The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae)


Abstract
:1. Introduction
2. Results
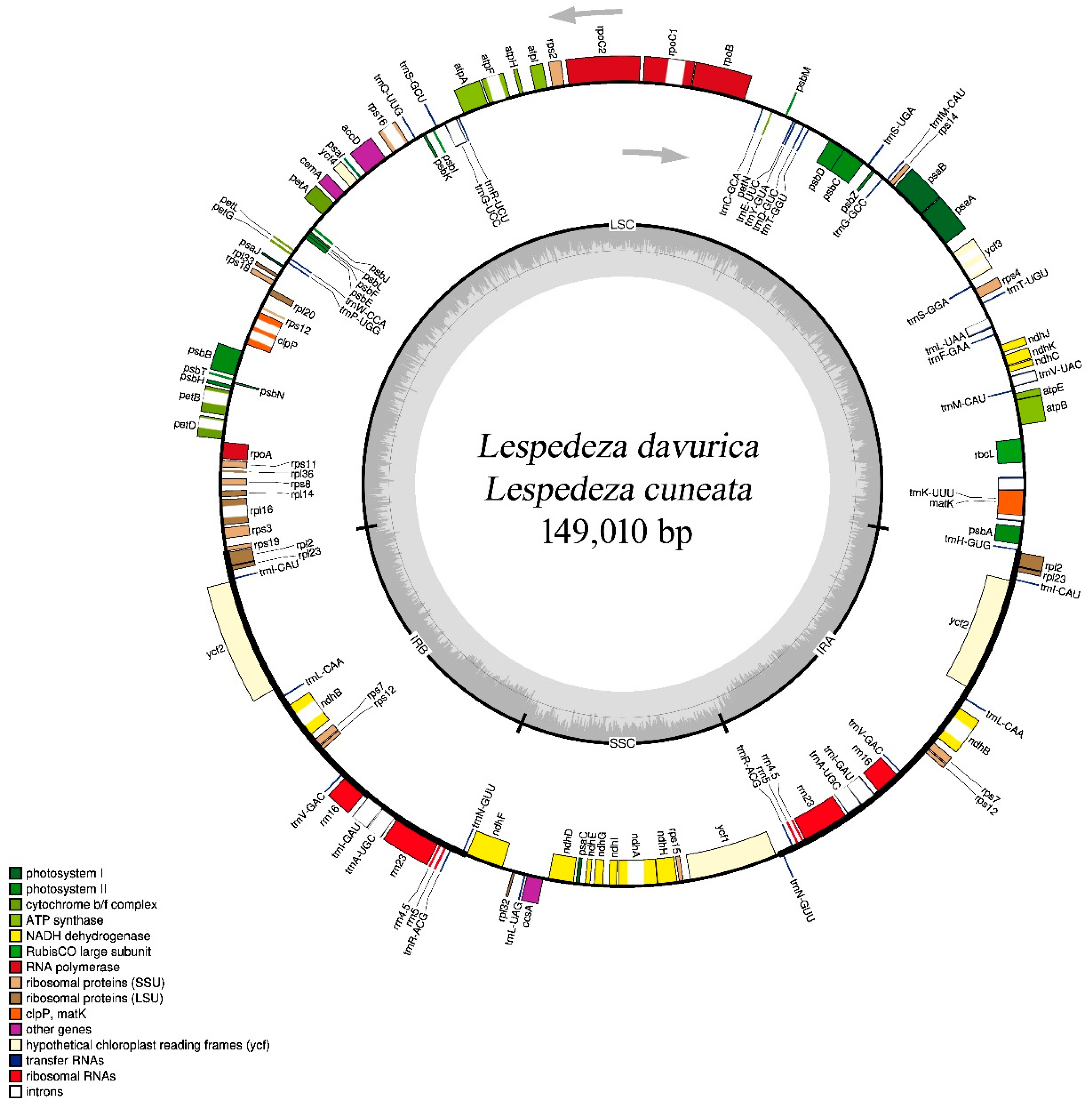
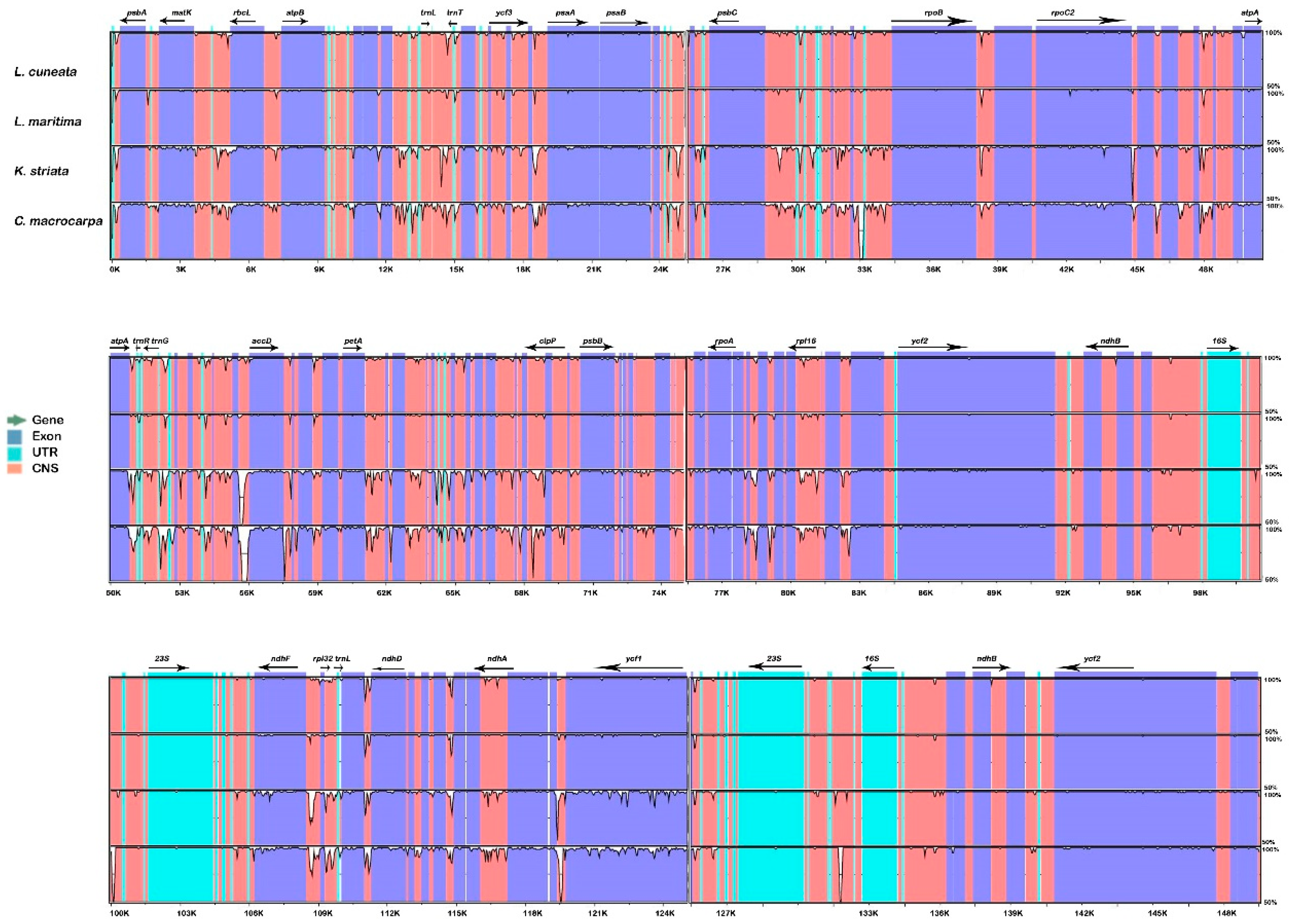
