the in-situ generation of bromine, however, above-stoichiometric amounts of exogenous oxidizers are required under harsh conditions [44]. The utilization of inherently safe bromine surrogates such as N-bromosuccinimide and pyridinium tribromide allows the controlled liberation of Br2. Nevertheless, their preparation is difficult, wasteful and costly. Moreover, their recycling as well as their production requires elemental bromine, and therefore these agents are considered low-atom economic [45,46].
Oxidative electrochemistry provides a fundamentally greener alternative compared to the traditional strategies, as electricity replaces the usually harmful and surplus redox reagents. The relatively low oxidation potential of bromides has inspired extensive studies for the electro-generation of bromine in different solvent systems [47,48,49]. The oxidation of bromide to bromine or to its stable tribromide salt is described as a two-electron transfer process (Equation (1)), which has been extensively studied in aprotic media due to the stabilization effect of solvent, facilitating the formation of both brominating species simultaneously [47,48,49]. The first oxidation potential corresponds to the direct formation of bromine (0.82 V in acetonitrile at a platinum disk electrode), which simultaneously forms stable tribromide complexes in the presence of a high bromide concentration in aprotic media (Equation (2)) [47]. Via a second oxidation potential, bromine is generated from the aforementioned tribromide species (1.2 V in acetonitrile at a platinum disk electrode) (Equation (3)) [47].
2Br− → Br2 + 2e−
Br2 + Br− ⇄ Br3−
2Br3− → 3Br2 + 2e−
Moreover, as bromine can be generated easily from its salts under electrochemical conditions, the traditional production of bromine via the chlorine oxidation process can be completely avoided.
Bromides are popular alternatives due to their proportionate pricing compared to traditional brominating agents such as HBr and Br2 (Table 1). In particular, NaBr and KBr provide an economically greener choice, as the abundance of sodium (2.36%) and potassium metal (2.09%) in the Earth´s crust is relatively high, with sodium being the sixth most abundant element [50]. In combination with their non-corrosive natures and long shelf-lives, they are desired substitutes for smaller research facilities that struggle with the burden of conventional brominating reagents and storage issues.
Organic bromide salts bridge the obligatory protic additive issue that is associated with alkali bromide salts, providing a simpler setup. Nevertheless, these agents are more expensive, and their preparation is elaborate and therefore low-atom economic.
Via the conscious choice of bromine substitution and the precise amendment of the electrochemical parameters, an intrinsically green, efficient and viable approach can be provided [4,52,53]. In particular, careful consideration has to be made regarding the counter electrode reactions [54,55,56,57]. In combination with sustainable electro-organic chemistry and green chemistry principles, it is highly desired that bromine from a “green source” can be generated synergically, for example under linear paired electrolysis conditions [58,59,60,61,62].
3. Electrochemical Bromofunctionalization of Alkenes
The electrochemical bromination and bromofunctionalization of alkenes are well-researched areas, as their products serve as highly valuable synthetic intermediates and important chemicals. Vicinal dibromides, bromohydrins and bromoesters serve as useful intermediates for pharmaceuticals and fine chemicals [29,63]. Moreover, the halogenated species can be simply further functionalized resulting in a plethora of elaborated products. Therefore, the simultaneous and selective installation of one or two functionalities across the alkenes has been extensively researched as the desired products can be obtained under a simplified set-up and “green” conditions. The electrochemical bromofunctionalization of alkenes is mostly represented by the utilization of bromide salt with a dual role as both a reagent and supporting electrolyte in aprotic media accompanied by a protic additive such as water or short-chained alcohols. These agents do not only provide the respective hydroxyl or ether functionalities but also compliment the cathodic reaction liberating H2, providing a “clean” by-product [54]. The employment of organobromines as halogenating agents has also become popular as it allows bromine to form as a result of an electrochemical redox process, excluding the need for the sacrificial half reaction. Nevertheless, these agents are usually harmful and therefore should be avoided.
3.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
Pioneering work in the field was demonstrated by Torii in the early 80s, reporting the bromofunctionalization of alkenes in the presence of aq. NaBr, which popularized the indirect electrochemical oxidation of olefins using stable and inexpensive metal halide salts [64,65,66,67,68]. This form of a bromine source has received particular attention due to its ability to tame hazardous and toxic molecular bromine in-situ under controlled conditions. It is worth mentioning that the complementary half-reaction´s electrode choice is usually a low-hydrogen-overpotential material such as platinum or nickel, which supports cathodic hydrogen evolution. Nevertheless, platinum group metals are depleting, costly and highly contaminating [69,70,71].
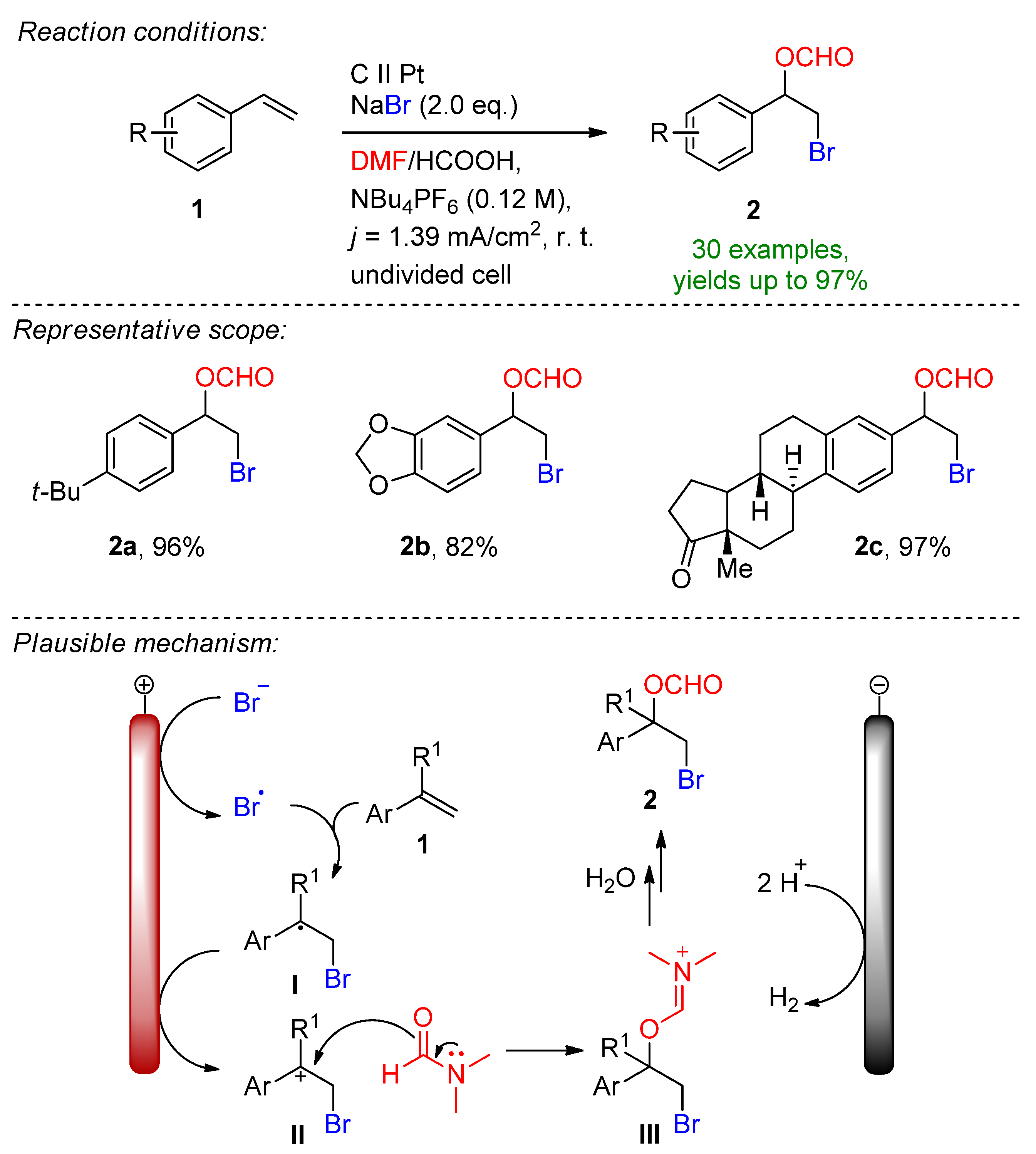
In 2019 Hu, Fang and Mei reported the electrochemical radical formyloxylation–bromination of various alkenes in a regio- and chemoselective fashion [72]. The implementation of stable NaBr as a radical bromine source makes this approach sustainable. DMF serves a dual role as both the solvent and the formyloxylation reagent, while acetic acid supports the cathodic reaction. Para-substituted electron-rich styrene derivatives afforded excellent yields, while electron-deficient and strongly electron-withdrawing groups prolonged the reaction time. The naturally derived safrole-type and estrone-type derivates also readily underwent electrochemical formyloxylation, proving the possibility for the late-stage functionalization of pharmaceutical scaffolds. In total, 30 examples were reported with excellent yields up to 97%. The scalability was proved via a gram-scale experiment (6.0 mmol) resulting in an isolated yield of 90%. The method was extended to the formyloxylation–chlorination and formyloxylation–trifluoromethylation of alkenes as well. The authors suggest that bromide is oxidized at the anode to bromo-radicals, which then rapidly combine with arylalkene 1 to form the benzyl radical I. Via a second oxidation, the forming benzylic carbocation II is subjected to nucleophilic attack by DMF to iminium intermediate III, which is subsequently hydrolyzed resulting in the formyloxylated brominated product 2 (Scheme 1).
The electrobiocatalytic bromolactonization was reported by Bormann and Holtmann on carbon-nanotube-modified gas-diffusion electrodes [73]. The paper reports the cathodic reduction of ambient oxygen to H2O2 at a reduced overpotential in a divided cell separated via a proton exchange membrane. Curvularia inaequalis (CiVCPO), a vanadium-dependent chloroperoxidase enzyme converts the forming peroxide in the presence of potassium bromide into hypobromite, which leads to the bromolactonization of 5-pentenoic acid. The reaction is complimented via the oxidation of water at a platinum anode. The superiority of the reaction lies in the oxygen-enriched, modified-carbon-nanotube diffusion electrode that significantly lowers the overpotential for peroxide formation, which is a sensible factor for the enzymatic catalytic reactivity [74].
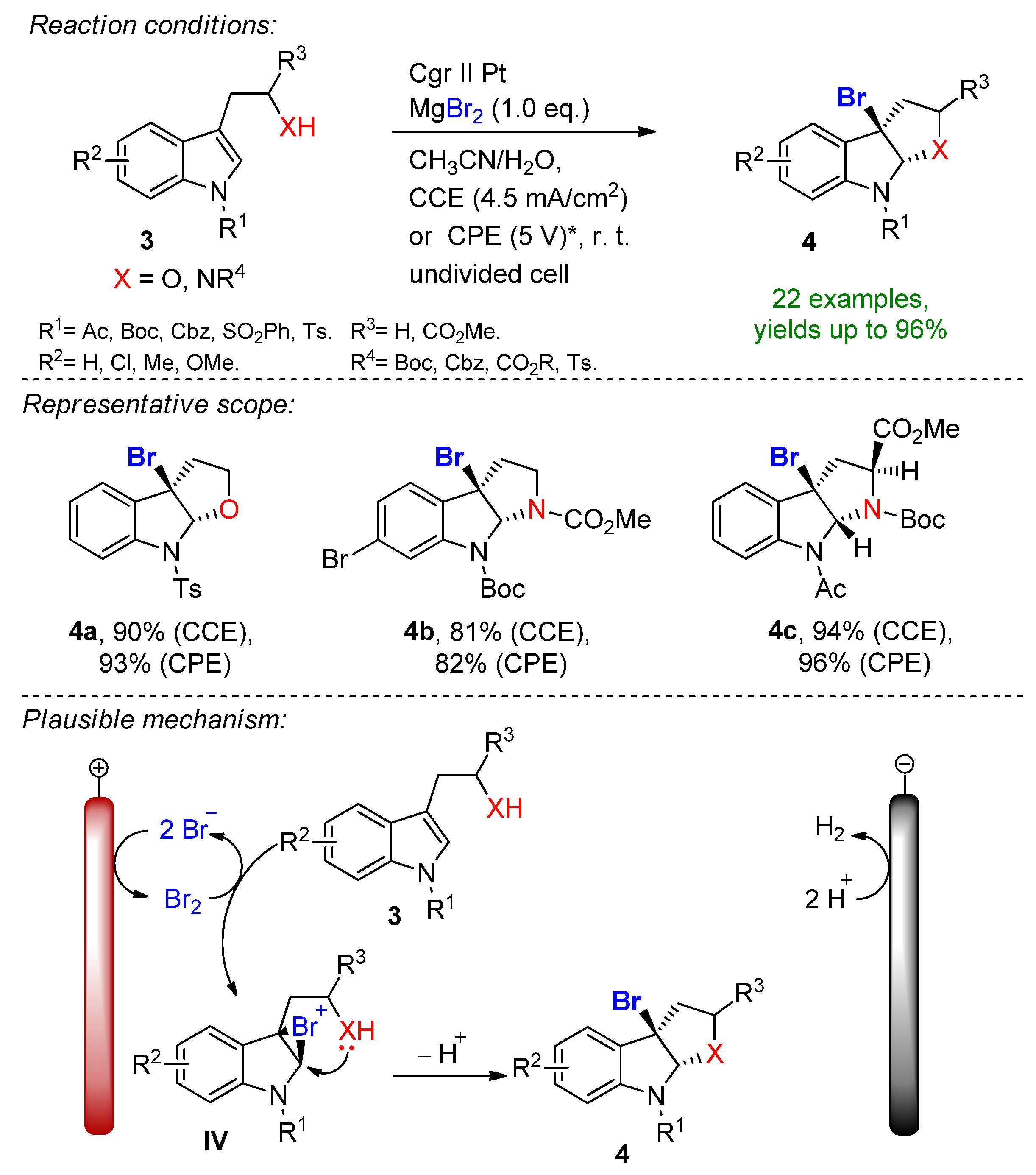
The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivatives was recently reported by Wu and Vincent [75]. This efficient protocol features MgBr2 with a dual role as a halogen source and supporting electrolyte. The reaction proceeds in an undivided cell under both constant current (CCE) and constant potential (CPE) conditions in the presence of 1 eq. MgBr2. Magnesium bromide is oxidized at the carbon anode into bromine in an acetonitrile/water solution under ambient conditions. It is worth mentioning that under CPE, the constant potential window is maintained via measuring the potential against a reference electrode. Here, the constant terminal voltage of the cell was determined via measuring the constant terminal voltage between the electrodes, which is not an adequate parameter for CPE conditions. The dearomative reaction proceeds via the formation of bromonium intermediate IV, followed by intramolecular cyclization to form the corresponding brominated derivatives with excellent yields (Scheme 2). This simple and environmentally friendly set up featured 22 examples with up to 96% isolated yields and a broad scope of functional group tolerance.
