De novo Sequencing and Analysis of Salvia hispanica Tissue-Specific Transcriptome and Identification of Genes Involved in Terpenoid Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials & Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction and Illumina Sequencing
2.3. Bioinformatic Analysis
2.4. Clustering
2.5. Phylogenetic Analysis
2.6. Hormone and Stress Treatments
2.7. cDNA Synthesis & qPCR Analysis
3. Results and Discussion
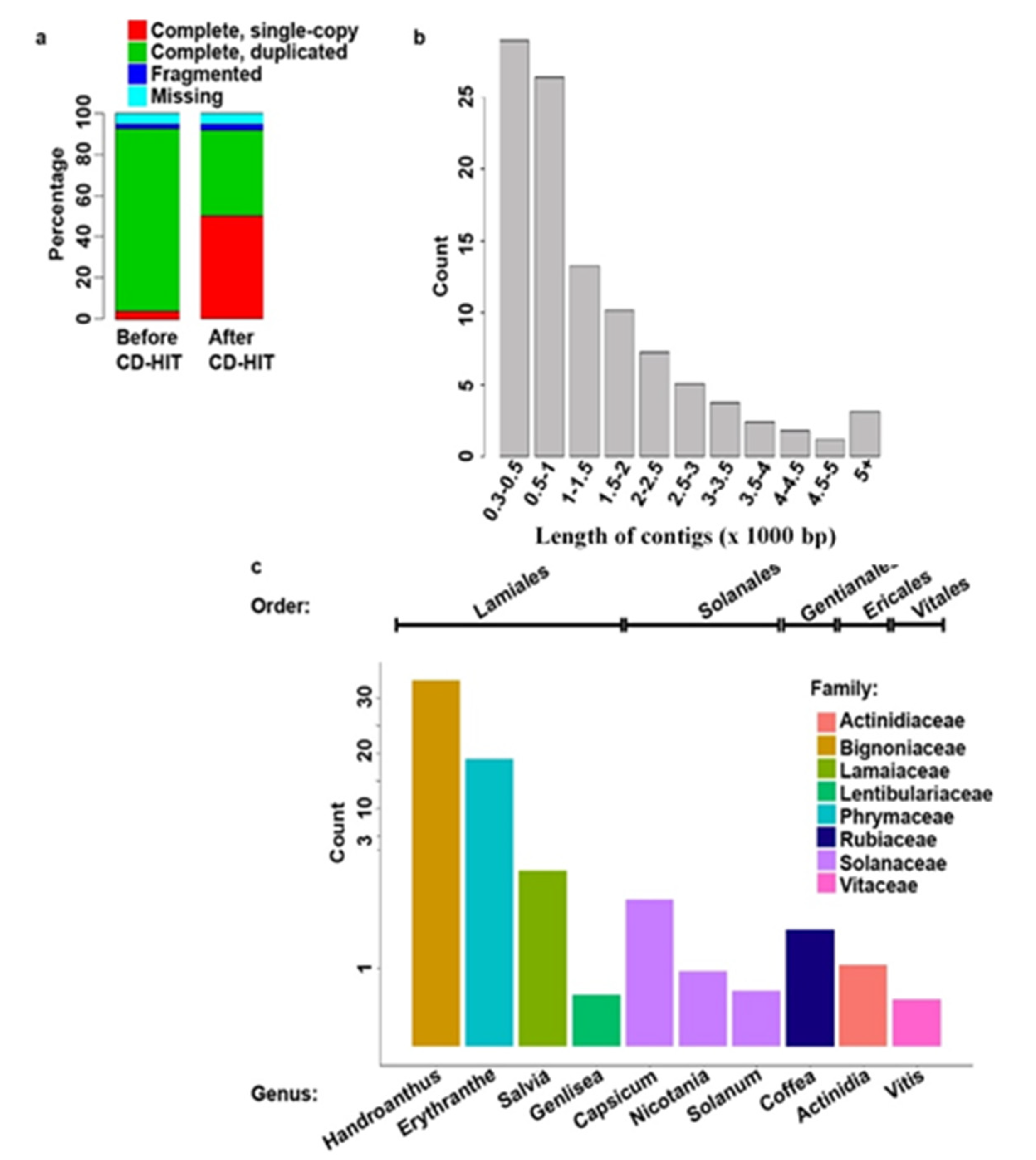
3.1. This Sequencing and de novo Assembly
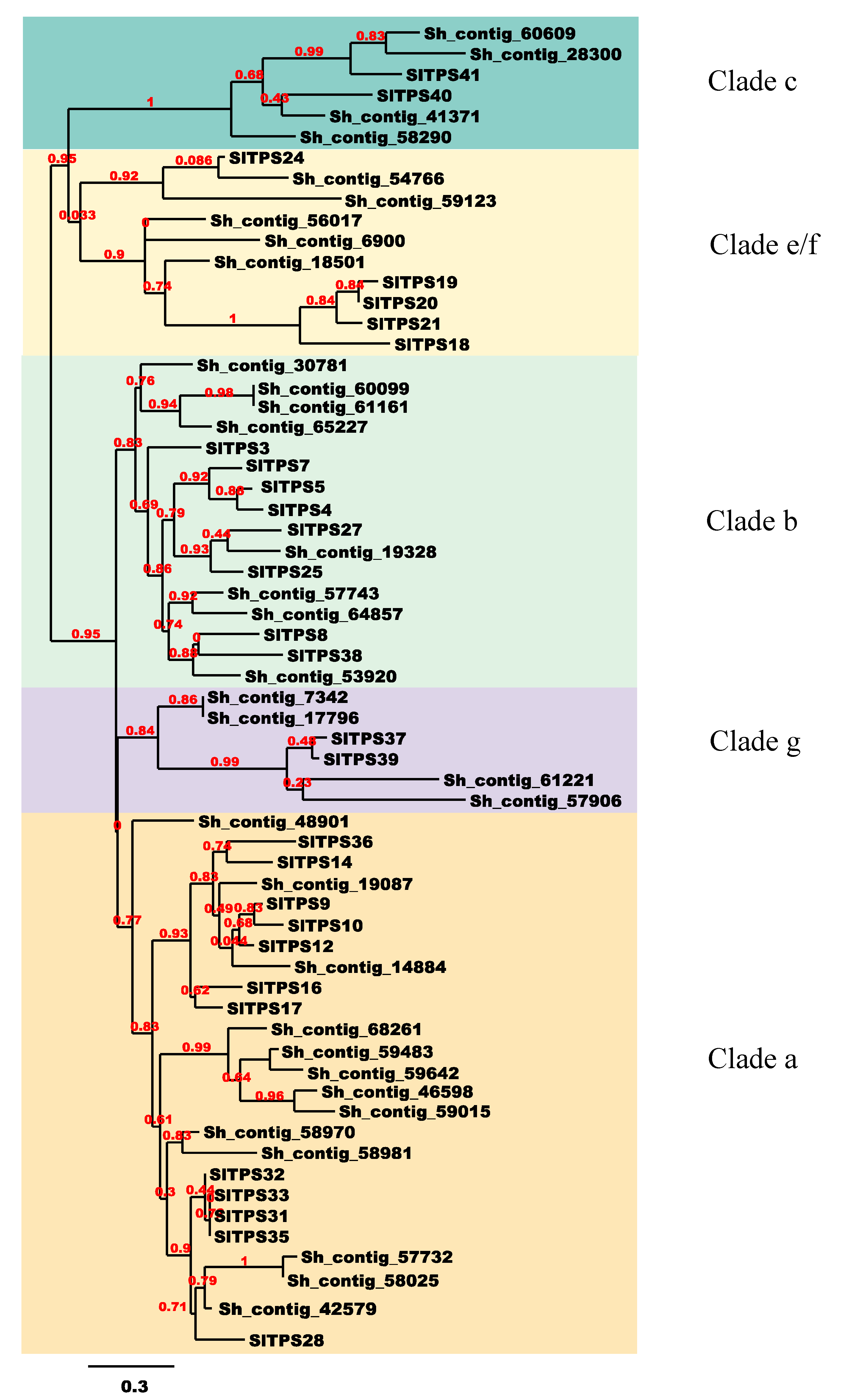
3.2. Annotation and Phylogenetic Analysis
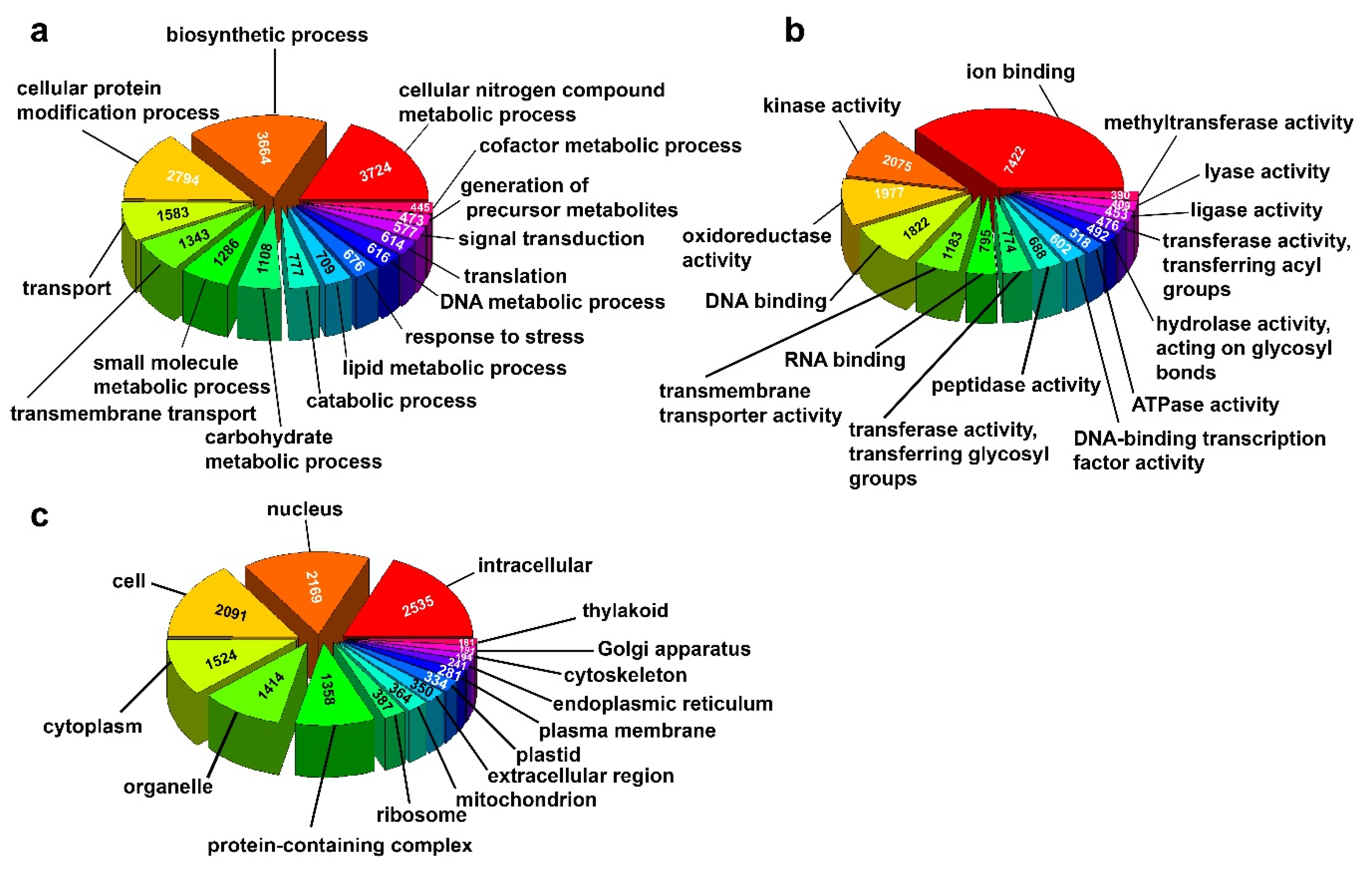
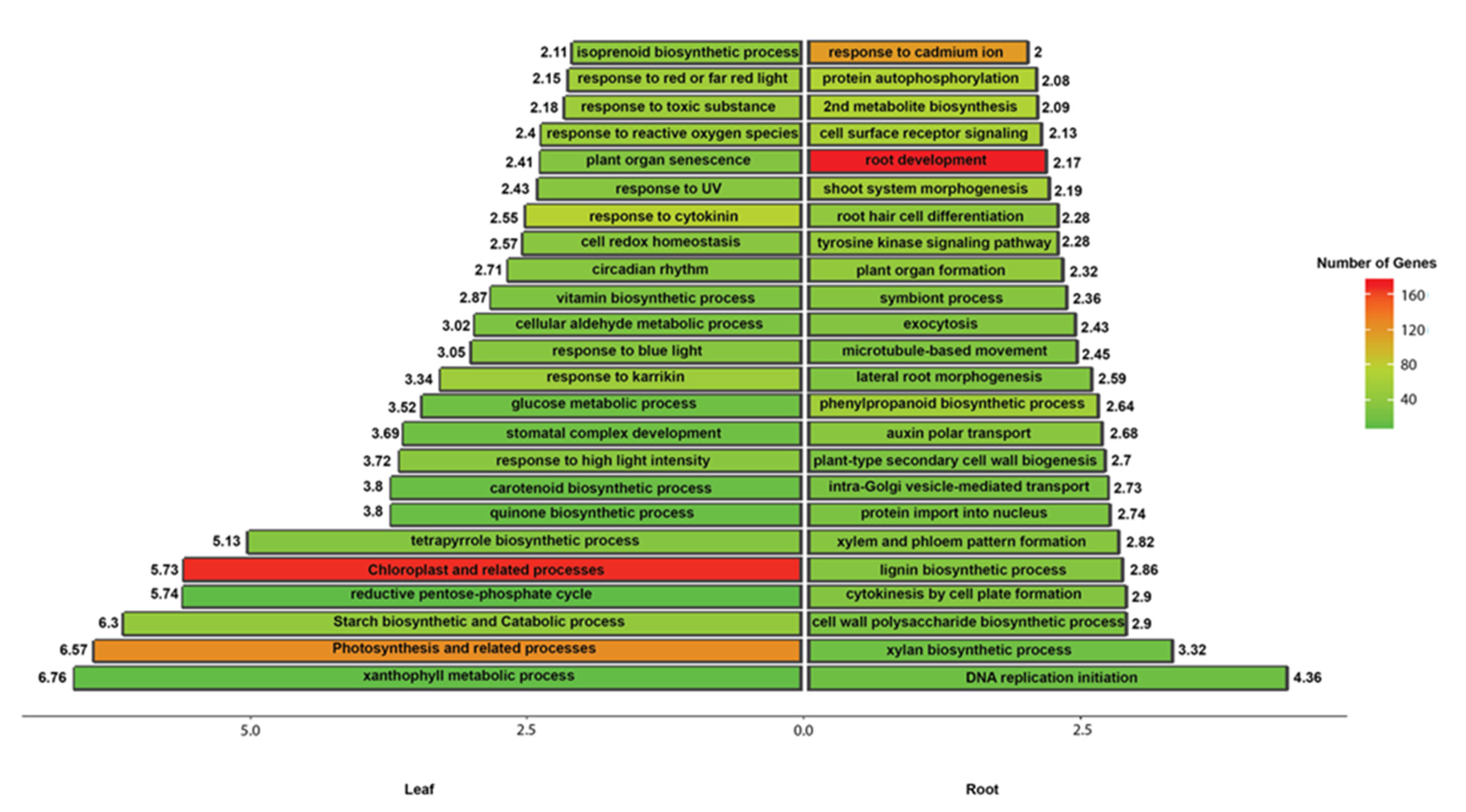
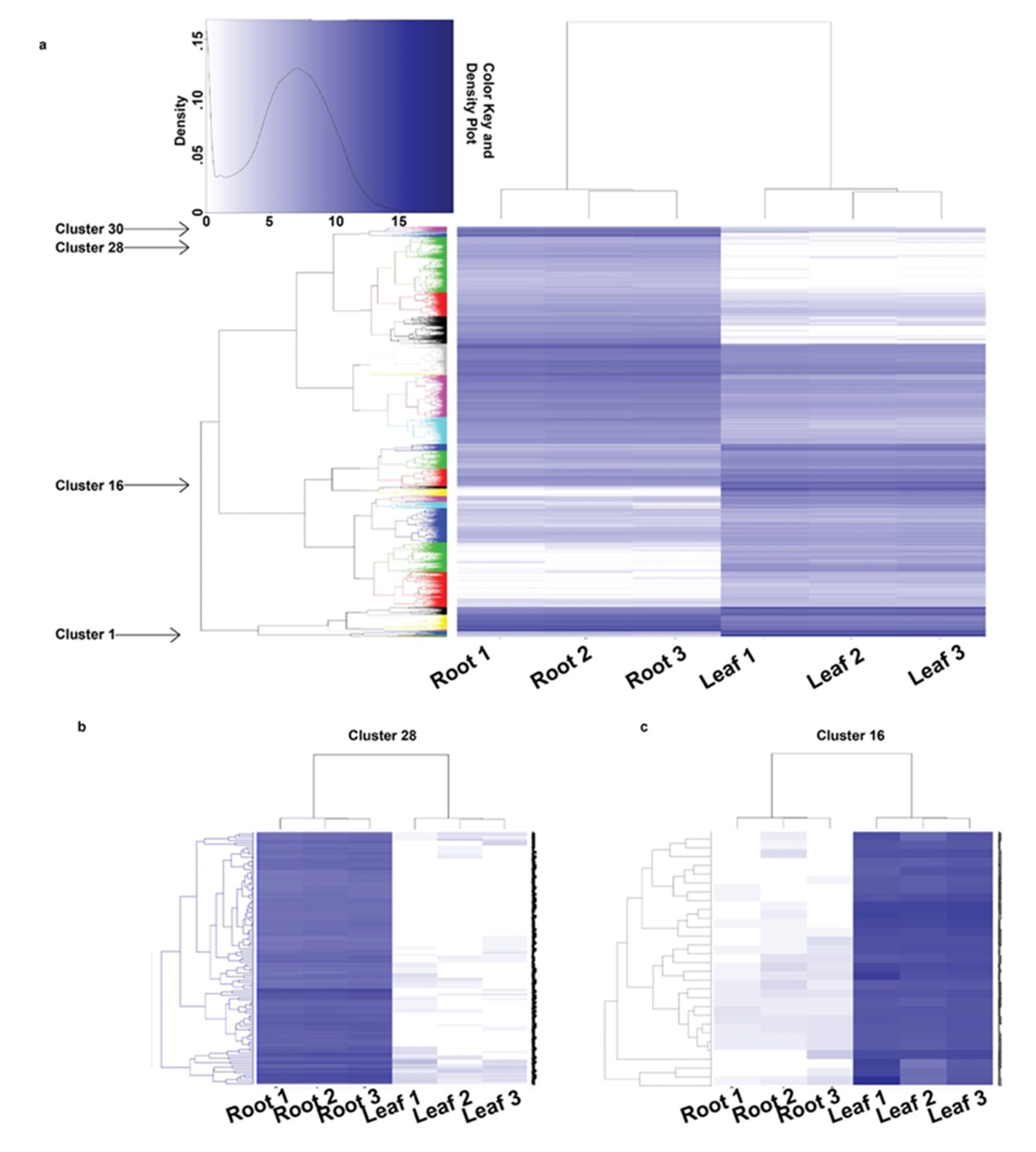
3.3. Differential Gene Expression and GO Enrichment Analysis
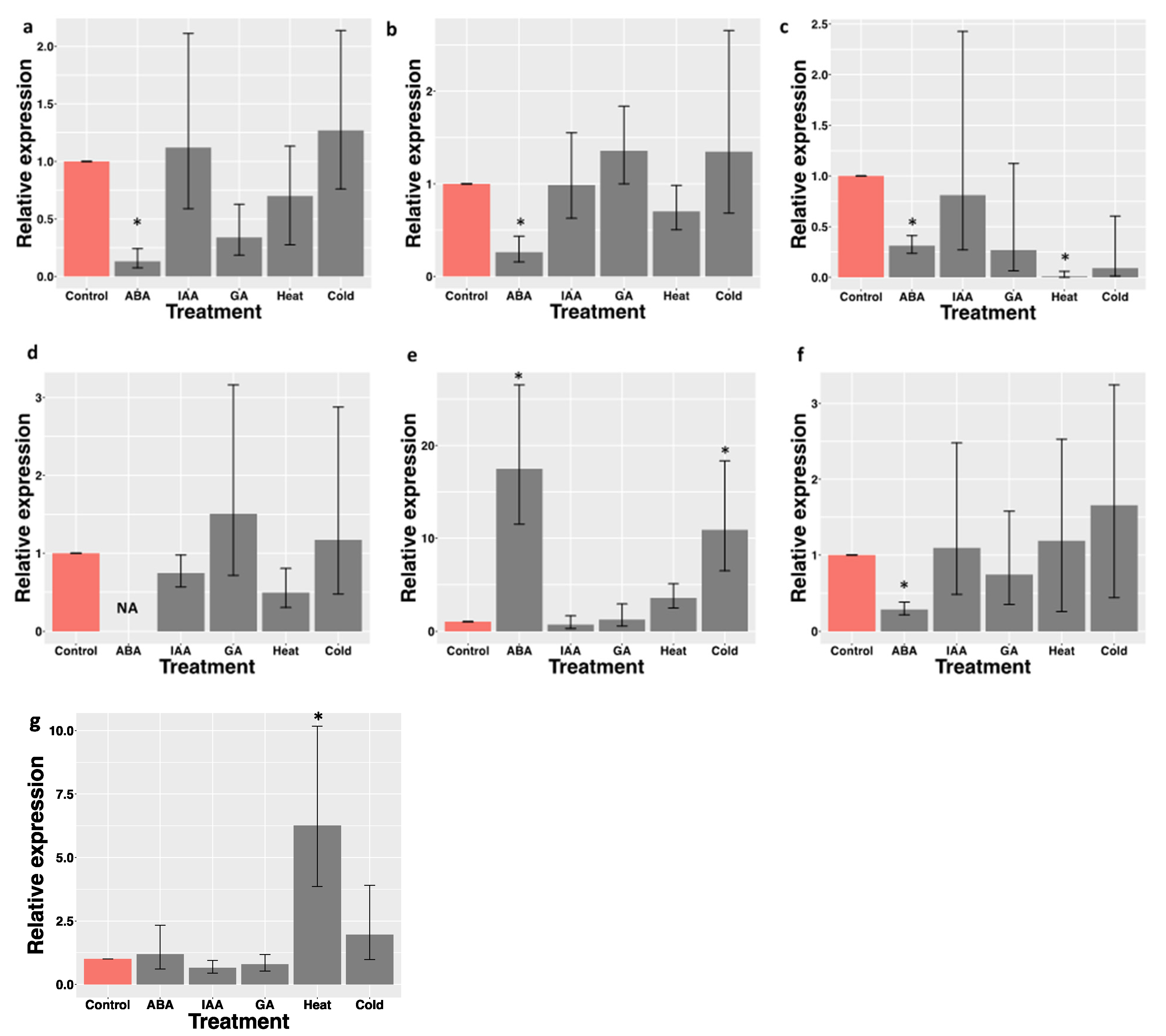
3.4. Enrichment of Vitamin Biosynthetic Genes and RT-qPCR Validation of Gene Expression
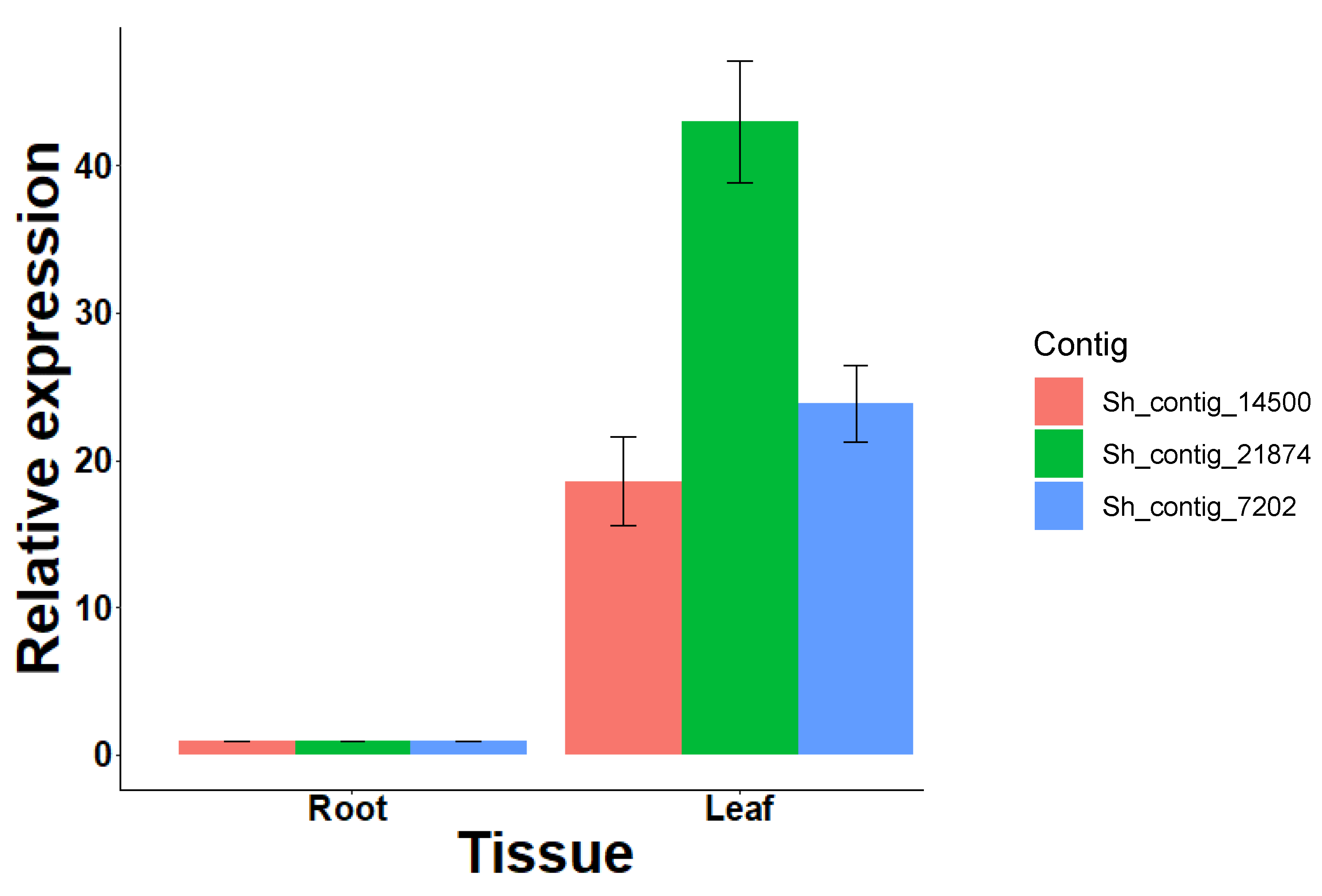
3.5. S. hispanica Terpene Synthase Family Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ayerza, R.; Coates, W. Chia: Rediscovering a Forgotten Crop of the Aztecs; University of Arizona Press: Tucson, AZ, USA, 2005. [Google Scholar]
- Cahill, J.P. Ethnobotany of Chia, Salvia hispanica L. (Lamiaceae). Econ. Bot. 2003, 57, 604–618. [Google Scholar] [CrossRef]
- Reyes-Caudillo, E.; Tecante, A.; Valdivia-Lopez, M.A. Dietary fibre content and antioxidant activity of phenolic compounds present in Mexican Chia (Salvia hispanica L.) seeds. Food Chem. 2008, 107, 656–663. [Google Scholar] [CrossRef]
