A Genetically Encoded Bioluminescence Intracellular Nanosensor for Androgen Receptor Activation Monitoring in 3D Cell Models

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Plasmids
2.2. Plasmids Construction
2.3. Comparison of TK and SV40 Promoters
2.4. 2D Cell Culture, Transfection, and PPI Assay
2.5. 3D Cell Culture, Transfection, and PPI Assay
2.6. 2D Live Cell Imaging
3. Results
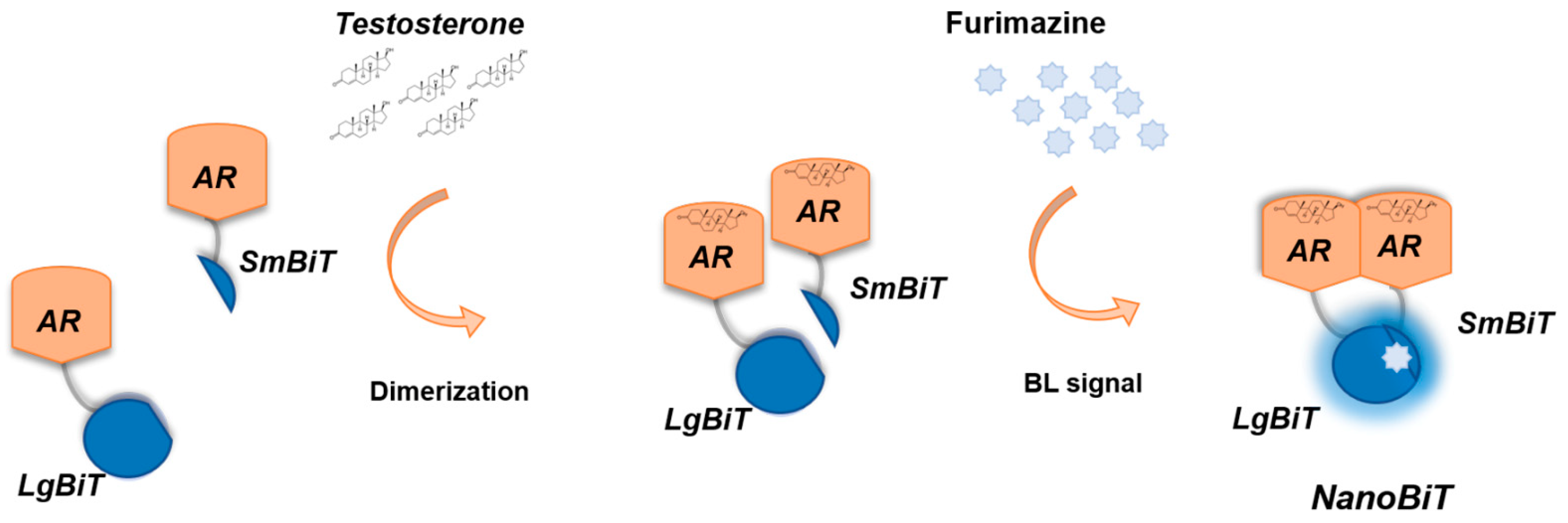
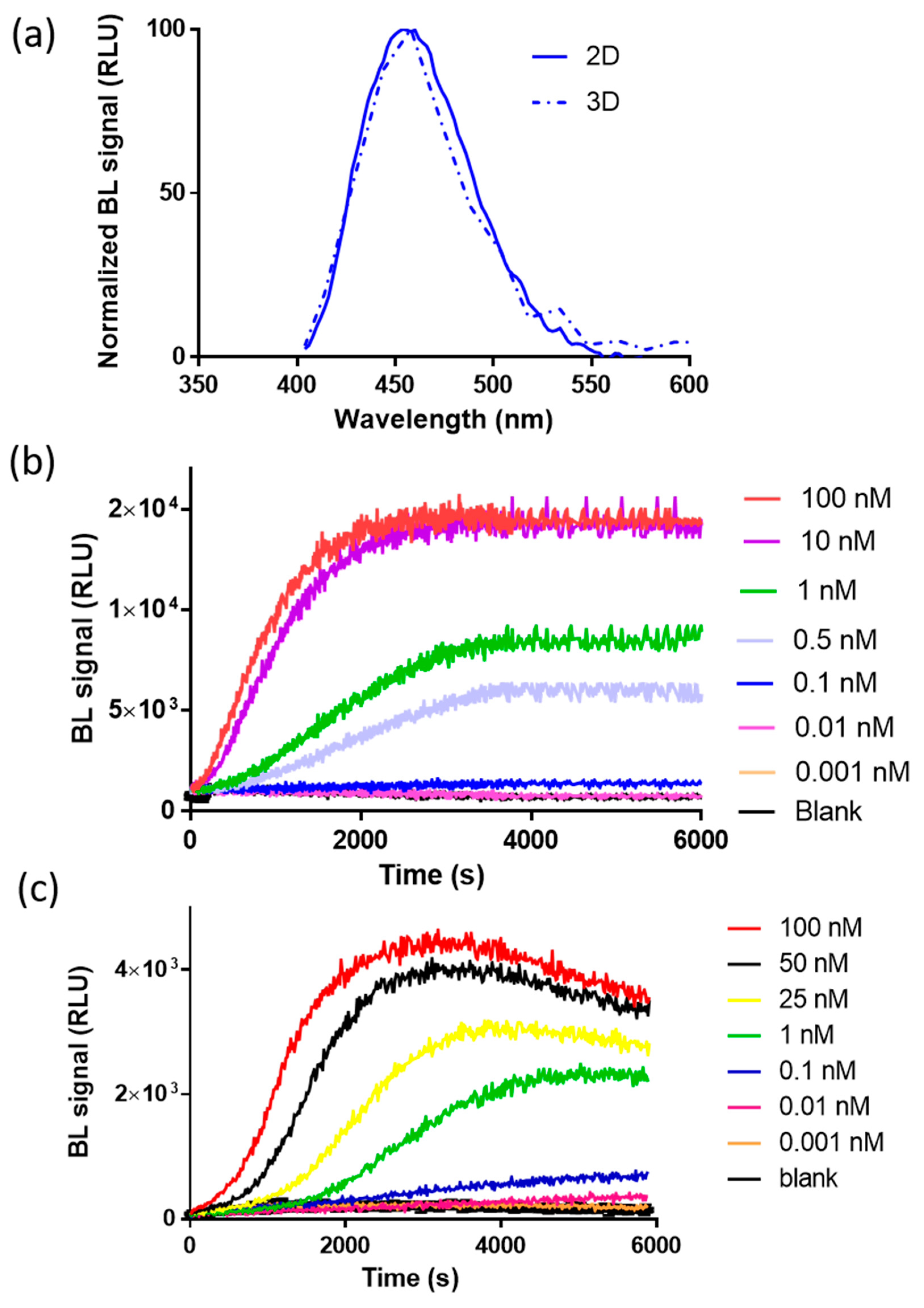
3.1. Characterization of the NanoBiT Reporter Expressed in 2D and 3D Cell Models
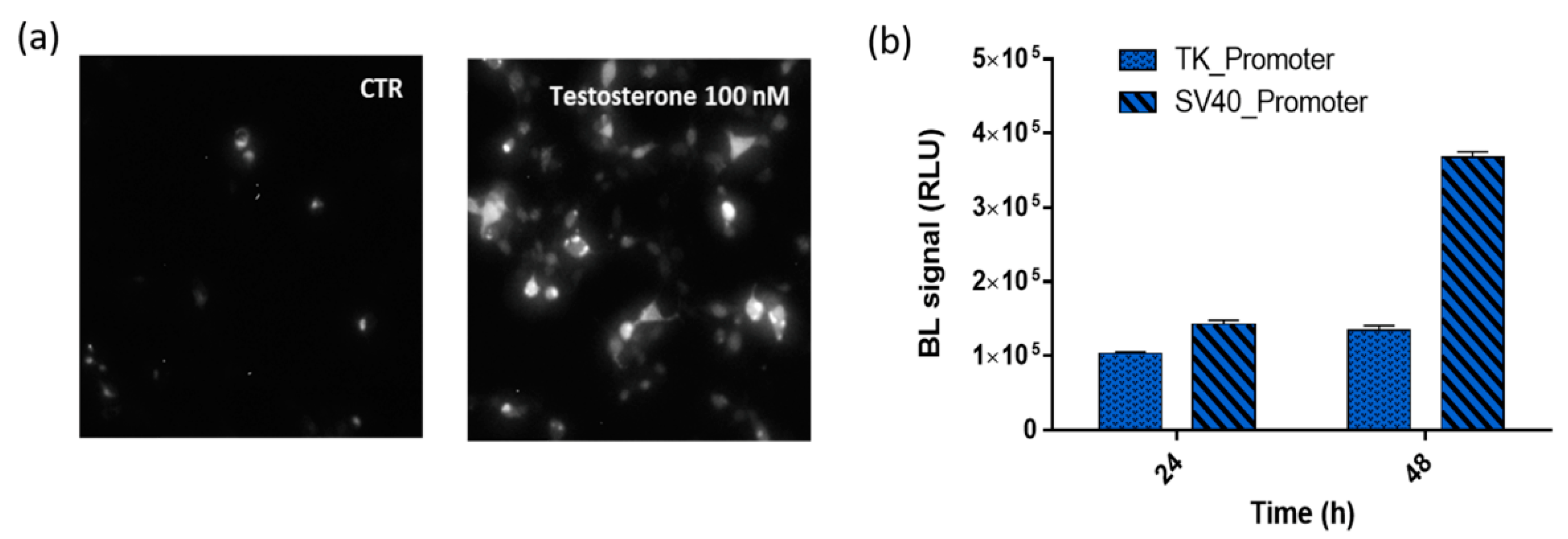
3.2. Comparison of Chimeric Protein Expression under TK and SV40 Promoters
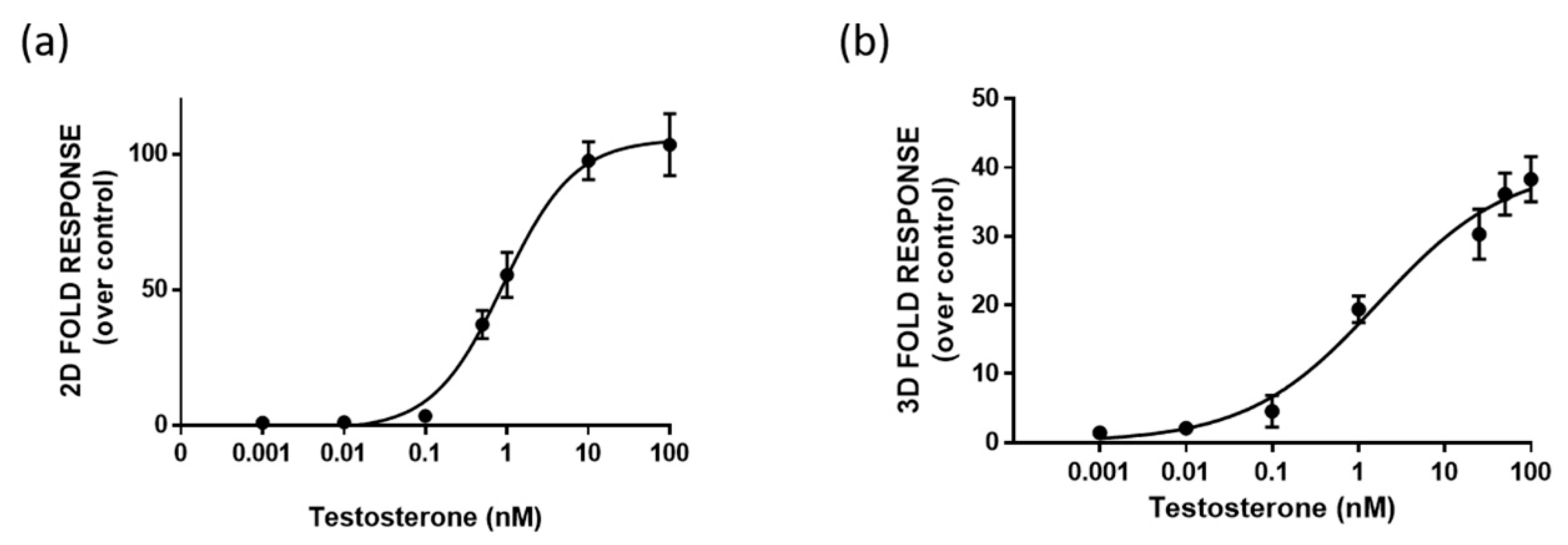
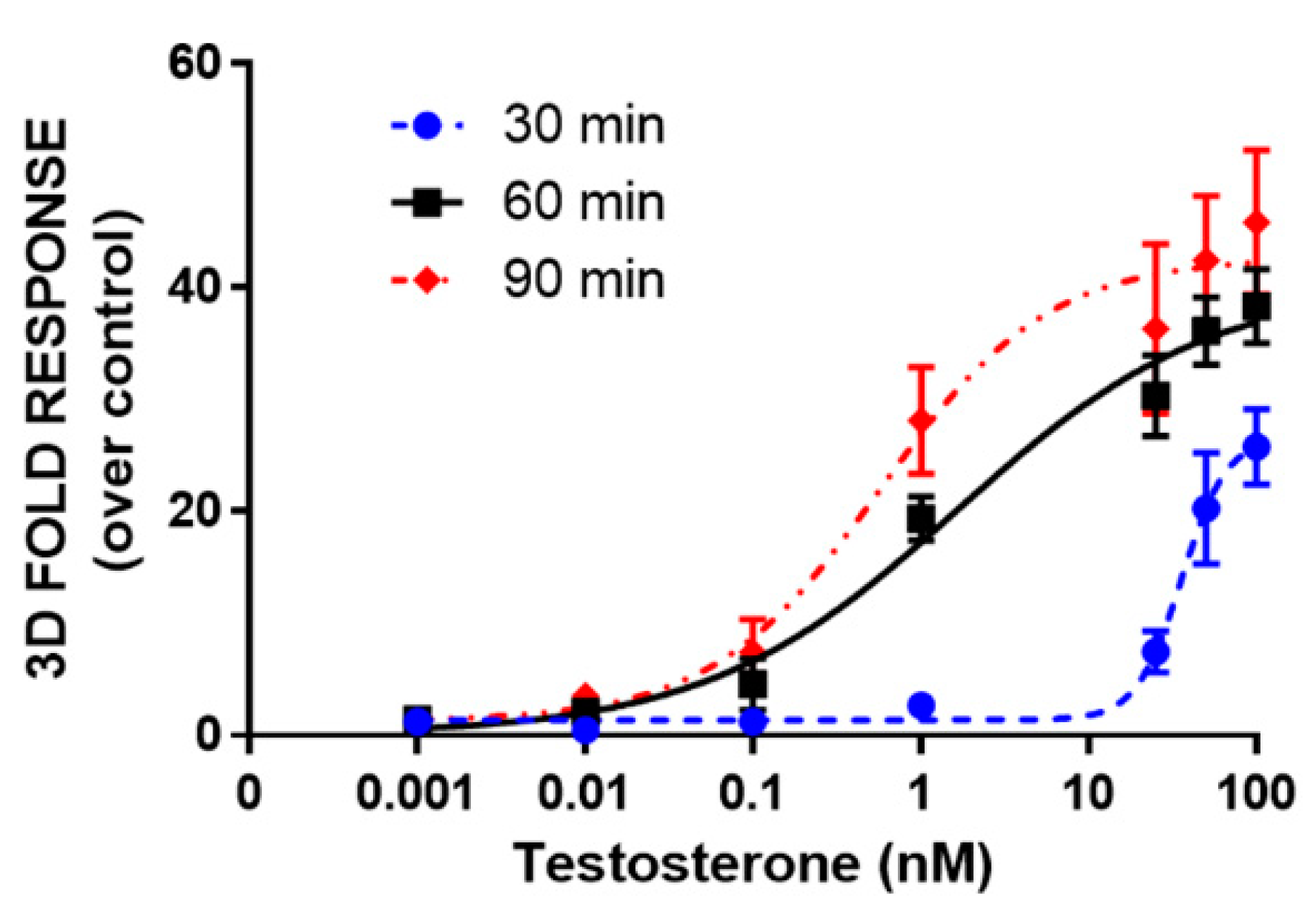
3.3. 3D Bioluminescent Assay for Androgen Receptor Activation Monitoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Misawa, N.; Kafi, A.K.; Hattori, M.; Miura, K.; Masuda, K.; Ozawa, T. Rapid and high-sensitivity cell-based assays of protein-protein interactions using split click beetle luciferase complementation: An approach to the study of G-protein-coupled receptors. Anal. Chem. 2010, 82, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current Challenges and Opportunities in Designing Protein-Protein Interaction Targeted Drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar] [PubMed]