The synthetic utility was demonstrated via the electrochemical bromocyclization and further functionalization of L-tryptophan-derived diketopiperazine (5) to (−)-epi-amauromine (7a) and (+)-novoamauromine (7b), which was reported to be superior to the conventional procedure using stoichiometric NBS (Scheme 3) [76].
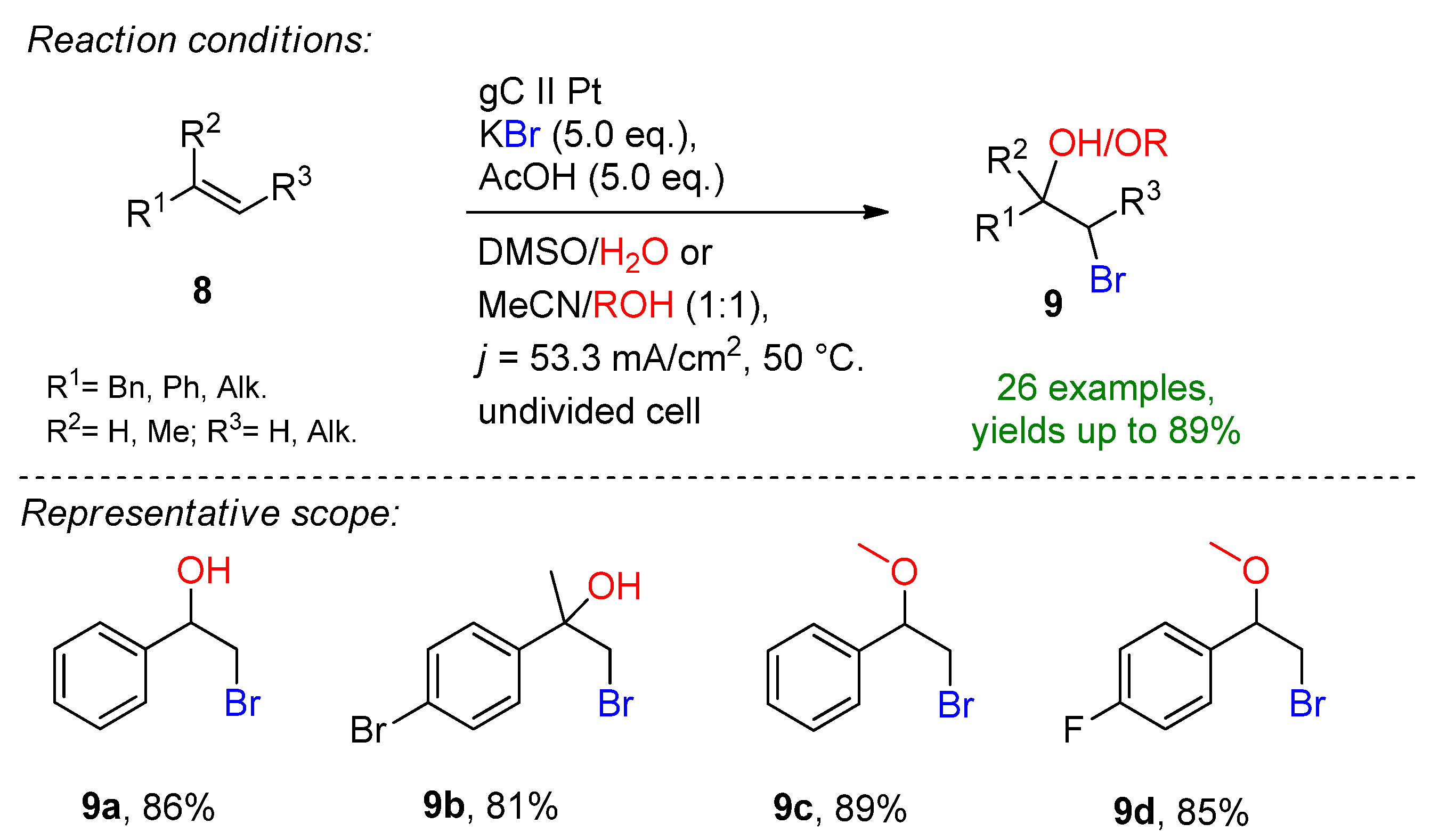
Solvent stabilizing effects have been reported in electrochemical synthesis, for example using hexafluoroisopropanol (HFIP) [77,78,79,80,81,82]. Yoshida demonstrated the formation of α-bromocarbonyls and bromohydrins via a low-temperature DMSO-stabilized halogen cation pool method [83,84]. Based on these reports, a sophisticated technique for the electrochemical bromohydrin and bromohydrin ether formation was reported in a chemoselective fashion (Scheme 4) [85]. The anodic oxidation of potassium bromide on a glassy carbon electrode in the presence of 10 eq. acetic acid in DMSO/H2O allowed the corresponding bromohydrins to be produced in excellent yields. By changing the solvent system to acetonitrile/alcohol, bromohydrin ethers were obtained. The key additive of this approach is the addition of acetic acid or trifluoroacetic acid. In the absence of the additive, the yield drops dramatically. The reaction is complemented with hydrogen evolution at a platinum cathode.
Sun and co-workers reported an elegant way of on-site bromination and hydrogenation in a simultaneous fashion [86]. The paired electrolysis takes place in a H-type cell setup divided by a Nafion membrane. This method demonstrates high halogenation flexibility and functional group tolerance, as the bromination reaction is spatially separated from the electrolysis event. The key principle is the use of bench-stable NaBr as a halogen source and the utilization of cathodic hydrogen evolution to achieve a high atom economy and energy efficiency. The anodically generated bromine gas from aqueous sodium bromide in acidic media is transferred into a separate compartment, where ideal conditions for the corresponding substrates can be set up. Likewise, cathodically generated hydrogen gas is used in the presence of Pt/C, demonstrating the synergic pairing of on-site bromination and hydrogenation. This is an excellent example for the utilization of the redox properties of the electrochemical cell.
Similarly, Hilt and his group published a linear paired electrolysis for the electrochemical dibromination of alkenes with the realization of 200% current efficiency for stoichiometric transformations in the presence of oxygen [87]. The dual role supporting electrolyte and bromide source NBu4Br is used as a brominating agent. Bromide undergoes direct oxidation at the anode to bromine and is produced mediated via reductively formed H2O2 determining the theoretical applied charge of 1 F (Scheme 5). The stable tribromide species are the results of the stabilizing effect of acetonitrile and the high concentration of bromide. Using this method, the authors reported 13 examples with good to excellent yields, providing 11c quantitatively with a current efficiency of 200%. The reaction proved to have a good functional group tolerance via performing the Glorius-test [88,89]. The method could be adopted to the bromination of arenes and for the iodination of alkenes as well.
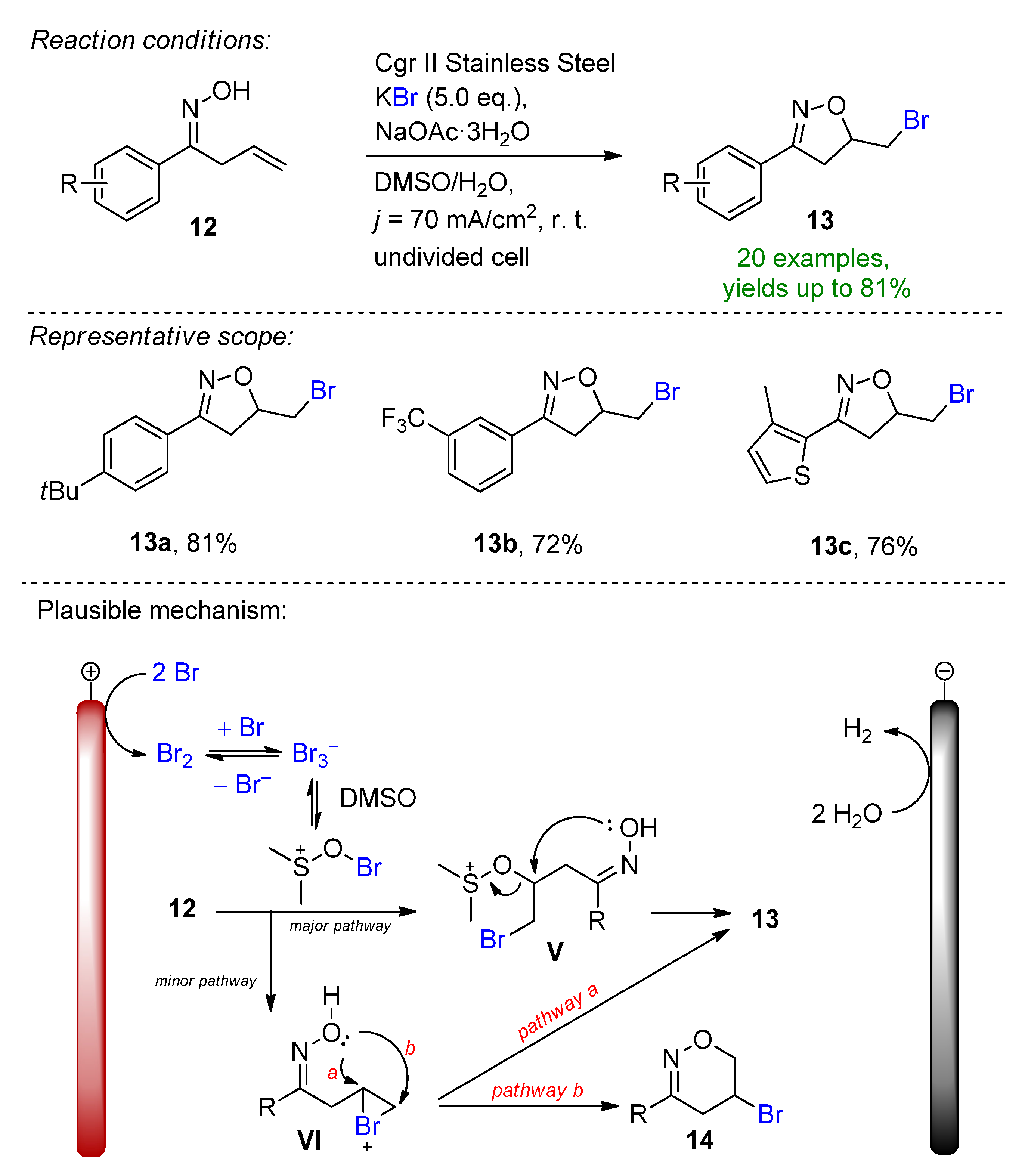
A highly regioselective electrochemical protocol for the synthesis of isoxazolines from β,γ-unsaturated ketoximes via cascade C–O and C–Br annulation was reported (Scheme 6) [90]. The stable inorganic potassium bromide has a dual role of serving as a bromine source as well as a supporting electrolyte. The key features of this method are the generation of DMSO-stabilized bromine in-situ and the employment of a sodium acetate base under high current density conditions to form the desired bromoethyl-substituted isoxazolines. The method demonstrated excellent functional group tolerance and a high selectivity, and the isoxazoline derivatives could be obtained with up to 81% isolated yields. The presence of the base as well as the electrode material choice is crucial to the success of the reaction. The scalability in both batch-type and flow electrolysis were demonstrated.
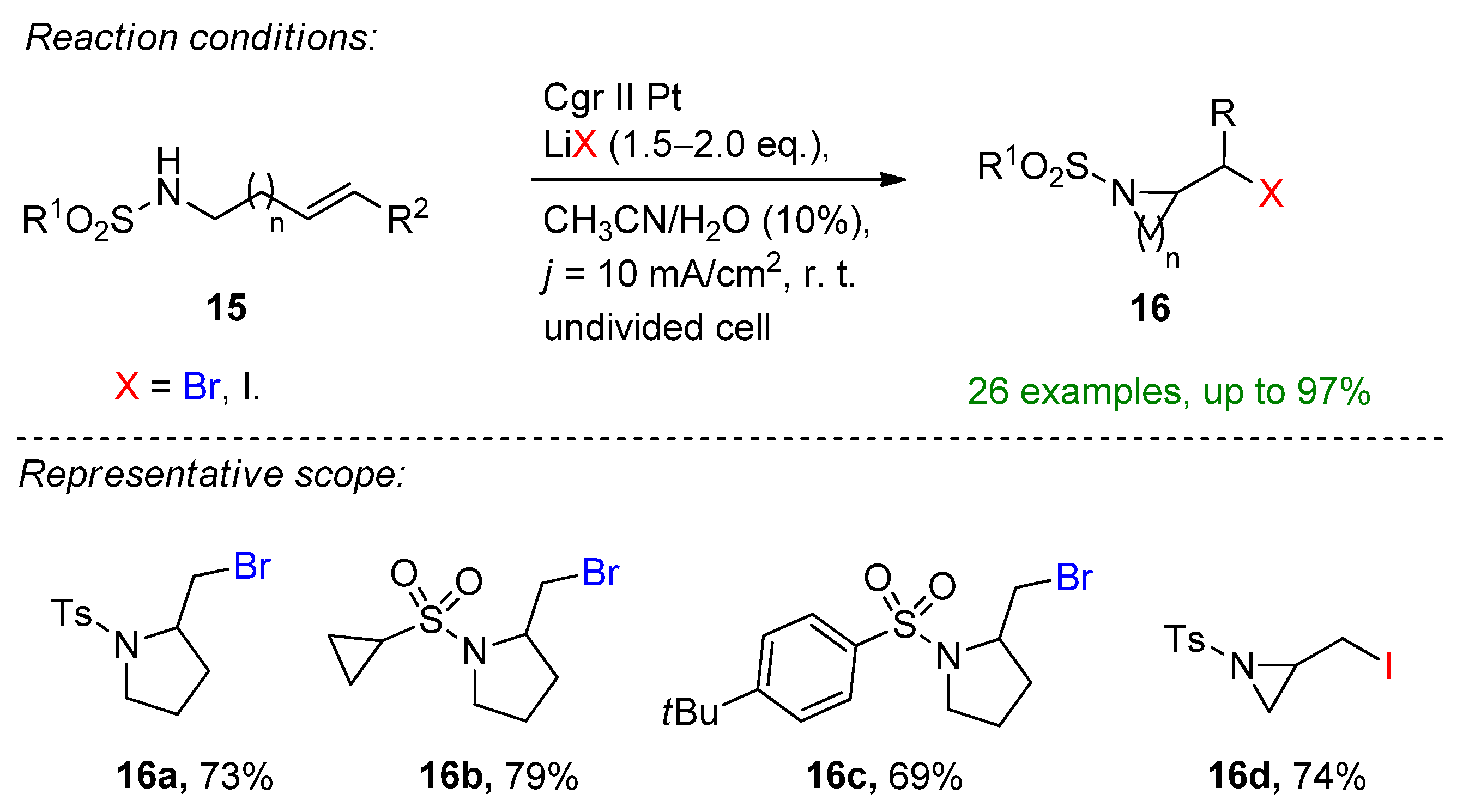
Yin and co-workers introduced an oxidant- and base-free electrochemical intramolecular halo-amination of unactivated alkenes to form diverse brominated N-heterocycles [91]. They provide a simple electrochemical protocol using bench-stable LiBr or LiI as a halide source and a supporting electrolyte furnishing a dual role. The reaction proceeded smoothly at room temperature, providing 26 examples combined with the iodo-cyclization in excellent yields. The reaction tolerates labile functional groups such as cyclopropyl, substituted aromatics and heterocycles. Moreover, in the presence of LiI, highly challenging N-heterocycles such as the three-membered aziridine or six-membered piperidine could be formed (Scheme 7).