- Sandoval-Oliveros, M.R.; Paredes-Loópez, O. Isolation and characterization of proteins from Chia seeds (Salvia hispanica L.). J. Agric. Food Chem. 2013, 61, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Ayerza, R.; Coates, W. Protein content, oil content and fatty acid profiles as potential criteria to determine the origin of commercially grown chia (Salvia hispanica L.). Ind. Crops Prod. 2011, 34, 1366–1371. [Google Scholar] [CrossRef]
- Ouzounidou, G.; Skiada, V.; Papadopoulou, K.K.; Stamatis, N.; Kavvadias, V.; Eleftheriadis, E.; Gaitis, F. Effects of soil pH and arbuscular mycorrhiza (AM) inoculation on growth and chemical composition of chia (Salvia hispanica L.) leaves. Braz. J. Bot. 2015, 38, 487–495. [Google Scholar] [CrossRef]
- Elshafie, H.S.; Aliberti, L.; Amato, M.; De Feo, V.; Camele, I. Chemical composition and antimicrobial activity of chia (Salvia hispanica L.) essential oil. Eur. Food Res. Technol. 2018, 244, 1675–1682. [Google Scholar] [CrossRef]
- Pascual-Villalobos, M.; Correal, E.; Molina, E.; Martínez, J. Evaluación y Selección de Especies Vegetales Productoras de CompuestosNaturales con Actividad Insecticid; Centro de Investigación y DesarrolloAgroalimentario (CIDA): Murcia, Spain, 1997. [Google Scholar]
- Ahmed, M.; Ting, I.P.; Scora, R.W. Leaf oil composition of Salvia hispanica L. from three geographical areas. J. Essent. Oil Res. 1994, 6, 223–228. [Google Scholar] [CrossRef]
- Cheng, A.X.; Lou, Y.G.; Mao, Y.B.; Lu, S.; Wang, L.J.; Chen, X.Y. Plant terpenoids: Biosynthesis and ecological functions. J. Integr. Plant Biol. 2007, 49, 179–186. [Google Scholar] [CrossRef]