- Biewenga, L.; Rosier, B.J.H.M.; Merkx, M. Engineering with NanoLuc: A playground for the development of bioluminescent protein switches and sensors. Biochem. Soc. Trans. 2020, 48, 2643–2655. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Li, Z.; Li, H. Analysis of Protein-Protein Interactions by Split Luciferase Complementation Assay. Curr. Protoc. Toxicol. 2019, 82, e90. [Google Scholar] [CrossRef] [PubMed]
- Cevenini, L.; Calabretta, M.M.; Calabria, D.; Roda, A.; Michelini, E. Luciferase Genes as Reporter Reactions: How to Use Them in Molecular Biology? In Bioluminescence: Fundamentals and Applications in Biotechnology; Thouand, G., Marks, R., Eds.; Springer: Cham, Switzerland, 2015; Volume 154, pp. 3–17. [Google Scholar]
- Johnsson, N.; Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 10340–10344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romei, M.G.; Boxer, S.G. Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu. Rev. Biophys. 2019, 48, 19–44. [Google Scholar] [CrossRef]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein–protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Smith, M.C.P.; Luker, G.D.; Gammon, S.T.; Piwnica-Worms, H.; Piwnica-Worms, D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [Google Scholar] [CrossRef] [Green Version]
- Paulmurugan, R.; Gambhir, S.S. Combinatorial library screening for developing an improved split-firefly luciferase fragment-assisted complementation system for studying protein-protein interactions. Anal. Chem. 2007, 79, 2346–2453. [Google Scholar]