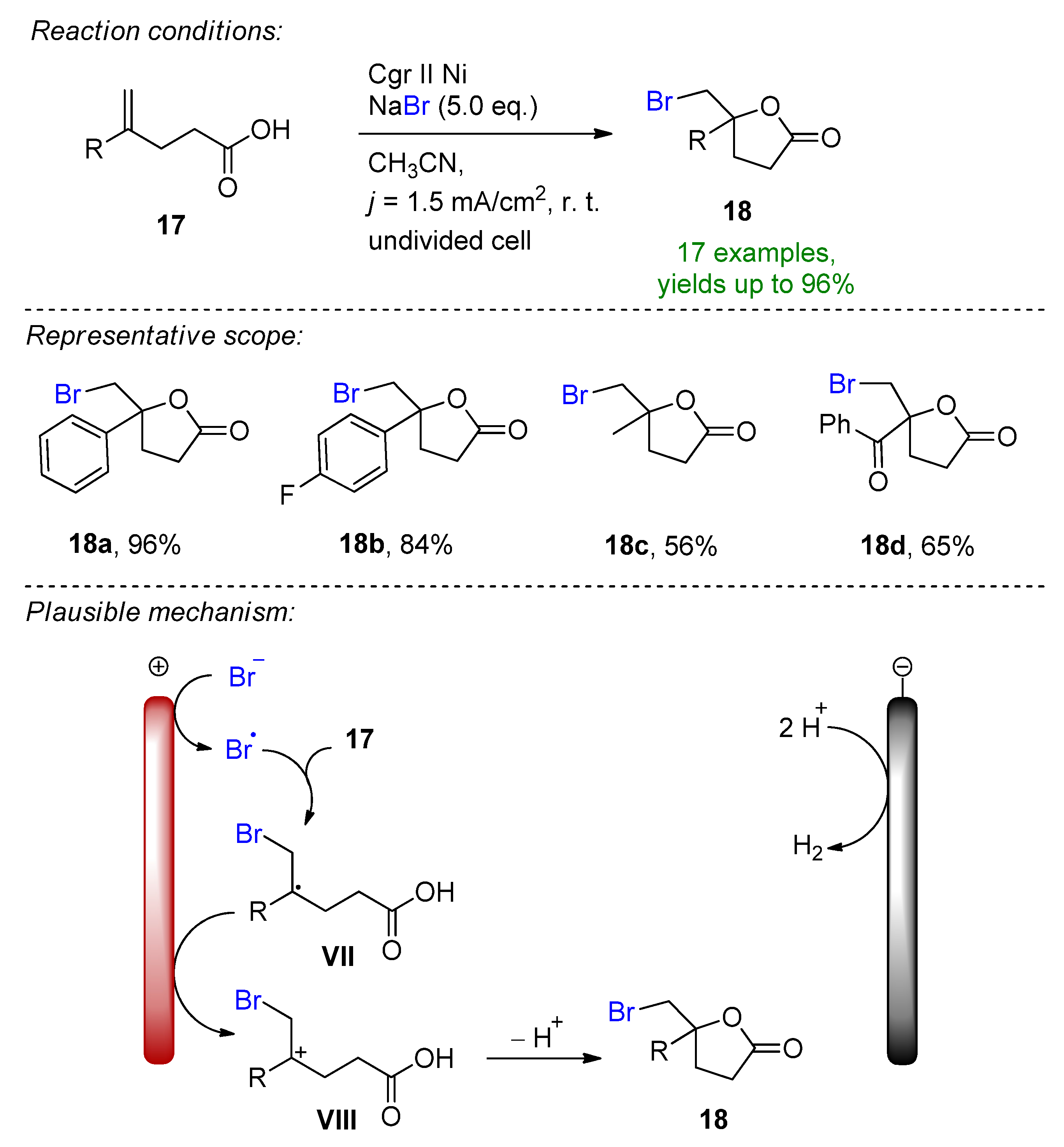
Similarly, the electrochemical oxidative bromolactonization of unsaturated carboxylic acids was reported for the first time [92]. This environmentally friendly approach features sodium bromide as the halogen source in acetonitrile, which is oxidized at the carbon rod anode to give the corresponding bromine radical, which directly reacts with the alkene of the carboxylic acid to form intermediate V (Scheme 8). After subsequent oxidation to the stabilized cationic intermediate VI and cyclization, the corresponding bromoethylated γ-lactones could be formed with excellent yields. Control experiments also supported the proposed mechanism, as in the presence of radical scavengers or molecular bromine, the yield dropped dramatically. The authors propose hydrogen evolution as a cathodic counter-reaction at a nickel electrode.
The electrochemical bromination of electron-deficient alkenes in quinones, coumarins, quinoxalines and 1,3-diketones has also been reported [93]. The synthetically useful organohalides could be obtained using bromides on graphite felt in combination with a platinum cathode. The key feature of this bromination method is the in-situ formation of HBr from KBr and H2SO4, which is anodically oxidized to bromine. Control experiments with 2,6-di-tert-butyl-4-methylphenol suggest a radical pathway by the formation of the halogen.
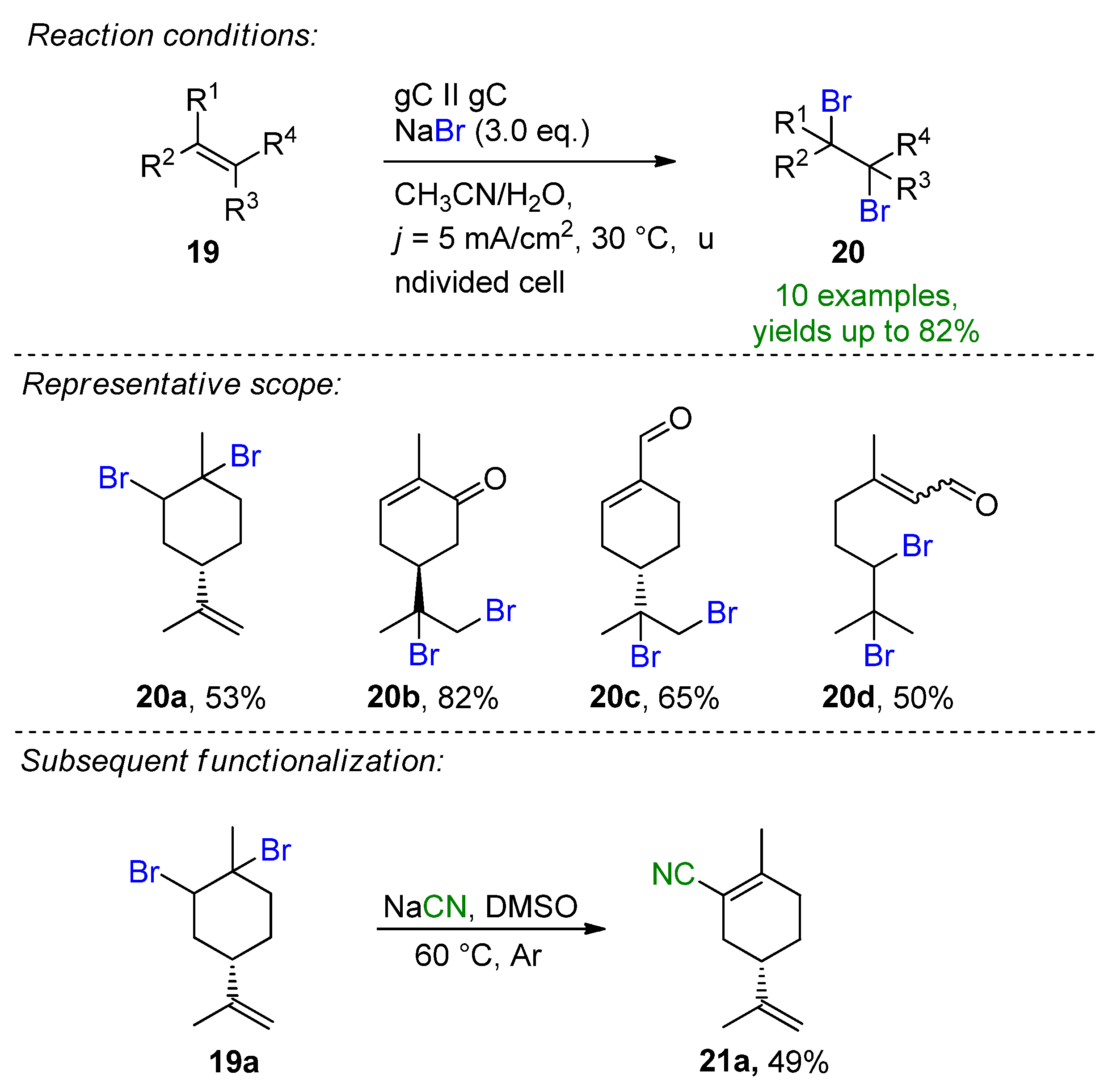
Recently, Waldvogel et al. published the selective electrochemical bromination of terpenes and naturally derived alkenes [94]. The challenging halofunctionalization of renewable feedstock was demonstrated by the employment of inexpensive and bench-stable NaBr with a dual role of being both a bromine source and supporting electrolyte. The test substrate, limonene, could be brominated selectively under ambient conditions giving the brominated derivative in a 53% yield. The optimization procedure showed that the careful consideration of the MeCN/H2O solvent system as well as the NaBr as a bromide source are important features of the method. Other inorganic or organic bromide salts resulted in a diminished yield or no conversion. The method could be successfully extended to linear and cyclic monoterpenes, terpenoids and phenylpropanoids to give 10 desired vicinal 1,2-dibromo derivatives, with the bromination of carvone representing the best yield of 82% (20b, Scheme 9). A slight change in the electrochemical parameters provided the tetrabrominated limonene derivative in a 74% isolated yield. The reaction proved to be scalable and a synthetic utility could be demonstrated via subsequent functionalization to the α,β-unsaturated nitrile derivative 21a for the first time. Cyclic voltammetry studies support the oxidative bromine formation in-situ, which is complimented by the liberation of H2 at the cathode.
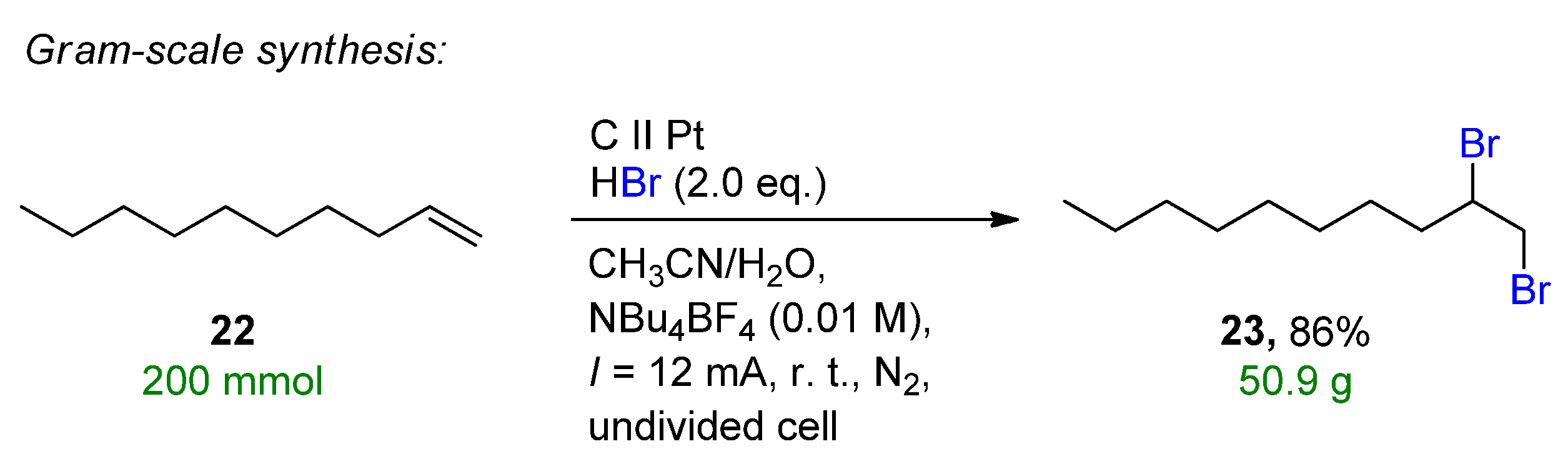
Another form of metal-free in-situ bromine generation is the use of HBr as a bromine source. Lei et al. reported an electrochemical oxidative clean halogenation of alkenes using HBr on a carbon anode and platinum cathode, providing 23 examples with up to 89% isolated yields under constant current conditions [95]. The method provides a general halogenation system of HX/MX including the chlorination and bromination of various heteroarenes, arenes, alkenes, aliphatic hydrocarbons and alkynes as well. Under the optimal conditions, the gram-scale synthesis of the electrochemical dibromination of dodecane (22) allowed the formation of 1,2-dibromododecane (23) with a yield of 86% and 50.9 g isolated clean product (Scheme 10).