- Dudareva, N.; Pichersky, E.; Gershenzon, J. Biochemistry of plant volatiles. Plant Physiol. 2004, 135, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Mumm, R.; Posthumus, M.A.; Dicke, M. Significance of terpenoids in induced indirect plant defence against herbivorous arthropods. Plant Cell Environ. 2008, 31, 575–585. [Google Scholar] [CrossRef]
- Pichersky, E.; Gershenzon, J. The formation and function of plant volatiles: Perfumes for pollinator attraction and defense. Curr. Opin. Plant Biol. 2002, 5, 237–243. [Google Scholar] [CrossRef]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant terpenoid synthases: Molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falara, V.; Akhtar, T.A.; Nguyen, T.T.H.; Spyropoulou, E.A.; Bleeker, P.M.; Schauvinhold, I.; Matsuba, Y.; Bonini, M.E.; Schilmiller, A.L.; Last, R.L.; et al. The tomato terpene synthase gene family. Plant Physiol. 2011, 157, 770–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Abel, C.; Sohrabi, R.; Petri, J.; Haupt, I.; Cosimano, J.; Gershenzon, J.; Tholl, D. Variation of herbivore-induced volatile terpenes among arabidopsis ecotypes depends on allelic differences and subcellular targeting of two terpene synthases, TPS02 and TPS03. Plant Physiol. 2010, 153, 1293–1310. [Google Scholar] [CrossRef] [Green Version]
- Aubourg, S.; Lecharny, A.; Bohlmann, J. Genomic analysis of the terpenoid synthase ( AtTPS) gene family of Arabidopsis thaliana. Mol. Genet. Genom. 2002, 267, 730–745. [Google Scholar] [CrossRef]