- Oladzad, A.; Nikkhah, M.; Hosseinkhani, S. Optimization of Experimental Variables Influencing Apoptosome Biosensor in HEK293T Cells. Sensors 2020, 20, 1782. [Google Scholar] [CrossRef] [Green Version]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Nguyen, H.T.; Yong, H.J.; Reyes-Alcaraz, A.; Lee, Y.N.; Park, H.K.; Na, Y.H.; Lee, C.S.; Ham, B.J.; Seong, J.Y.; et al. Establishment of a NanoBiT-Based Cytosolic Ca2+ Sensor by Optimizing Calmodulin-Binding Motif and Protein Expression Levels. Mol. Cells 2020, 43, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Kilchrist, K.V.; Tierney, J.W.; Duvall, C.L. Genetically Encoded Split-Luciferase Biosensors to Measure Endosome Disruption Rapidly in Live Cells. ACS Sens. 2020, 5, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, L.; Yang, L.; Ni, W.; Li, Y.; Wu, Y. Tandem reassembly of split luciferase-DNA chimeras for bioluminescent detection of attomolar circulating microRNAs using a smartphone. Biosens. Bioelectron. 2021, 173, 112824. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Otani, Y.; Umezawa, Y.; Tao, H. Bioluminescent indicator for determining protein-protein interactions using intramolecular complementation of split click beetle luciferase. Anal. Chem. 2007, 79, 4820–4826. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Sato, M.; Tao, H. Split Gaussia Luciferase-Based Bioluminescence Template for Tracing Protein Dynamics in Living Cells. Anal. Chem. 2009, 81, 67–74. [Google Scholar] [CrossRef]