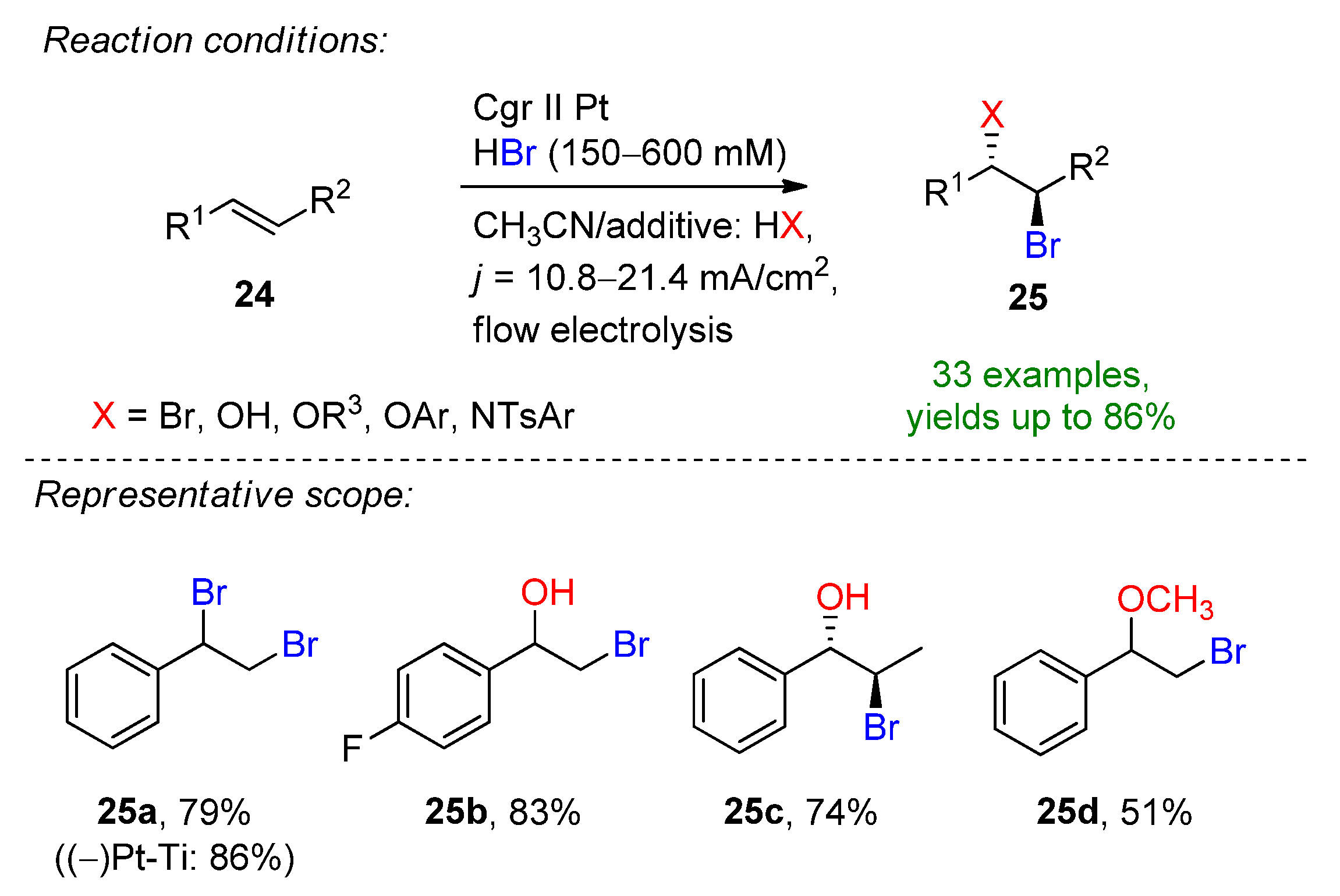
Encouraged by these results, the Wirth group described the electrochemical bromination and bromofunctionalization of activated and unactivated alkenes in a flow reactor under single-pass conditions [96]. The 600 µL reactor was equipped with platinum foil electrodes separated by a 500 µM fluorinated ethylene propilene (FEP) spacer. The small interelectrode distance allowed the omission of the addition of water and supporting electrolyte, which was described in previous studies. The optimized conditions for the dibromination of styrene were found in the presence of 6 eq. HBr in pure acetonitrile. The combination of a 0.4 mL/min flow rate, 4 F applied charge and platinum electrodes allowed the formation of 25a in a 79% isolated yield (Scheme 11). When the optimized conditions were applied using a platinum-coated titanium cathode, 25a could be obtained in an excellent yield of 86%. Via switching the co-solvent, the electrochemical bromohydrin formation could be also targeted. The applicability of this method was demonstrated on several aromatic and aliphatic alkenes. In total, 33 examples were provided exhibiting the formation of dibrominated, tetrabrominated, hydrobrominated and alkoxybrominated products with good to excellent yields. The scalability of the flow-procedure was also demonstrated via applying the conditions for 9.5 h providing 25a in an isolated yield of 65% and a productivity of 413 mg/h.
3.2. Organohalides as Bromine Sources
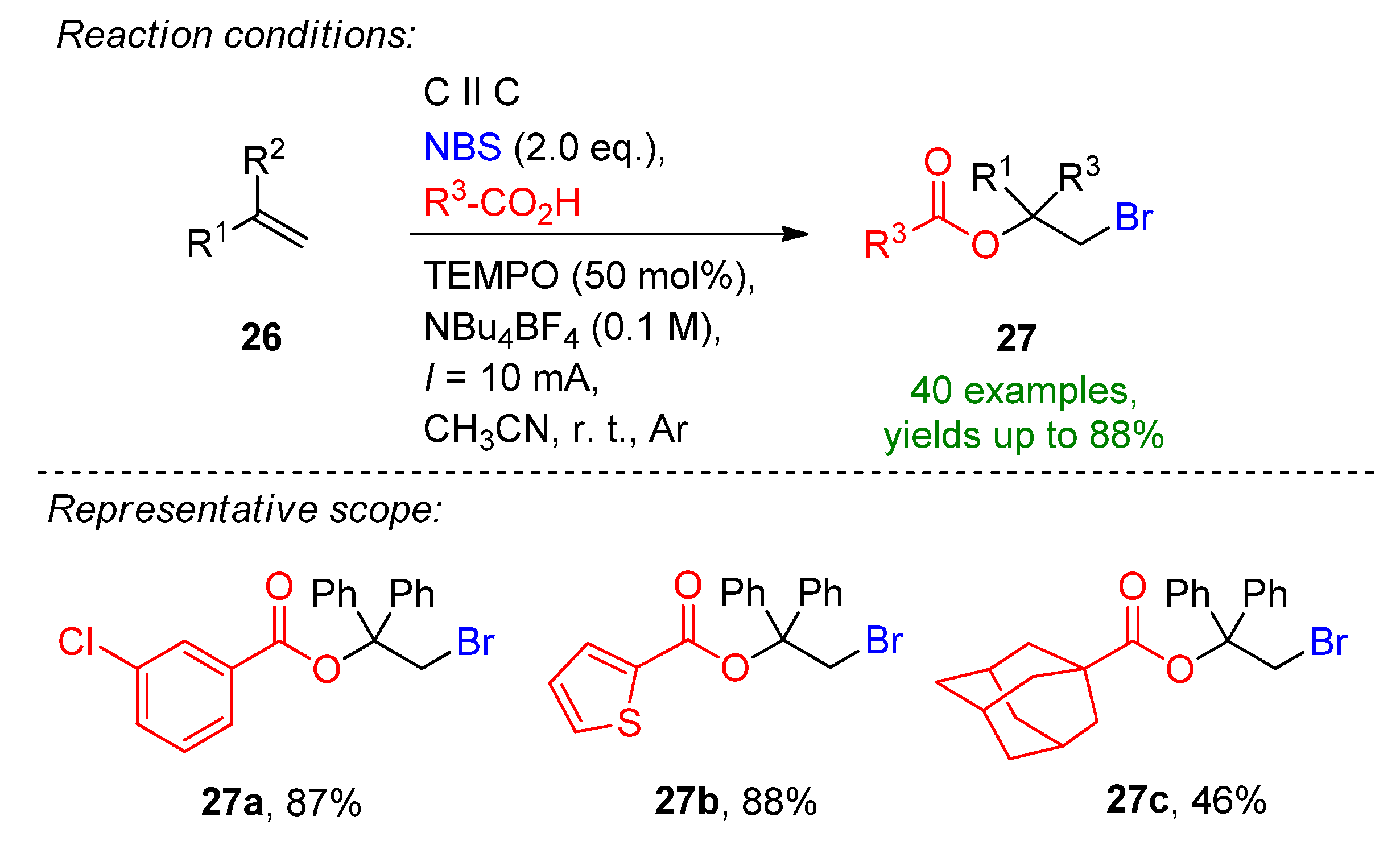
Li et al. reported a three-component, TEMPO-mediated 1,2-bromoesterification of alkenes with the aid of carboxylic acids and N-bromosuccinimide [97]. The method allows the simultaneous addition of a C–O and C–Br bond to form β-bromoalkyl esters, which are excellent intermediates for natural products and pharmaceutical agents [97,98]. The reaction proceeded well in the presence of 50 mol% TEMPO, 2 eq. of carboxylic acids and 2 eq. NBS to form 27b in an 88% isolated yield (Scheme 12). The optimized conditions were extended to a variety of carboxylic acids and alkenes, and a total of 40 examples were provided with excellent yields. Even challenging substrates such as adamantane-1-carboxylic acid and amino acids were tolerated. The reaction is complemented via H2 evolution at the cathode.
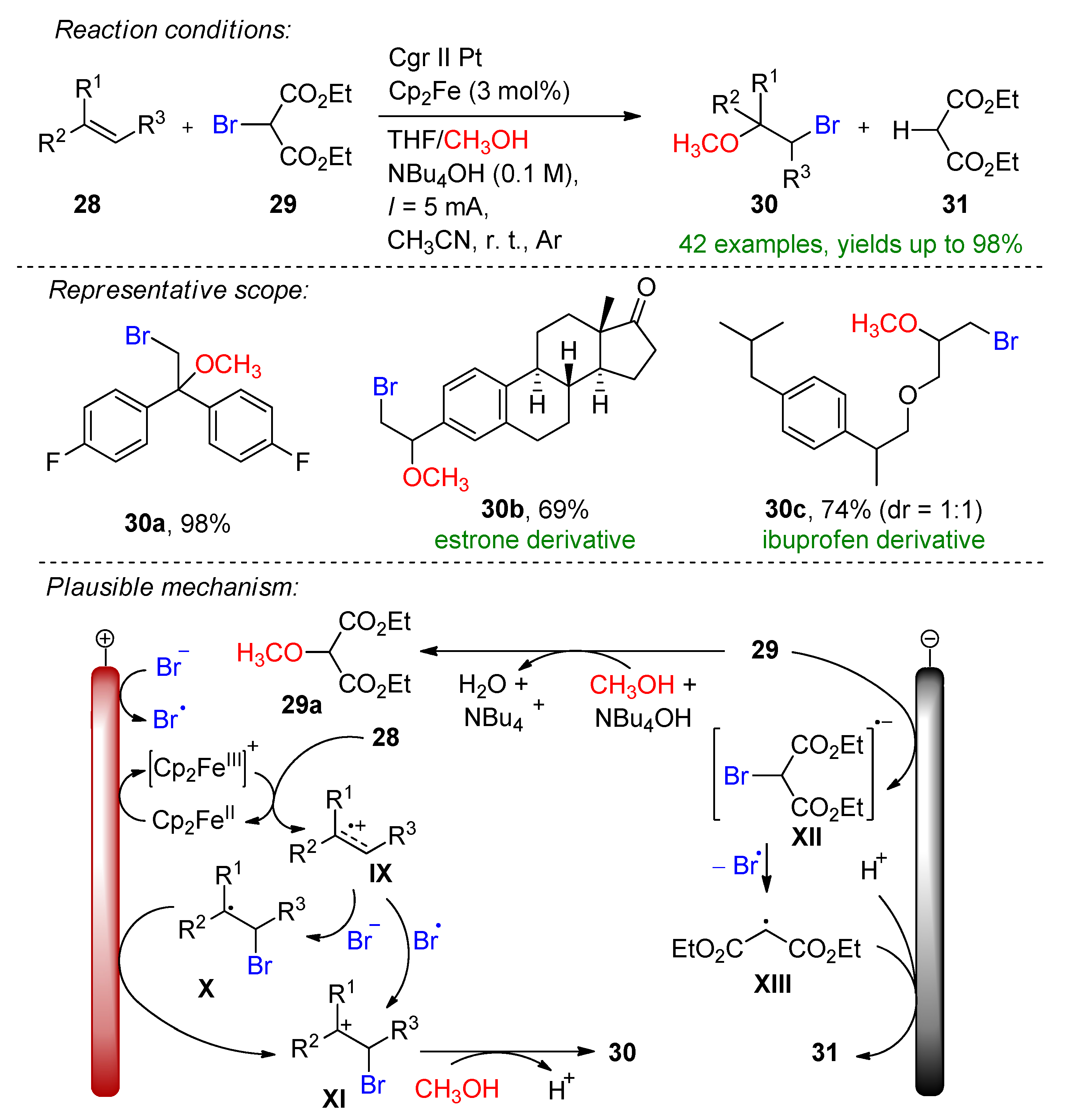
Paired electrolysis methods are generally preferred over a sacrificial approach [57]. Here, the same authors introduced electrochemical alkoxyhalogenation and organohalide dehalogenation within a convergent strategy [99]. Diethyl-2-bromomalonate (29) serves as the bromine source. The key promoters of this method are NBu4OH and Cp2Fe, as omitting one or both decreases the yield. The reaction mechanism was postulated via the mediated oxidation of alkene 28 by Cp2Fe+, which forms the radical cation IX. Intermediate IX is scavenged by the bromine radical or bromine that is either formed via the cathodic reduction of bromomalonate to XIII or via the SN2 reaction induced by the supporting electrolyte (Scheme 13).
The bromide could also be oxidized in-situ to form bromine. The formed alkyl cation XI is complemented via methanol to provide the desired product. The formation of malonic ester (31) is depicted via GC and GC-MS. The convergent strategy was demonstrated on various aryl alkenes bearing para-, meta- or both substituents and alkyl halides. The alkoxyhalogenation of bioactive molecules such as adamantene, estrone and ibuprofen derivatives were also demonstrated. In combination, 43 examples were provided with up to a 98% yield.
A merging e-shuttle reaction for the retro-dihalogenation reaction was developed by Waldvogel and Morandi, enhancing the already existing halogenation reactions (Scheme 14) [62].
The attraction of this elegant, electrochemically promoted shuttle reaction is the use of non-corrosive vicinal 1,2-organodihalides as halogen promoters, which eliminate the use of low-atom -efficient halogenating agents and surrogates. Readily available, inexpensive, non-oxidizing 1,2-dibromoethane serves as the bromine source. The aforementioned organohalide undergoes reductive dehalogenation at the cathode and is transferred to an acceptor alkene, providing a convergent application for the reductive dehalogenation and formation of fine chemicals in one step. The reaction proceeds readily on cheap graphite electrodes in the presence of 1 v/v% HFIP in acetonitrile. The additive suppresses the oxidation of alkene and promotes dehalogenation at the cathode. Via this method, the dibromination, dichlorination, thiobromination and thiochlorination of alkenes was facilitated. The method demonstrates an extensive functional group tolerance as alkenes with alcohols, ethers, silylethers, sulfones, phosphones, aromatics and alkyne functional groups equally remained intact. Moreover, the electrochemical dehalogenation of persistent and toxic hexachlorocyclohexane (HCH) to benzene was also described. Various lindane-contaminated soil samples were subjected to the optimized conditions to yield the corresponding benzene and vicinal 1,2-dichloride in excellent yields.