- Miller, D.J.; Allemann, R.K. Sesquiterpene synthases: Passive catalysts or active players? Nat. Prod. Rep. 2011, 29, 60–71. [Google Scholar] [CrossRef]
- Schnee, C.; Kollner, T.G.; Held, M.; Turlings, T.C.; Gershenzon, J.; Degenhardt, J. The products of a single maize sesquiterpene synthase form a volatile defense signal that attracts natural enemies of maize herbivores. Proc. Natl. Acad. Sci. USA 2006, 103, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Tang, K.; Li, P. Plant metabolic engineering strategies for the production of pharmaceutical terpenoids. Front. Plant Sci. 2016, 7, 1647. [Google Scholar] [CrossRef] [Green Version]
- Amato, M.; Caruso, M.C.; Guzzo, F.; Galgano, F.; Commisso, M.; Bochicchio, R.; Labella, R.; Favati, F. Nutritional quality of seeds and leaf metabolites of Chia (Salvia hispanica L.) from Southern Italy. Eur. Food Res. Technol. 2015, 241, 615–625. [Google Scholar] [CrossRef]
- Kumar, R.; Ichihashi, Y.; Kimura, S.; Chitwood, D.H.; Headland, L.R.; Peng, J.; Maloof, J.N.; Sinha, N.R. A High-Throughput Method for Illumina RNA-Seq Library Preparation. Front. Plant Sci. 2012, 3, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P.; et al. AgBase: A functional genomics resource for agriculture. BMC Genom. 2006, 7, 229. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, T. Dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnes, G.R. Gplots: Various R Programming Tools for Plotting Data. Available online: http://cranr-projectorg/web/packages/gplots/indexhtml (accessed on 15 March 2018).