- Paulmurugan, R.; Gambhir, S.S. An intramolecular folding sensor for imaging estrogen receptor-ligand interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 15883–15888. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Arts, R.; Merkx, M. Ratiometric Bioluminescent Sensor Proteins Based on Intramolecular Split Luciferase Complementation. ACS Sens. 2019, 4, 20–25. [Google Scholar] [CrossRef]
- Van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein–protein interactions in living cells. J. Cell Biol. 2007, 177, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Brancato, V.; Oliveira, J.M.; Correlo, V.M.; Reis, R.L.; Kundu, S.C. Could 3D models of cancer enhance drug screening? Biomaterials 2020, 232, 119744. [Google Scholar] [CrossRef]
- Calabretta, M.M.; Montali, L.; Lopreside, A.; Michelini, E.; Roda, A. High-Throughput Bioluminescence Imaging and Reporter Gene Assay with 3D Spheroids from Human Cell Lines. In Bioluminescent Imaging; Humana: New York, NY, USA, 2020; Volume 2081, pp. 3–14. [Google Scholar]
- Langan, L.M.; Dodd, N.J.; Owen, S.F.; Purcell, W.M.; Jackson, S.K.; Jha, A.N. Direct Measurements of Oxygen Gradients in Spheroid Culture System Using Electron Parametric Resonance Oximetry. PLoS ONE 2016, 11, e0149492. [Google Scholar]
- Anada, T.; Fukuda, J.; Sai, Y.; Suzuki, O. An oxygen-permeable spheroid culture system for the prevention of central hypoxia and necrosis of spheroids. Biomaterials 2012, 33, 8430–8441. [Google Scholar] [CrossRef] [PubMed]
- Cevenini, L.; Calabretta, M.M.; Lopreside, A.; Branchini, B.R.; Southworth, T.L.; Michelini, E.; Roda, A. Bioluminescence Imaging of Spheroids for High-Throughput Longitudinal Studies on 3D Cell Culture Models. Photochem. Photobiol. 