While the electrochemical dibromination and bromofunctionalization of alkenes are mainly characterized by the application of metal bromide salts as potential bromine sources, there are more and more methods that provide the electrochemical generation of bromine in a synergistic operation. The advantage of the former is the utilization of bench-stable and safe bromide salts. These, unfortunately, usually require an obligatory protic additive for solubility reasons and, except for organic salts, are not metal-free. Replacing the alkali bromide salts with usually toxic organobromine compounds overcomes the solubility issues, providing a simpler setup. These procedures allow the elimination or the utilization of the sacrificial half-reaction as bromine can be generated in a linear-paired fashion [62,87].
4. Electrochemical Bromofunctionalization of Alkynes
Although there have been substantially less protocols reported, alkynes still represent a popular electrochemical target functionality [100]. In terms of halofunctionalization, they have also received significant attention, as the forming organohalides are excellent synthons for heterocyclic molecules and other synthetic intermediates [101,102].
4.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
Wang et al. reported the electrochemical oxyhalogenation of alkynes in the presence of KBr or HCl to form the corresponding α,α-dihaloacetoketone derivatives [103]. The reaction proceeds readily in a divided cell equipped with a cation exchange membrane to form the desired products in good to excellent yields. LiClO4 serves as an additional supporting electrolyte and platinum electrodes promote the oxidation of halides at the anode and the liberation of hydrogen at the cathode. The authors suggest the in-situ formation of HOBr and HOCl as powerful halogenating agents.
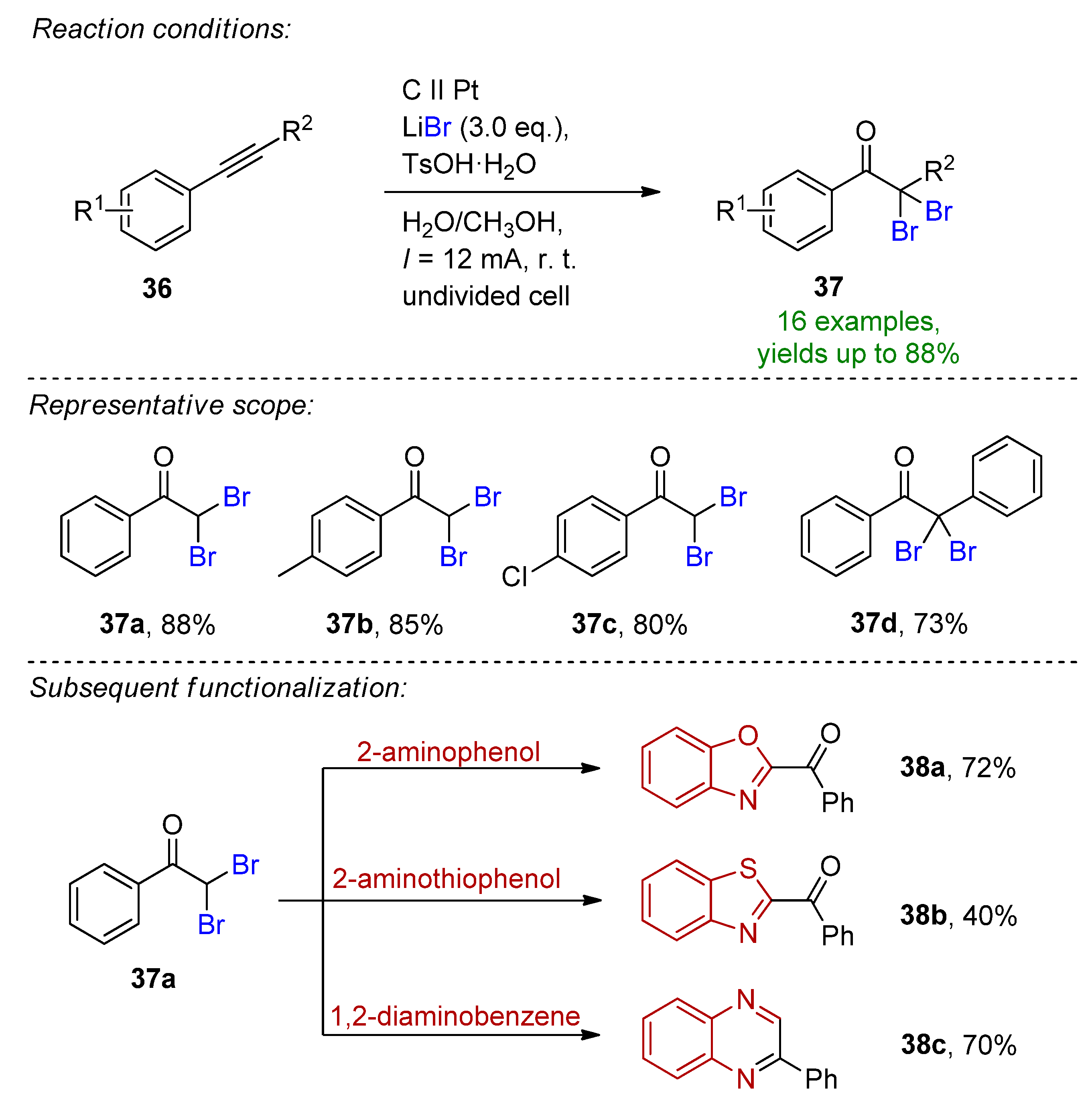
An improved electrochemical method for the oxidative functionalization of arylalkynes was published under undivided cell conditions [104]. The desired α,α-dibromoacetophenone derivatives readily formed in the presence of 3 eq. LiBr and p-toluenesulfonic acid monohydrate in aqueous media including 10% methanol. H2O (100%) as well as H2O/MeOH (50%) resulted in reduced yields, suggesting that water had a significant role in the reaction and the co-solvent was beneficial as it promoted the solvation of phenylacetylene. The combination of LiBr and p-TsOH·H2O was obligatory to the success of the reaction as switching either the bromide salt or the acid resulted in diminished yields. Under the optimized conditions, 16 different α,α-dibromoacetophenones were presented with yields up to 88%. The reaction proved to be scalable both in a batch and in-flow setup, and the synthetic applicability of the products were presented in the synthesis of three different heterocyclic compounds (Scheme 15).
A highly stereoselective, three-component annulation–sulfonylation method of 1,6-enynes to 1-indanones was reported in the presence of NaI or NaBr [105]. The test substrate, 1,6-enyne 39 was readily converted to the corresponding bromosulfonylated product 40 in the presence of 2 eq. of NaBr and 3 eq. of 4-methylbenzenesulfonylhydrazine as the sulfonyl donor (Scheme 16). The halide salts play a triple role of an electrolyte, redox catalysts and halogenating reagents. The reaction performed best at platinum electrodes in a solvent mixture consisting of THF/H2O (1:1). The highly selective nature of the reaction was postulated to rise from the electrochemically formed arylsulfonyl halide intermediate, that undergoes homolytic fission to the arylsulfonyl radical and bromine radical. The latter is oxidized at the anode to a bromonium ion. The resulting arylsulfonyl radical is scavenged by 39 to form intermediate XIV, which then, due to steric hindrance, selectively forms the Z isomer.
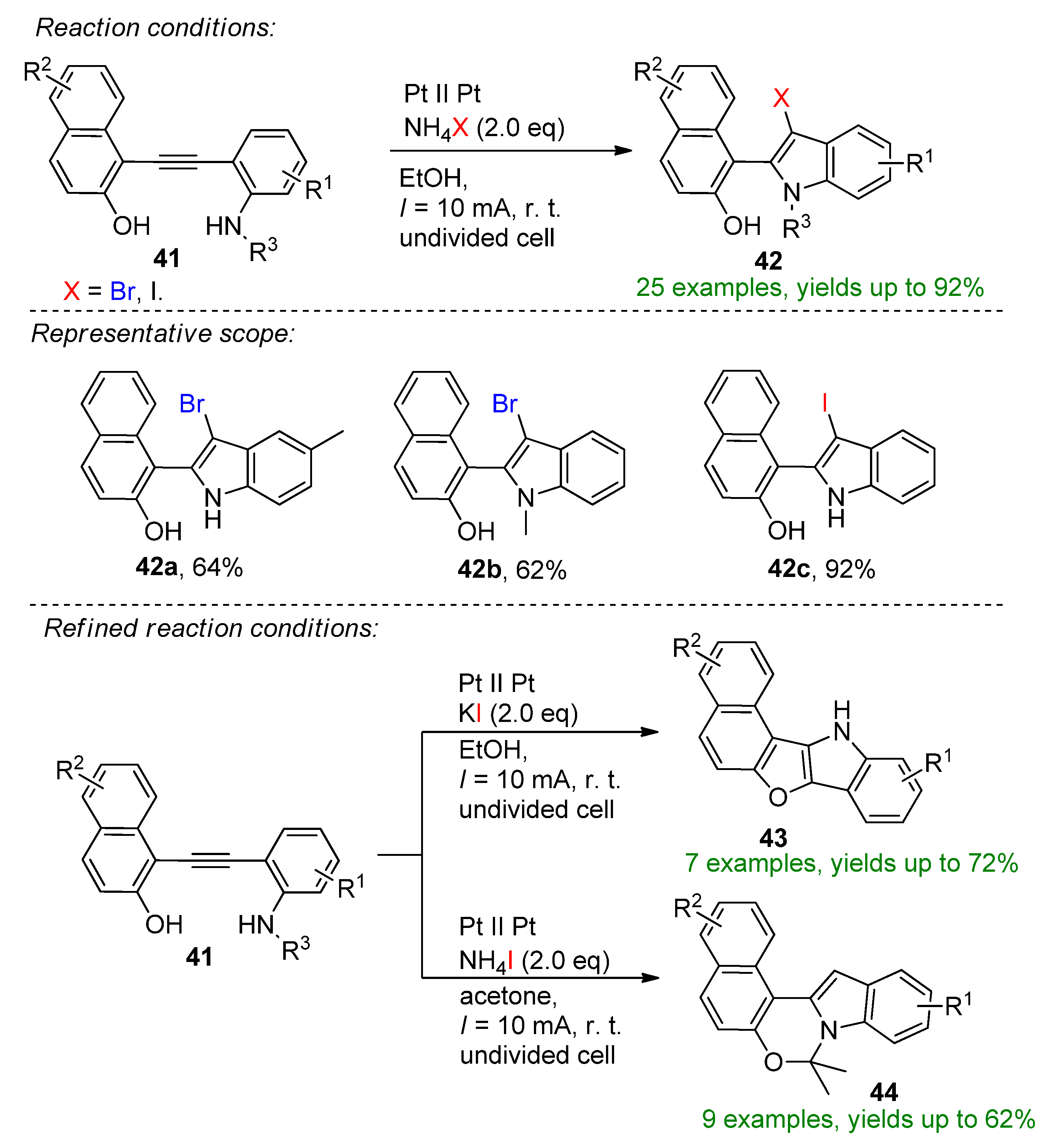
The direct halogenation of o-arylalkynylanilines to C3-halogenated indoles using NH4Br both as electrolyte and the halogen source was reported [106]. The reaction proceeded smoothly on platinum electrodes in undivided cell conditions using ethanol as solvent. The reaction also proceeded to yield to corresponding 3-iodoindoles in the presence of ammonium iodide, providing access to skeletally diverse indole derivatives with yields up to 92%. Slightly changing the electrochemical parameters allowed the formation of unexpected indole derivatives. Changing the halide source to KI favored the generation of pentacyclic indole (43), while in the presence of NH4I in acetone solvent, 44 was formed establishing the diverse applicability of the electrochemical method (Scheme 17).
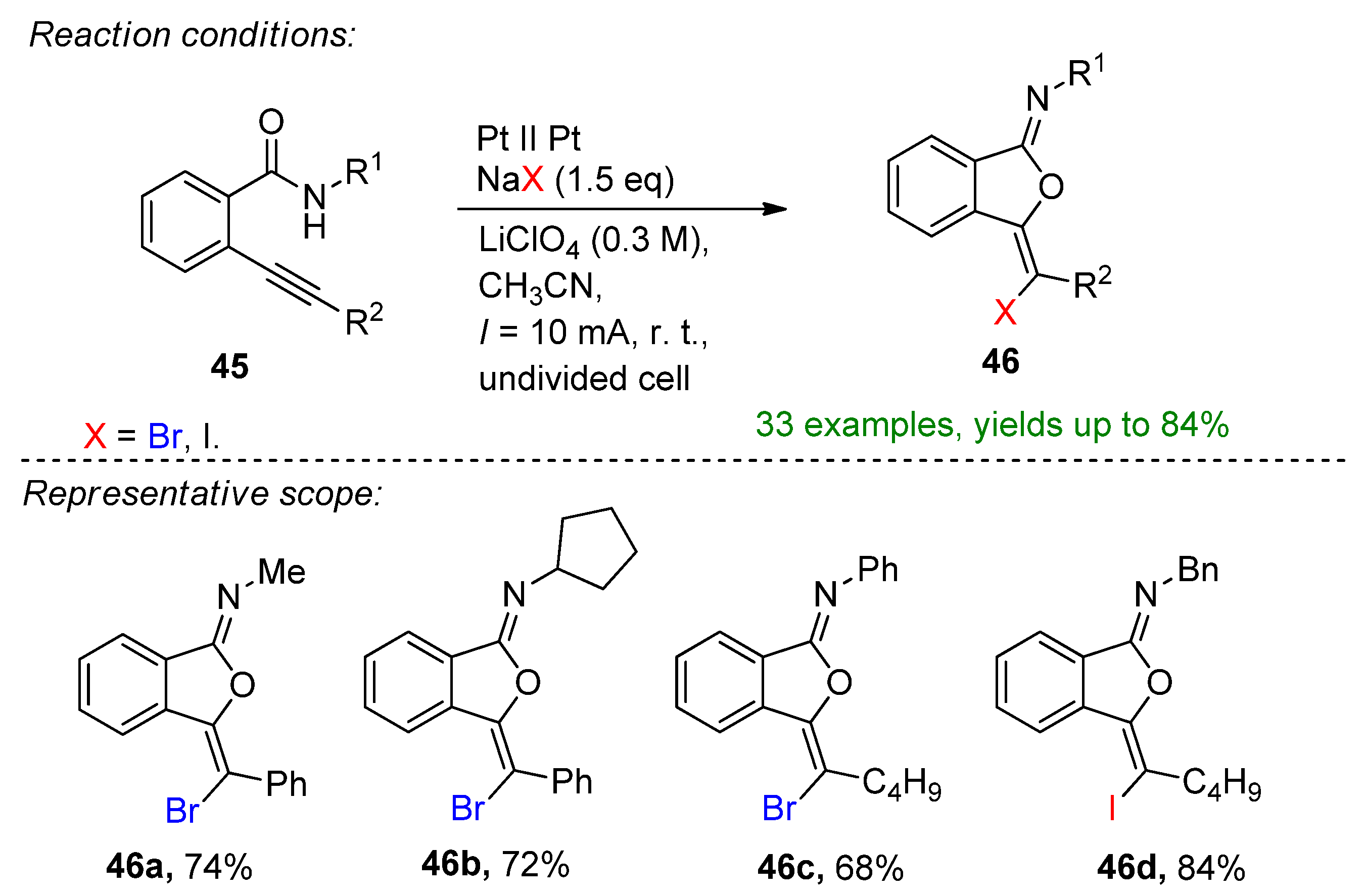
The oxidative 5-exo-dig-oxo-cyclization of o-alkynylbenzamides for the synthesis of isobenzofuran-1-imines using NaBr was reported (Scheme 18) [107]. Using additional LiClO4 as an electrolyte at platinum electrodes at room temperature gave the brominated derivatives with up to a 77% isolated yield. The choice of additional supporting electrolyte as well as the solvent were the key features of this electrochemical method. Changing LiClO4 or the absence of the supporting electrolyte had detrimental effect on the reaction. Using 1.5 eq. NaI as a halogen source gave the corresponding 3-iodo-isobenzofuran-1-imines with excellent yields. The combined halogenation process provided 33 examples with up to an 84% isolated yield.