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Gopalam, R.; Rupwate, S.D.; Tumaney, A.W. Selection and validation of appropriate reference genes for quantitative real-time PCR analysis in Salvia hispanica. PLoS ONE 2017, 12, e0186978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, X.G.; Armbruster, P.A. Comparative performance of transcriptome assembly methods for non-model organisms. BMC Genom. 2016, 17, 523. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Bushway, A.A.; Belyea, P.R.; Bushway, R.J. Chia seed as a source of oil, polysaccharide, and protein. J. Food Sci. 1981, 46, 1349–1351. [Google Scholar] [CrossRef]
- Marcinek, K.; Krejpcio, Z. Chia seeds (Salvia hispanica): Health promoting properties and therapeutic applications—A review. Rocz. Panstw. Zakl. Hig. 2017, 68, 123–129. [Google Scholar]
- Dong, H.; Beer, S.V. Riboflavin induces disease resistance in plants by activating a novel signal transduction pathway. Phytopathology 2000, 90, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Goyer, A. Thiamine in plants: Aspects of its metabolism and functions. Phytochemistry 2010, 71, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.B.; Basset, G.J.; Borel, P.; Carrari, F.; DellaPenna, D.; Fraser, P.D.; Hellmann, H.; Osorio, S.; Rothan, C.; Valpuesta, V.; et al. Vitamin deficiencies in humans: Can plant science help? Plant Cell 2012, 24, 395–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Liu, B.; Li, M.; Gao, X.; Fang, Q.; Liu, C.; Ding, H.; Wang, L.; Gao, X. Identification and characterization of terpene synthase genes accounting for volatile terpene emissions in flowers of Freesia x hybrida. J. Exp. Bot. 2018, 69, 