2.1. Comparison of Lespedeza cp Genome Structures
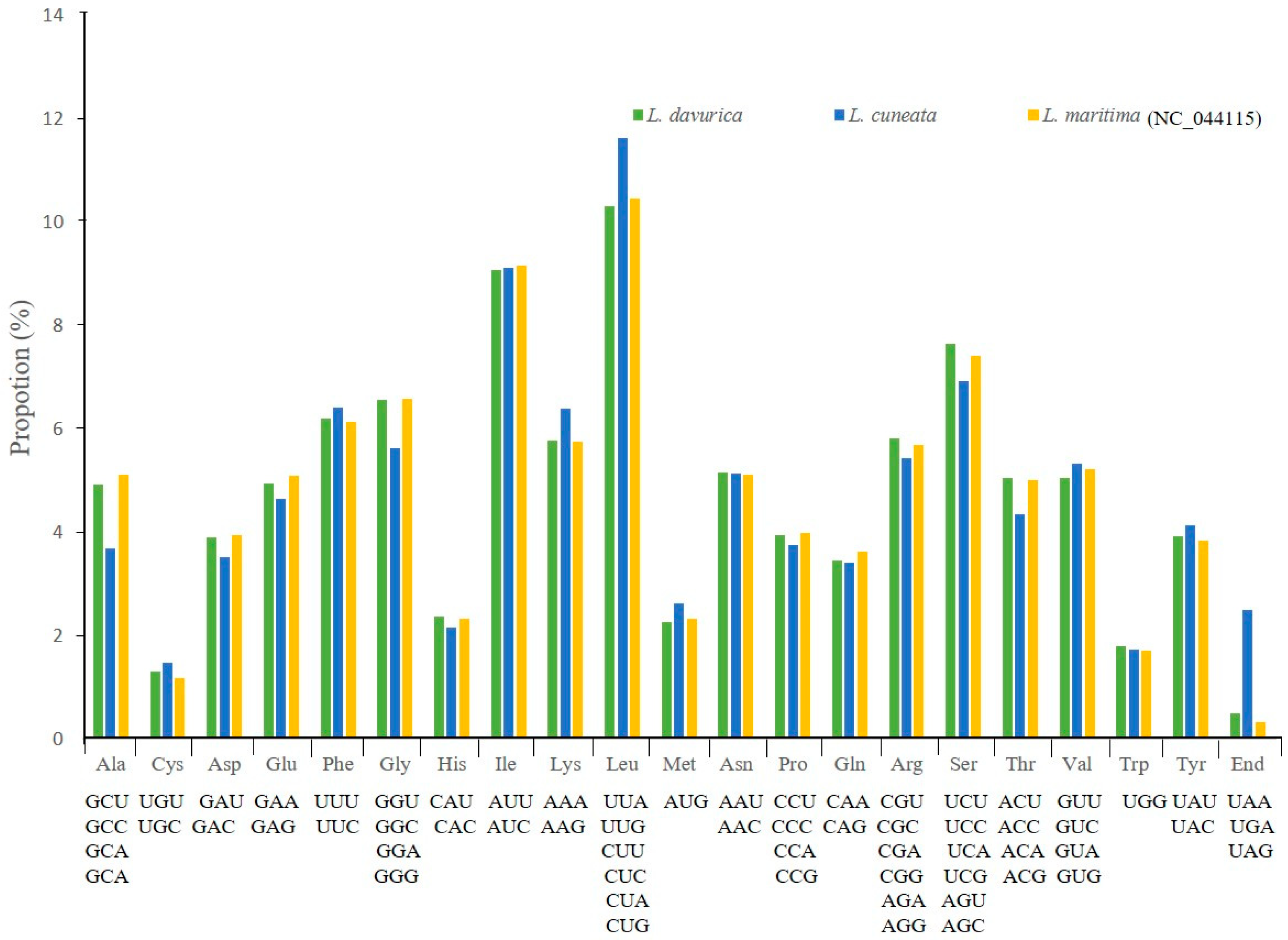