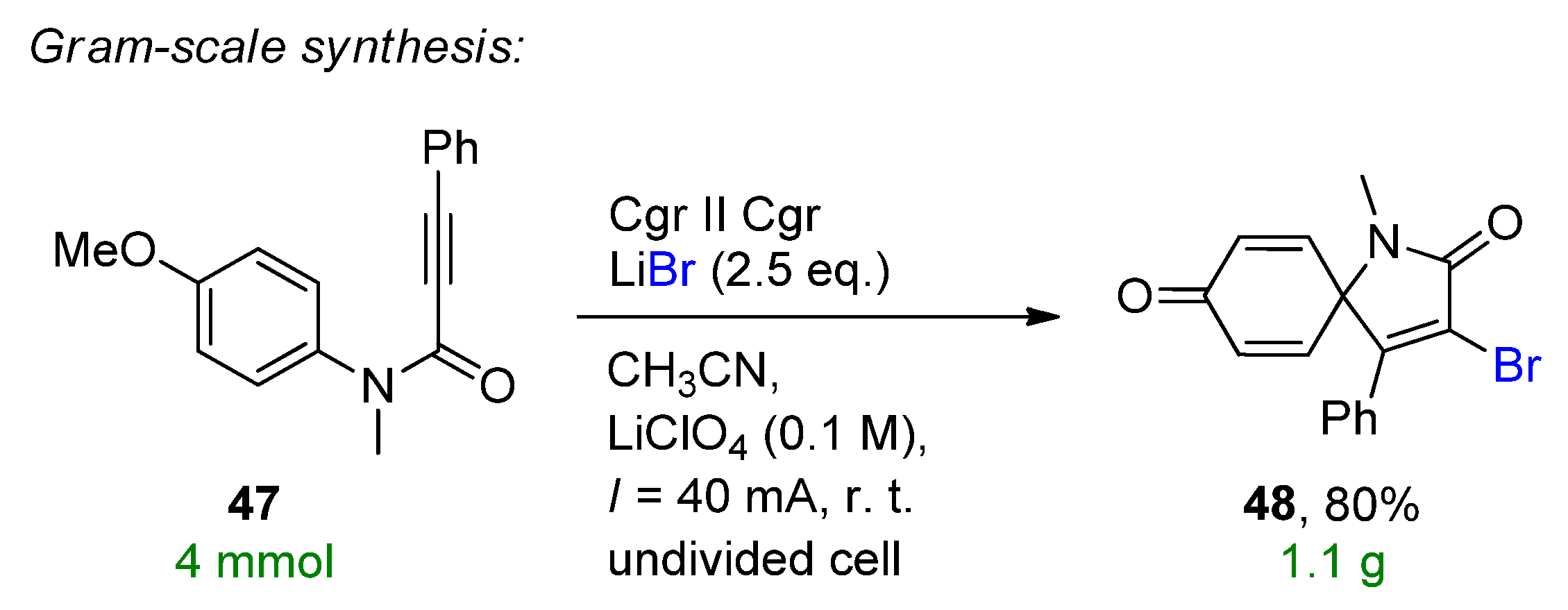
The electrochemical dearomative spirocyclization of N-aryl alkynamides to the corresponding spiro[4,5]trienones was reported [108]. The highly selective reaction was catalyzed by inexpensive and bench-stable lithium halides on graphite electrodes. The model substrate showed high affinity in the presence of 2 eq. LiBr to form the 3-bromoderivative. Extension of the scope provided 20 different brominated examples with up to a 98% isolated yield. Substitutions both on the nitrogen as well as the aniline were tolerated with only noticeable changes in the yield in the presence of electron-withdrawing groups on the nitrogen. In absence of the methoxy group as well as the halogen, no formation of the desired product was detected. Similarly, the chloro- and iodo-derivatives could be also obtained with moderate to good yields. The synthetic utility was proved by a 32-fold scale-up to provide 48 with a yield of 80% (Scheme 19).
4.2. Organohalides as Bromine Sources
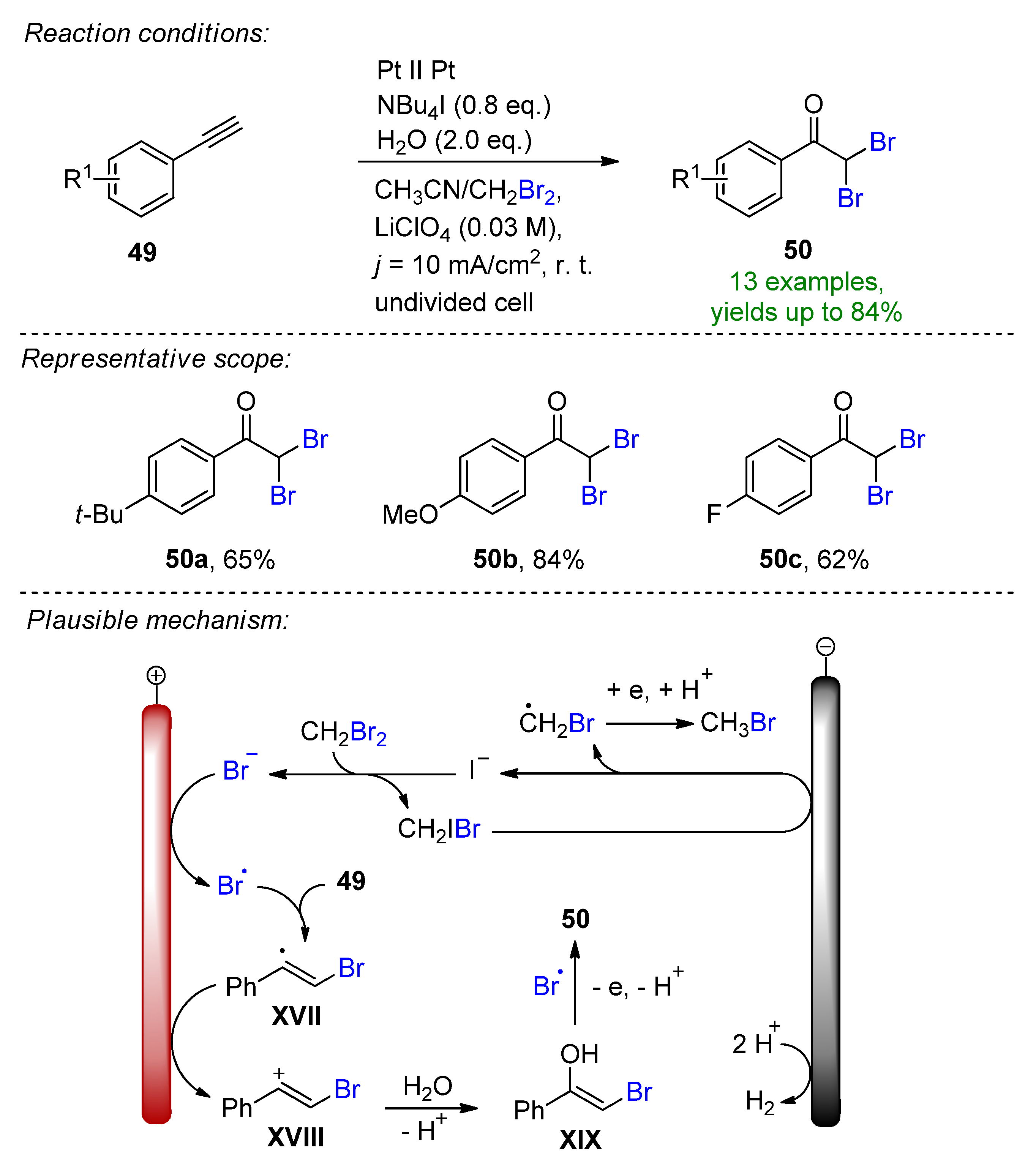
The first electrocatalytic oxydihalogenation of alkynes using organohalides (CH2Br2, CHCl3, CH2Cl2, Cl–CH2–CH2–Cl) with water as an eco-friendly oxygen source and NBu4I as a catalyst was reported [109]. The reaction also worked for the formation of dichloroacetophenone using different organochlorine sources. The mechanism was previsioned by the SN2 substitution of the CH2Br2 by iodide, which then underwent oxidation at the anode to a bromine radical that was subsequently trapped by the alkene 49 (Scheme 20). The resulting intermediate XVII was oxidized to carbocation XVIII, which then, upon reaction with water, formed the corresponding enol, and further combination with bromoradical and subsequent oxidation resulted in the product 50. The procedure could be extended to provide 13 examples with up to 84% isolated yields.
5. Conclusions
The electrochemical bromination and bromofunctionalization of alkenes and alkynes have proved to be emerging hot topics in the last years. Via the anodic oxidation of readily available and bench-stable bromides, bromine can be generated under controlled parameters to form the desired brominated products and intermediates in a highly sustainable fashion without the use of catalysts or exogenous oxidants. The utilization of alkali bromide salts in a dual role as both a reagent and supporting electrolyte augments electrochemical bromination protocols over the traditional ones, providing a greener and accessible alternative for the scientific community. Furthermore, their non-toxic and non-corrosive nature provides a solution for the handling and storage issues that are indispensable requirements for Br2 or HBr, thus opening a more sustainable and atomically economic way for the electrochemical generation of bromine and bromofunctionalization reactions.
While the electrochemical methods are represented via the utilization of metal bromide as brominating agents, there are more and more procedures that use organobromines in a paired electrolysis fashion, replacing the usual H2 evolution as a cathodic counterpart. Although the exchange of bench-stable and safe bromide salts to organobromine compounds might raise questions, this approach could potentially open a field and draw interest for the elimination of persistent halogenated organic pollutants, such as hexachlorocyclohexane or hexabromocyclododecane. Since the cathodic removal of bromo substituents is a common synthetic tool, which has even been applied to pharmaceutical intermediates, the basis for an electrically driven circular economy for bromine has been virtually established [110,111,112].
Author Contributions
L.G.G. and S.R.W. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Forschungsinitiative des Landes Rheinland-Pfalz in frame of SusInnoScience.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
L.G.G. and S.R.W. highly appreciate the support from the Forschungsinitiative des Landes Rheinland-Pfalz in frame of SusInnoScience.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M. Global Warming of 1.5 °C. Available online: https://www.ipcc.ch/sr15/ (accessed on 25 August 2022).