4249–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagegowda, D.A. Plant volatile terpenoid metabolism: Biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett. 2010, 584, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Jayaprakasha, G.K.; Jagan Mohan Rao, L.; Sakariah, K.K. Volatile constituents from Cinnamomum zeylanicum fruit stalks and their antioxidant activities. J. Agric. Food Chem. 2003, 51, 4344–4348. [Google Scholar] [CrossRef]
- Mockute, D.; Bernotiene, G.; Judzentiene, A. The essential oil of Origanum vulgare L. ssp. vulgare growing wild in vilnius district (Lithuania). Phytochemistry 2001, 57, 65–69. [Google Scholar] [CrossRef]
- Orav, A.; Stulova, I.; Kailas, T.; Muurisepp, M. Effect of storage on the essential oil composition of Piper nigrum L. fruits of different ripening states. J. Agric. Food Chem. 2004, 52, 2582–2586. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.Z.; Xie, X.Q.; Altmann, K.H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Legault, J.; Pichette, A. Potentiating effect of beta-caryophyllene on anticancer activity of alpha-humulene, isocaryophyllene and paclitaxel. J. Pharm. Pharmacol. 2007, 59, 1643–1647. [Google Scholar] [CrossRef]
- Tholl, D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 2006, 9, 297–304. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Miner, D.P.; Larson, M.; McDowell, E.; Gang, D.R.; Wilkerson, C.; Last, R.L. Studies of a biochemical factory: Tomato trichome deep expressed sequence tag sequencing and proteomics. Plant Physiol. 