2017, 93, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Campana, C.; Rege, J.; Turcu, A.F.; Pezzi, V.; Gomez-Sanchez, C.E.; Robins, D.M.; Rainey, W.E. Development of a novel cell based androgen screening model. J. Steroid Biochem. Mol. Biol. 2016, 156, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Michelini, E.; Cevenini, L.; Mezzanotte, L.; Leskinen, P.; Virta, M.; Karp, M.; Roda, A. A sensitive recombinant cell-based bioluminescent assay for detection of androgen-like compounds. Nat. Protoc. 2008, 3, 1895–1902. [Google Scholar] [CrossRef]
- Bovee, T.H.F.; Bor, G.; Heskamp, H.H.; Lasaroms, J.J.; Sanders, M.B.; Nielen, M.W.F. Validation and application of a yeast bioassay for screening androgenic activity in calf urine and feed. Anal. Chim. Acta 2009, 637, 225–234. [Google Scholar] [CrossRef]
- Hu, D.G.; Hickey, T.E.; Irvine, C.; Wijayakumara, D.D.; Lu, L.; Tilley, W.D.; Selth, L.A.; Mackenzie, P.I. Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm. Cancer 2014, 5, 61–71. [Google Scholar] [CrossRef]
- Lambrechts, D.; Roeffaers, M.B.J.; Goossens, K.; Hofkens, J.; Van De Putte, T.; Schrooten, J.; Van Oosterwyck, H. A Causal Relation between Bioluminescence and Oxygen to Quantify the Cell Niche. PLoS ONE 2014, 9, e97572. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Hassan, J.; Barkin, J. Testosterone deficiency syndrome: Benefits, risks, and realities associated with testosterone replacement therapy. Can. J. Urol. 2016, 23, 20–30. [Google Scholar] [PubMed]