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic electrosynthesis: Applications in complex molecule synthesis. ChemElectroChem 2019, 6, 4067–4092. [Google Scholar] [CrossRef]
- Hilt, G. Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 2020, 7, 395–405. [Google Scholar] [CrossRef]
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020, 11, 12375–12592. [Google Scholar] [CrossRef]
- Novaes, L.F.T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J.M.; Lin, S. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. [Google Scholar] [CrossRef]
- Beil, S.B.; Pollok, D.; Waldvogel, S.R. Reproducibility in electroorganic synthesis—Myths and misunderstandings. Angew. Chem. Int. Ed. 2021, 60, 14750–14759. [Google Scholar] [CrossRef]
- Yoshida, J.; Shimizu, A.; Hayashi, R. Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev. 2017, 118, 4702–4730. [Google Scholar] [CrossRef]
- Röckl, J.L.; Pollok, D.; Franke, R.; Waldvogel, S.R. A decade of electrochemical dehydrogenative C,C-coupling of aryls. Acc. Chem. Res. 2020, 53, 45–61. [Google Scholar] [CrossRef]
- Waldvogel, S.R.; Lips, S.; Selt, M.; Riehl, B.; Kampf, C.J. Electrochemical arylation reactions. Chem. Rev. 2018, 118, 6706–6765. [Google Scholar] [CrossRef]
- Seidler, J.; Strugatchi, J.; Gärtner, T.; Waldvogel, S.R. Does electrifying organic synthesis pay off—The energy efficiency of electro-organic conversions. MRS Energy Sustain. 2020, 7, E42. [Google Scholar] [CrossRef]
- Margarita, C.; Lundberg, H. Recent advances in asymmetric catalytic electrosynthesis. Catalysts 2020, 10, 982. [Google Scholar] [CrossRef]
- Chicas-Baños, D.F.; Frontana-Uribe, B.A. Electrochemical generation and use in organic synthesis of C-,O-, and N-centered radicals. Chem. Rec. 2021, 21, 2538–2573. [Google Scholar] [CrossRef]
- Klüh, D.; Waldmüller, W.; Gaderer, M. Kolbe electrolysis for the conversion of carboxylic acids to valuable products—A process design study. Clean Technol. 2021, 3, 1–18. [Google Scholar] [CrossRef]
- Gleede, B.; Selt, M.; Franke, R.; Waldvogel, S.R. Developments in the dehydrogenative electrochemical synthesis of 3,3´,5,5´-tetramethyl-2,2´-biphenol. Chem. Eur. J. 2021, 27, 8252–8263. [Google Scholar] [CrossRef]
- Zheng, Y.; Shao, X.; Ramadoss, V.; Tian, L.; Wang, Y. Recent developments in photochemical and electrochemical decarboxylative C(sp3)-N bond formation. Synthesis 2020, 52, 1357–1368. [Google Scholar] [CrossRef]
- Listratova, A.V.; Sbei, N.; Voskressensky, L.G. Catalytic electrosynthesis of N,O-heterocycles—Recent advances. Eur. J. Org. Chem. 2020, 2020, 2012–2027. [Google Scholar] [CrossRef]
- Wirtanen, T.; Rodrigo, E.; Waldvogel, S.R. Recent advances in the electrochemical reduction of substrates involving N-O bonds. Adv. Synth. Catal. 2020, 362, 2088–2101. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Pajkert, R.; Wang, L.; Li, Z.; Röschenthaler, G.; Han, J. Chemistry of electrochemical oxidative reactions of sulfinate salts. Green Chem. 2020, 22, 3028–3059. [Google Scholar] [CrossRef]
- Blum, S.P.; Hofman, K.; Manolikakes, G.; Waldvogel, S.R. Advances in photochemical and electrochemical incorporation of sulfur dioxide for the synthesis of value-added compounds. Chem. Commun. 2021, 57, 8236–8249. [Google Scholar] [CrossRef]
- Sbei, N.; Martins, G.M.; Shirinfar, B.; Ahmed, N. Electrochemical phosphorylation of organic molecules. Chem. Rec. 2020, 20, 1530–1552. [Google Scholar] [CrossRef]
- Thang, S.; Liu, Y.; Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution: A green and sustainable way for bond formation. Chem 2018, 4, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Fang, P.; Mei, T. Recent Advances in organic electrochemical C-H functionalization. Chin. J. Chem. 2018, 36, 338–352. [Google Scholar] [CrossRef]
- Scheide, M.R.; Nicoleti, C.R.; Martins, G.M.; Braga, A.L. Electrohalogenation of organic compounds. Org. Biomol. Chem. 2021, 19, 2578–2602. [Google Scholar] [CrossRef]
- Kisukuri, C.M.; Fernandes, V.A.; Delgado, J.A.C.; Häring, A.P.; Paixão, M.W.; Waldvogel, S.R. Electrochemical installation of CFH2−, CF2H−, CF3−, and perfluoroalkyl groups into small organic molecules. Chem. Rec. 2021, 21, 2502–2525. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Tiz, D.B.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New halogen-containing drugs approved by FDA in 2021: An overview on their synthesis and pharmaceutical use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef]
- Bidleman, T.F.; Andersson, A.; Jantunen, L.M.; Kucklick, J.R.; Kylin, H.; Letcher, R.J.; Tysklind, M.; Wong, F. A review of halogenated natural products in Arctic, Subarctic and Nordic ecosystems. Emerg. Contam. 2019, 5, 89–115. [Google Scholar] [CrossRef]
- Häggblom, M.M.; Bossert, I.D. Halogenated organic compounds: A global perspective. In Dehalogenation: Microbial Processes and Environmental Applications; Springer: Boston, MA, USA, 2003; pp. 3–29. [Google Scholar] [CrossRef]
- Yuan, Y.; Lei, A. Is electrosynthesis always green and advantageous compared to traditional methods? Nat. Commun. 2020, 11, 802. [Google Scholar] [CrossRef] [Green Version]
- Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S.R. Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 2018, 57, 6018–6041. [Google Scholar] [CrossRef]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying organic synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef]
- Tang, H.; Jia, J.; Pan, Y. Halogen-mediated electrochemical organic synthesis. Org. Biomol. Chem. 2020, 18, 5315–5333. [Google Scholar] [CrossRef]
- Lian, F.; Xu, K.; Zeng, C. Indirect electrosynthesis with halogen ions as mediators. Chem. Rec. 2021, 21, 2290–2305. [Google Scholar] [CrossRef]
- Inokuchi, T.; Matsumoto, S.; Torii, S. Indirect electrooxidation of alcohols by a double mediatory system with two redox couples of [R2N+=O]/R2NO· and [Br· or Br+]/Br− in an organic-aqueous two-phase solution. J. Org. Chem. 1991, 56, 2416–2421. [Google Scholar] [CrossRef]
- Limborg, F.; Clauson-Kaas, N. A small cell for electrolytic alkoxylation of furans. Acta Chem. Scand. 1953, 7, 234–235. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Mendoza, E.; Guevara-Salazar, J.A.; Ramírez-Apan, M.T.; Frontana-Ulribe, B.A.; Cogordan, B.A.; Cárnedas, J. Electro-oxidation of hispanolone and anti-inflammatory properties of the obtained derivates. J. Org. Chem. 2005, 70, 4538–4541. [Google Scholar] [CrossRef]
- Salameh, E.; Tarawneh, A.; Al-Raggad, M. Origin of high bromide concentration in the water sources in Jordan and in the Dead Sea water. Arab. J. Geosci. 2016, 9, 414. [Google Scholar] [CrossRef]
- Fernández, L. Market Volume of Bromine Worldwide from 2015 to 2021, with a Forecast for 2022 to 2029. Available online: https://www.statista.com/statistics/1245240/bromine-market-volume-worldwide/ (accessed on 25 August 2022).
- Wu, N.; Herrmann, T.; Paepke, O.; Tickner, J.; Hale, R.; Harvey, E.; La Guardia, M.; McClean, M.D.; Webster, T.F. Human exposure to PBDEs: Association of PBDE body burdens with food consumption and house dust concentrations. Environ. Sci. Technol. 2007, 41, 1584–1589. [Google Scholar] [CrossRef]
- Hales, B.F.; Robaire, B. Effects of brominated and organophospate ester flame retardants on male reproduction. Andrology 2020, 8, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Addis, D.R.; Molyvdas, A.; Ambalavanan, N.; Matalon, S.; Jilling, T. Halogen exposure injury in the developing lung. Ann. N. Y. Acad. Sci. 2020, 1480, 30–43. [Google Scholar] [CrossRef]
- Addis, D.R.; Aggarwal, S.; Lazrak, A.; Jilling, T.; Matalon, S. Halogen-induced chemical injury to the mammalian cardiopulmonary systems. Physiology 2021, 36, 272–291. [Google Scholar] [CrossRef]
- Saikira, I.; Borah, A.J.; Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev. 2016, 116, 6837–7042. [Google Scholar] [CrossRef]
- Eissen, M.; Lenoir, D. Electrophilic bromination of alkenes: Environmental, health and safety aspects of new alternative methods. Chem. Eur. J. 2008, 14, 9830–9841. [Google Scholar] [CrossRef]
- Van Kerrebroeck, R.; Horsten, T.; Stevens, C.V. Bromide oxidation: A safe strategy for electrophilic brominations. Eur. J. Org. Chem. 2022, in press. [CrossRef]
- Tariq, Z.M. Electrochemistry of Br−/Br2 Redox Couple in Acetonitrile, Methanol and Mix Media of Acetonitrile–Methanol: An Insight into Redox Behavior of Bromide on Platinum (Pt) and Gold (Au) Electrode. Phys. Chem. 2019, 234, 295–312. [Google Scholar] [CrossRef]
- Halász, D.; Visy, C.; Szűcs, A.; Novák, M. Bromide ion oxidation of various Pt surfaces. React. Kinet. Catal. Lett. 1992, 48, 177–188. [Google Scholar] [CrossRef]
- Vojinovic, V.; Mentus, S.; Komnenic, V.J. Bromide oxidation and bromine reduction in propylene carbonate. Electroanal. Chem. 2003, 547, 109–113. [Google Scholar] [CrossRef]
- Haynes, W.M. Abundance of elements in the earth’s crust and in the sea. In CRC Handbook of Chemistry and Physics; CRC Press: Boulder, CO, USA, 2016; pp. 14–17. ISBN 978-1-4822-0868-9. [Google Scholar]
- Sigma Aldrich. Available online: https://www.sigmaaldrich.com/DE/en (accessed on 14 September 2022).
- Kingston, C.; Palkowitz, M.D.; Takahira, Y.; Vantourout, J.C.; Peters, B.K.; Kawamata, Y.; Baran, P.S. A survival guide for the “Electro-curious”. Acc. Chem. Rev. 2020, 53, 72–83. [Google Scholar] [CrossRef]
- Schotten, C.; Nicholls, T.P.; Bourne, R.A.; Kapur, N.; Nguen, B.N.; Williams, C.E. Making electrochemistry easily accessible to the synthetic chemist. Green Chem. 2020, 22, 3358–3375. [Google Scholar] [CrossRef]
- Wu, T.; Nguyen, B.H.; Daugherty, M.C.; Moeller, K.D. Paired electrochemical reactions and the on-site generation of a chemical reagent. Angew. Chem. Int. Ed. 2019, 58, 3562–3565. [Google Scholar] [CrossRef]
- Wu, T.; Moeller, K.D. Organic electrochemistry: Expanding the scope of paired reactions. Angew. Chem. Int. Ed. 2021, 60, 12883–12890. [Google Scholar] [CrossRef]
- Marken, F.; Cresswell, A.J.; Bull, S.T. Recent advances in paired electrosynthesis. Chem. Rec. 2021, 21, 2585–2600. [Google Scholar] [CrossRef]
- Klein, M.; Waldvogel, S.R. Counter electrode reactions—Important stumbling blocks on the way to a working electro-organic synthesis. Angew. Chem. Int. Ed. 2022. accepted. [Google Scholar] [CrossRef]
- Clark, J.H. Green Chemistry: Challenges and opportunities. Green Chem. 1998, 1, 1–8. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Frontana-Ulribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic synthesis. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical- and less classical solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Roeckl, J.L.; Waldvogel, S.R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514. [Google Scholar] [CrossRef]
- Salom-Rolg, X.; Bauder, C. Recent applications in the use of sulfoxides as chiral auxiliaries for the asymetric synhesis of natural and biologically active products. Synthesis 2020, 52, 964–978. [Google Scholar] [CrossRef]
- Torii, S.; Uneyama, K.; Ueda, K. Electrochemical procedure for a practical preparation of piperonal from isosafrole. J. Org. Chem. 1984, 49, 1830–1832. [Google Scholar] [CrossRef]
- Uneyama, K.; Masatsugu, Y.; Torii, S. Electrochemical epoxidation and carbon-carbon bond cleavage for the preparation of 3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-8-carboxylic acid from 3-methyl-2-phenyl-8-(1-prophenyl)-4H-1-benzopyran-4-one. Bull. Chem. Soc. Jpn. 1985, 58, 2361–2365. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, T.; Matsumoto, S.; Tsuji, M.; Torii, S. Electrohalogenation of propargyl acetates and amides to form the 1,1-dibromo-2-oxo functionality and a facile synthesis of furaneol. J. Org. Chem. 1992, 57, 5023–5027. [Google Scholar] [CrossRef]
- Inês, M.; Mendonça, A.J.; Esteves, A.P.; Mendonça, D.I.; Medeiros, M.J. Electroepoxidation of natural and synthetic alkenes mediated by sodium bromide. Comptes Rendus Chim. 2009, 12, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Kulangiappar, K.; Ramaprakash, M.; Vasudevan, D.; Raju, T. Electrochemical bromination of cyclic and acyclic enes using biphasic electrolysis. Synth. Commun. 2016, 46, 145–153. [Google Scholar] [CrossRef]
- Yang, C. An impending platinum crisis and its implications for the future of the automobile. Energy Policy 2009, 37, 1805–1808. [Google Scholar] [CrossRef]
- Das, K.K.; Reddy, R.C.; Bagoji, I.B.; Das, S.; Bagali, S.; Mullur, L.; Khodnapur, J.P.; Biradar, M.S. Primary concept of nickel toxicity—An overview. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Savignan, L.; Faucher, S.; Chéry, P.; Lespes, G. Platinum group elements contamination in soils: Review of the current state. Chemosphere 2021, 271, 129517. [Google Scholar] [CrossRef]
- Sun, X.; Ma, H.; Mei, T.; Fang, P.; Hu, Y. Electrochemical radical formyloxylation-bromination, -chlorination, and -trifluoromethylation of alkenes. Org. Lett. 2019, 21, 3167–3171. [Google Scholar] [CrossRef]
- Bormann, S.; Van Schie, M.M.C.H.; De Almeida, T.P.; Zhang, W.; Stöckl, M.; Ulber, R.; Hollman, F.; Holtmann, D. H2O2 production at low overpotential for electroenzymatic halogenation reactions. ChemSusChem 2019, 12, 4759–4763. [Google Scholar] [CrossRef] [Green Version]
- Wever, R.; Renirie, R.; Hollmann, F. Vanadium chloroperoxidases as versatile biocatalysts. In Vanadium Catalysis; Sudradhar, M., Pombeiro, A.J.L., da Silva, J.A.L., Eds.; Royal Society of Chemistry: London, UK, 2021; Chapter 24; ISBN 978-1-78801-857-9. [Google Scholar]
- Wu, J.; Abou-Hamdan, H.; Guillot, R.; Kouklovsky, C.; Vincent, G. Electrochemical synthesis of 3a-bromofuranoindolines and 3a-bromopyrroloindolines mediated by MgBr2. Chem. Commun. 2020, 56, 1713–1716. [Google Scholar] [CrossRef]
- Hakamata, H.; Sato, S.; Ueda, H.; Tokuyama, H. AgNTf2-Mediated Allylation with Allylsilanes at C3a-Position of Hexahydropyrroloindoles: Application to Total Syntheses of Amauromine Alkaloids. Org. Lett. 2017, 19, 5308–5311. [Google Scholar] [CrossRef]
- Ramos-Villaseñor, J.M.; Rodríguez-Cárdenas, E.; Díaz, C.E.B.; Frontana-Uribe, B.A. Review—Use of 1,1,1,3,3,3–hexafluoro–2–propanol (HFIP) Co-Solvent Mixtures in Organic Electrosynthesis. J. Electrochem. Soc. 2020, 167, 155509. [Google Scholar] [CrossRef]