2010, 153, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colby, S.M.; Crock, J.; Dowdle-Rizzo, B.; Lemaux, P.G.; Croteau, R. Germacrene C synthase from Lycopersicon esculentum cv. VFNT cherry tomato: cDNA isolation, characterization, and bacterial expression of the multiple product sesquiterpene cyclase. Proc. Natl. Acad. Sci. USA 1998, 95, 2216–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilmiller, A.L.; Schauvinhold, I.; Larson, M.; Xu, R.; Charbonneau, A.L.; Schmidt, A.; Wilkerson, C.; Last, R.L.; Pichersky, E. Monoterpenes in the glandular trichomes of tomato are synthesized from a neryl diphosphate precursor rather than geranyl diphosphate. Proc. Natl. Acad. Sci. USA 2009, 106, 10865–10870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Chen, F.; Wang, X.; Rajapakse, N.C. Effect of methyl jasmonate on secondary metabolites of sweet basil (Ocimum basilicum L.). J. Agric. Food Chem. 2006, 54, 2327–2332. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Chen, F.; Kim, H.J. Chemical changes and overexpressed genes in sweet basil (Ocimum basilicum L.) upon methyl jasmonate treatment. J. Agric. Food Chem. 2007, 55, 706–713. [Google Scholar] [CrossRef]
- Rodriguez-Saona, C.; Crafts-Brandner, S.J.; Pare, P.W.; Henneberry, T.J. Exogenous methyl jasmonate induces volatile emissions in cotton plants. J. Chem. Ecol. 2001, 27, 679–695. [Google Scholar] [CrossRef]
- Alonso, R.; Berli, F.J.; Bottini, R.; Piccoli, P. Acclimation mechanisms elicited by sprayed abscisic acid, solar UV-B and water deficit in leaf tissues of field-grown grapevines. Plant Physiol. Biochem. 2015, 91, 56–60. [Google Scholar] [CrossRef]