- Matsumoto, A.M.; Bremner, W.J. Serum testosterone assays--accuracy matters. J. Clin. Endocrinol. Metab. 2004, 89, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekström, L.; Cevenini, L.; Michelini, E.; Schulze, J.; Ethörngren, J.-O.; Belanger, A.; Guillemette, C.; Egarle, M.; Roda, A.; Rane, A. Testosterone challenge and androgen receptor activity in relation to UGT2B17 genotypes. Eur. J. Clin. Investig. 2013, 43, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Piper, T.; Heimbach, S.; Adamczewski, M.; Thevis, M. An in vitro assay approach to investigate the potential impact of different doping agents on the steroid profile. Drug Test. Anal. 2020. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabretta, M.M.; Lopreside, A.; Montali, L.; Cevenini, L.; Roda, A.; Michelini, E. A Genetically Encoded Bioluminescence Intracellular Nanosensor for Androgen Receptor Activation Monitoring in 3D Cell Models. Sensors 2021, 21, 893. https://doi.org/10.3390/s21030893
Calabretta MM, Lopreside A, Montali L, Cevenini L, Roda A, Michelini E. A Genetically Encoded Bioluminescence Intracellular Nanosensor for Androgen Receptor Activation Monitoring in 3D Cell Models. Sensors. 2021; 21(3):893. https://doi.org/10.3390/s21030893
Chicago/Turabian StyleCalabretta, Maria Maddalena, Antonia Lopreside, Laura Montali, Luca Cevenini, Aldo Roda, and Elisa Michelini. 2021. "A Genetically Encoded Bioluminescence Intracellular Nanosensor for Androgen Receptor Activation Monitoring in 3D Cell Models" Sensors 21, no. 3: 893. https://doi.org/10.3390/s21030893