- Eberson, L.; Hartshorn, M.P.; Persson, O. 1,1,1,3,3,3-Hexafluoropropan-2-ol as a solvent for the generation of highly persistent radical cations. J. Chem. Soc. Perkin Trans. 2 1995, 1735–1744. [Google Scholar] [CrossRef]
- Eberson, L.; Hartshorn, M.P.; Persson, O. Generation of solutions of highly persistent radical cations by 4-tolylthallium(III) bis(trifluoroacetate) in 1,1,1,3,3,3-hexafluoropropan-2-ol. J. Chem. Soc. Chem. Commun. 1995, 1131–1132. [Google Scholar] [CrossRef]
- Eberson, L.; Persson, O.; Hartshorn, M.P. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew. Chem. Int. Ed. 1995, 34, 2268–2269. [Google Scholar] [CrossRef]
- Schulz, L.; Waldvogel, S.R. Solvent control in electro-organic synthesis. Synlett 2018, 30, 275–286. [Google Scholar] [CrossRef]
- Röckl, J.L.; Dörr, M.; Waldvogel, S.R. Electrosynthesis 2.0 in 1,1,1,3,3,3-Hexafluoroisopropanol/Amine Mixtures. ChemElectroChem 2020, 7, 3686–3694. [Google Scholar] [CrossRef]
- Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. Halogen and Chalcogen Cation Pools Stabilized by DMSO. Versatile Reagents for Alkene Difunctionalization. J. Am. Chem. Soc. 2013, 135, 16070–16073. [Google Scholar] [CrossRef]
- Shimizu, A.; Hayashi, R.; Ashikari, Y.; Nokami, T.; Yoshida, J. Switching the reaction pathways of electrochemically generated β-haloalkoxysulfonium ions—Synthesis of halohydrins and epoxides. Beilstein J. Org. Chem. 2015, 11, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Bityukov, O.V.; Vil’, V.A.; Nikishin, G.I.; Terent’ev, A.O. Alkene, Bromide, and ROH—How To Achieve Selectivity? Electrochemical Synthesis of Bromohydrins and Their Ethers. Adv. Synth. Catal. 2021, 363, 3070–3078. [Google Scholar] [CrossRef]
- Shang, X.; Liu, X.; Sun, Y. Flexible on-site halogenation paired with hydrogenation using halide electrolysis. Green Chem. 2021, 23, 2037–2043. [Google Scholar] [CrossRef]
- Strehl, J.; Abraham, M.L.; Hilt, G. Linear Paired Electrolysis—Realising 200 % Current Efficiency for Stoichiometric Transformations—The Electrochemical Bromination of Alkenes. Angew. Chem. Int. Ed. 2021, 60, 9996–10000. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.H.; Quach, L.; Paulisch, T.; Glorius, F. Visible-light-induced, metal-free carbene insertion into B-H bonds between acylsilanes and pinacolborane. J. Am. Chem. Soc. 2019, 141, 16227–16231. [Google Scholar] [CrossRef]
- Kale, A.P.; Nikolaienko, P.; Smirnova, K.; Rueping, M. Intramolecular Electrochemical Oxybromination of Olefins for the Synthesis of Isoxazolines in Batch and Continuous Flow. Eur. J. Org. Chem. 2021, 2021, 3496–3500. [Google Scholar] [CrossRef]
- He, Y.; Qin, X.; He, X.; Wu, X.; Yin, Z. Practical Synthesis of Halogenated N-Heterocycles via Electrochemical Anodic Oxidation of Unactivated Alkenes. Eur. J. Org. Chem. 2021, 2021, 5831–5834. [Google Scholar] [CrossRef]
- Kim, R.; Ha, J.; Woo, J.; Kim, D.Y. Electrochemical oxidative bromolactonization of unsaturated carboxylic acids with sodium bromide: Synthesis of bromomethylated γ-lactones. Tetrahedron Lett. 2022, 88, 153567. [Google Scholar] [CrossRef]
- Yu, D.; Ji, R.; Sun, Z.; Li, W.; Liu, Z. Electrochemical chlorination and bromination of electron-deficient C-H bonds in quinones, coumarins, quinoxalines and 1,3-diketones. Tetrahedron Lett. 2021, 86, 153514. [Google Scholar] [CrossRef]
- Gombos, L.G.; Werner, L.; Schollmeyer, D.; Martínez-Huitle, C.A.; Waldvogel, S.R. Selective Electrochemical Dibromination of Terpenes and Naturally Derived Olefins. Eur. J. Org. Chem. 2022. accepted. [Google Scholar] [CrossRef]
- Yuan, Y.; Yao, A.; Zheng, Y.; Gao, M.; Zhou, Z.; Qiao, J.; Hu, J.; Ye, B.; Zhao, J.; Wen, H.; et al. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303. [Google Scholar] [CrossRef]
- Seitz, J.; Wirth, T. Electrochemical bromofunctionalization of alkenes in a flow reactor. Org. Biomol. Chem. 2021, 19, 6892–6896. [Google Scholar] [CrossRef]
- Wan, C.; Song, R.; Li, J.H. Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide. J. Org. Lett. 2019, 21, 2800–2803. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ouyang, X.; Hu, M.; Xie, Y.; Li, J. Copper-Catalyzed Intermolecular Aminoalkylation of Alkenes with α-Bromoalkyl Esters and Amines toward Pyrrolidin-2-ones. Adv. Synth. Catal. 2017, 359, 2564–2570. [Google Scholar] [CrossRef]
- Zhang, T.; Luo, M.; Li, Y.; Song, R.; Li, J. Electrochemical Alkoxyhalogenation of Alkenes with Organohalides as the Halide Sources via Dehalogenation. Org. Lett. 2020, 22, 7250–7254. [Google Scholar] [CrossRef] [PubMed]
- Bortolami, M.; Petrucci, R.; Rocco, D.; Scarano, V.; Chiarotto, I. Alkynes as Building Blocks, Intermediates and Products in the Electrochemical Procedures Since 2000. ChemElectroChem 2021, 8, 3604–3613. [Google Scholar] [CrossRef]
- Chung, W.; Vanderwal, C.D. Stereoselective Halogenation in Natural Product Synthesis. Angew. Chem. Int. Ed. 2016, 55, 4365–4434. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Santhi, J.; Baire, B. The α,α-Dihalocarbonyl Building Blocks: An Avenue for New Reaction Development in Organic Synthesis. Chem. Eur. J. 2020, 26, 7145–7175. [Google Scholar] [CrossRef]
- Li, Z.; Sun, Q.; Qian, P.; Hu, K.; Zha, Z.; Wang, Z. Electrochemical synthesis of α,α-dihaloacetophenones from terminal alkyne derivatives. Chin. Chem. Lett. 2020, 31, 1855–1858. [Google Scholar] [CrossRef]
- Wang, D.; Wan, Z.; Zhang, H.; Lei, A. Electrochemical Oxidative Functionalization of Arylalkynes: Access to α,α-Dibromo Aryl Ketones. Adv. Synth. Catal. 2021, 363, 1022–1027. [Google Scholar] [CrossRef]
- Zhang, T.; Hao, W.; Wang, R.; Wang, S.; Tu, S.; Jiang, B. Electrocatalytic three-component annulation-halosulfonylation of 1,6-enynes toward 1-indanones using sodium halides as both halogen sources and electrolytes. Green Chem. 2020, 22, 4259–4269. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, S.; Hao, W.; Dong, G.; Tu, S.; Jiang, B. Tunable Electrocatalytic Annulations of o-Arylalkynylanilines: Green and Switchable Syntheses of Skeletally Diverse Indoles. J. Org. Chem. 2021, 86, 15886–15896. [Google Scholar] [CrossRef]
- Reddy, M.B.; Peri, R.; Bhagavathiachari, M.; Anandhan, R. Electrochemical synthesis of isobenzofuran-1-imines using oxidative halocyclization of o-alkynylbenzamides. Org. Biomol. Chem. 2021, 19, 6792–6796. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Kong, X.; Yang, J.; Li, G.; Xu, B.; Chen, Q. Electrochemical Oxidative Halogenation of N-Aryl Alkynamides for the Synthesis of Spiro[4.5]trienones. J. Org. Chem. 2021, 86, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, Y.; Luo, J.; Wang, F.; Cao, X.; Huang, S. Electrochemical Oxidative Oxydihalogenation of Alkynes for the Synthesis of α,α-Dihaloketones. Org. Lett. 2020, 22, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Gütz, C.; Selt, M.; Bäzinger, M.; Bucher, C.; Römelt, C.; Hecken, N.; Gallou, F.; Galvão, T.R.; Waldvogel, S.R. Novel cathode material for cathodic dehalogenation of 1,1,-dibromo cyclopropane derivatives. Chem. Eur. J. 2015, 21, 13878–13882. [Google Scholar] [CrossRef] [PubMed]
- Gütz, C.; Bäzinger, M.; Bucher, C.; Galvão, T.R.; Waldvogel, S.R. Development and scale-up of electrochemical dehalogenation for the synthesis of a key intermediate for NS5A inhibitors. Org. Process Res. Dev. 2015, 19, 1428–1433. [Google Scholar] [CrossRef]
- Gütz, C.; Grimaudo, V.; Holtkamp, M.; Hartmer, M.; Werra, J.; Frensemeier, L.; Kehl, A.; Karst, U.; Broekmann, P.; Waldvogel, S.R. Leaded Bronze—An Innovative Lead Substitute for Cathodic Electrosynthesis. ChemElectroChem 2018, 5, 247–252. [Google Scholar] [CrossRef]
Scheme 1.
Electrochemical radical formyloxylation bromination of alkenes [72].
Scheme 1.
Electrochemical radical formyloxylation bromination of alkenes [72].

Scheme 2.
The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivates utilizing MgBr2. * Constant terminal voltage of the cell was observed between a graphite anode and platinum plate cathode [75]. Cgr = graphite.
Scheme 2.
The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivates utilizing MgBr2. * Constant terminal voltage of the cell was observed between a graphite anode and platinum plate cathode [75]. Cgr = graphite.

Scheme 3.
Electrochemical formation and subsequent functionalization of 6a and 6b [76]. * Constant terminal voltage of the cell was observed between a graphite anode and a platinum cathode [75]. Cgr = graphite.

Scheme 4.
Electrochemical bromohydrin and bromohydrin ether formation [85]. gC = glassy carbon.
Scheme 4.
Electrochemical bromohydrin and bromohydrin ether formation [85]. gC = glassy carbon.

Scheme 5.
Linear paired electrolysis for the dibromination of alkenes [87]. gC = glassy carbon.
Scheme 5.
Linear paired electrolysis for the dibromination of alkenes [87]. gC = glassy carbon.

Scheme 6.
Electrochemical formation of isoxazolines with the aid of KBr [90]. Cgr = graphite.
Scheme 6.
Electrochemical formation of isoxazolines with the aid of KBr [90]. Cgr = graphite.

Scheme 7.
Electrochemical intramolecular haloamination with the aid of LiBr and LiI [91]. Cgr = graphite.
Scheme 7.
Electrochemical intramolecular haloamination with the aid of LiBr and LiI [91]. Cgr = graphite.

Scheme 8.
Electrochemical bromolactonization of unsaturated carboxylic acids [92]. Cgr = graphite.
Scheme 8.
Electrochemical bromolactonization of unsaturated carboxylic acids [92]. Cgr = graphite.

Scheme 9.
Electrochemical dibromination of terpenes and terpenoids [94]. gC = glassy carbon.
Scheme 9.
Electrochemical dibromination of terpenes and terpenoids [94]. gC = glassy carbon.

Scheme 10.
Electrochemical dibromination of 22 on a gram scale [95]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 10.
Electrochemical dibromination of 22 on a gram scale [95]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 11.
Electrochemical bromination and bromofunctionalization of alkenes in flow electrolysis [96]. Cgr = graphite.
Scheme 11.
Electrochemical bromination and bromofunctionalization of alkenes in flow electrolysis [96]. Cgr = graphite.

Scheme 12.
The three-component, TEMPO-mediated 1,2-bromoesterification of alkenes [97]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 12.
The three-component, TEMPO-mediated 1,2-bromoesterification of alkenes [97]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 13.
The electrochemical alkoxyhalogenation of alkenes within a convergent paired electrolysis method [99]. CCE conditions were described with current (I), as the immersed electrode area was not specified. Cgr = graphite.
Scheme 13.
The electrochemical alkoxyhalogenation of alkenes within a convergent paired electrolysis method [99]. CCE conditions were described with current (I), as the immersed electrode area was not specified. Cgr = graphite.

Scheme 14.
Merging e-shuttle for the retro-dibromination and bromofunctionalization of alkenes [62]. Cgr = graphite.
Scheme 14.
Merging e-shuttle for the retro-dibromination and bromofunctionalization of alkenes [62]. Cgr = graphite.

Scheme 15.
Electrochemical α,α-dibromoacetophenone formation with the aid of LiBr in an undivided cell [104]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 15.
Electrochemical α,α-dibromoacetophenone formation with the aid of LiBr in an undivided cell [104]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 16.
Electrochemical three-component annulation–sulfonylation of 1,6-enynes to 1-indanones [105]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 16.
Electrochemical three-component annulation–sulfonylation of 1,6-enynes to 1-indanones [105]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 17.
Electrochemical halogenation of arylalkylanilines to C3-halogenated indoles [106]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 17.
Electrochemical halogenation of arylalkylanilines to C3-halogenated indoles [106]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 18.
The oxidative 5-exo-dig-oxo-cyclization of o-alkynylbenzamides to isobenzofuran-1-imines using NaBr and NaI [107]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Scheme 18.
The oxidative 5-exo-dig-oxo-cyclization of o-alkynylbenzamides to isobenzofuran-1-imines using NaBr and NaI [107]. CCE conditions were described with current (I), as the immersed electrode area was not specified.

Scheme 19.
Gram-scale synthesis of spiro[4,5]trienone 48 [108]. CCE conditions were described with current (I), as the immersed electrode area was not specified. Cgr = graphite.
Scheme 19.
Gram-scale synthesis of spiro[4,5]trienone 48 [108]. CCE conditions were described with current (I), as the immersed electrode area was not specified. Cgr = graphite.

Scheme 20.
Electrocatalytic oxydihalogenation of alkynes in the presence of dibromoethane [109].
Scheme 20.
Electrocatalytic oxydihalogenation of alkynes in the presence of dibromoethane [109].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Price comparison of bromide salts with classical brominating reagents [51].
Table 1.
Price comparison of bromide salts with classical brominating reagents [51].
| Reagents 1 | Bulk Price 2 | Molar Price 3 |
|---|---|---|
| KBr | €95/kg | €11.31/mol |
| NaBr | €135/kg | €13.93/mol |
| LiBr | €294/kg | €25.53/mol |
| NH4Br | €220/kg | €21.55/mol |
| NEt4Br | €121/kg | €25.43/mol |
| NBu4Br | €831/kg | €267.89/mol |
| N-Bromosuccinimide | €140/kg | €24.92/mol |
| Pyridinium tribromide | €362/kg | €115.77/mol |
| HBr (48%) | €151/L | €8.20/mol |
| Bromine | €264/L | €13.57/mol * |
1 Prices were obtained from Sigma Aldrich website: https://www.sigmaaldrich.com/DE/en (accessed on 14 September 2022). 2 Bulk prices are given in EUR per kg or L. 3 Molar prices are calculated from bulk, prices are given in EUR per mol. * Refers to molar price of Br2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gombos, L.G.; Waldvogel, S.R. Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond. Sustain. Chem. 2022, 3, 430-454. https://doi.org/10.3390/suschem3040027
AMA Style
Gombos LG, Waldvogel SR. Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond. Sustainable Chemistry. 2022; 3(4):430-454. https://doi.org/10.3390/suschem3040027
Chicago/Turabian StyleGombos, Lilla G., and Siegfried R. Waldvogel. 2022. "Electrochemical Bromofunctionalization of Alkenes and Alkynes—To Sustainability and Beyond" Sustainable Chemistry 3, no. 4: 430-454. https://doi.org/10.3390/suschem3040027