- Kochan, E.; Balcerczak, E.; Szymczyk, P.; Sienkiewicz, M.; Zielinska-Blizniewska, H.; Szymanska, G. Abscisic acid regulates the 3-Hydroxy-3-methylglutaryl CoA Reductase gene promoter and ginsenoside production in Panax quinquefolium hairy root cultures. Int. J. Mol. Sci. 2019, 20, 1310. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Liu, R.; Liaw, S.Y.; Chan, H.S.; Tsay, H.S. Enhanced tanshinone production in hairy roots of ‘Salvia miltiorrhiza Bunge’ under the influence of plant growth regulators in liquid culture. Bot. Stud. 2011, 52, 435–443. [Google Scholar]
- Mansouri, H.; Asrar, Z.; Amarowicz, R. The response of terpenoids to exogenous gibberellic acid in Cannabis sativa L. at vegetative stage. Acta Physiol. Plant. 2010, 33, 1085–1091. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wimberley, J.; Cahill, J.; Atamian, H.S. De novo Sequencing and Analysis of Salvia hispanica Tissue-Specific Transcriptome and Identification of Genes Involved in Terpenoid Biosynthesis. Plants 2020, 9, 405. https://doi.org/10.3390/plants9030405
Wimberley J, Cahill J, Atamian HS. De novo Sequencing and Analysis of Salvia hispanica Tissue-Specific Transcriptome and Identification of Genes Involved in Terpenoid Biosynthesis. Plants. 2020; 9(3):405. https://doi.org/10.3390/plants9030405
Chicago/Turabian StyleWimberley, James, Joseph Cahill, and Hagop S. Atamian. 2020. "De novo Sequencing and Analysis of Salvia hispanica Tissue-Specific Transcriptome and Identification of Genes Involved in Terpenoid Biosynthesis" Plants 9, no. 3: 405. https://doi.org/10.3390/plants9030405