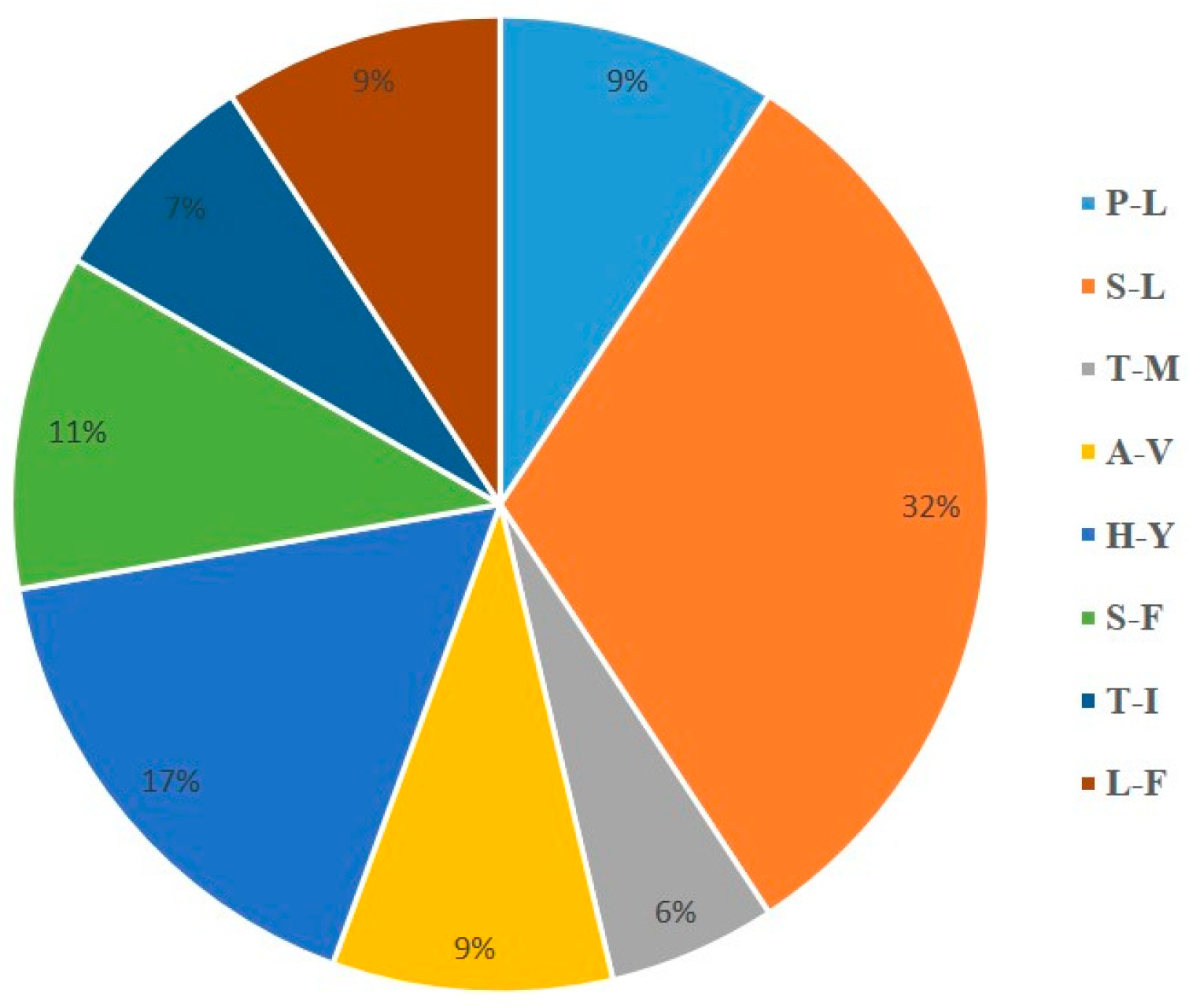
2.2. Codon Usage Bias and Prediction of RNA Editing Sites
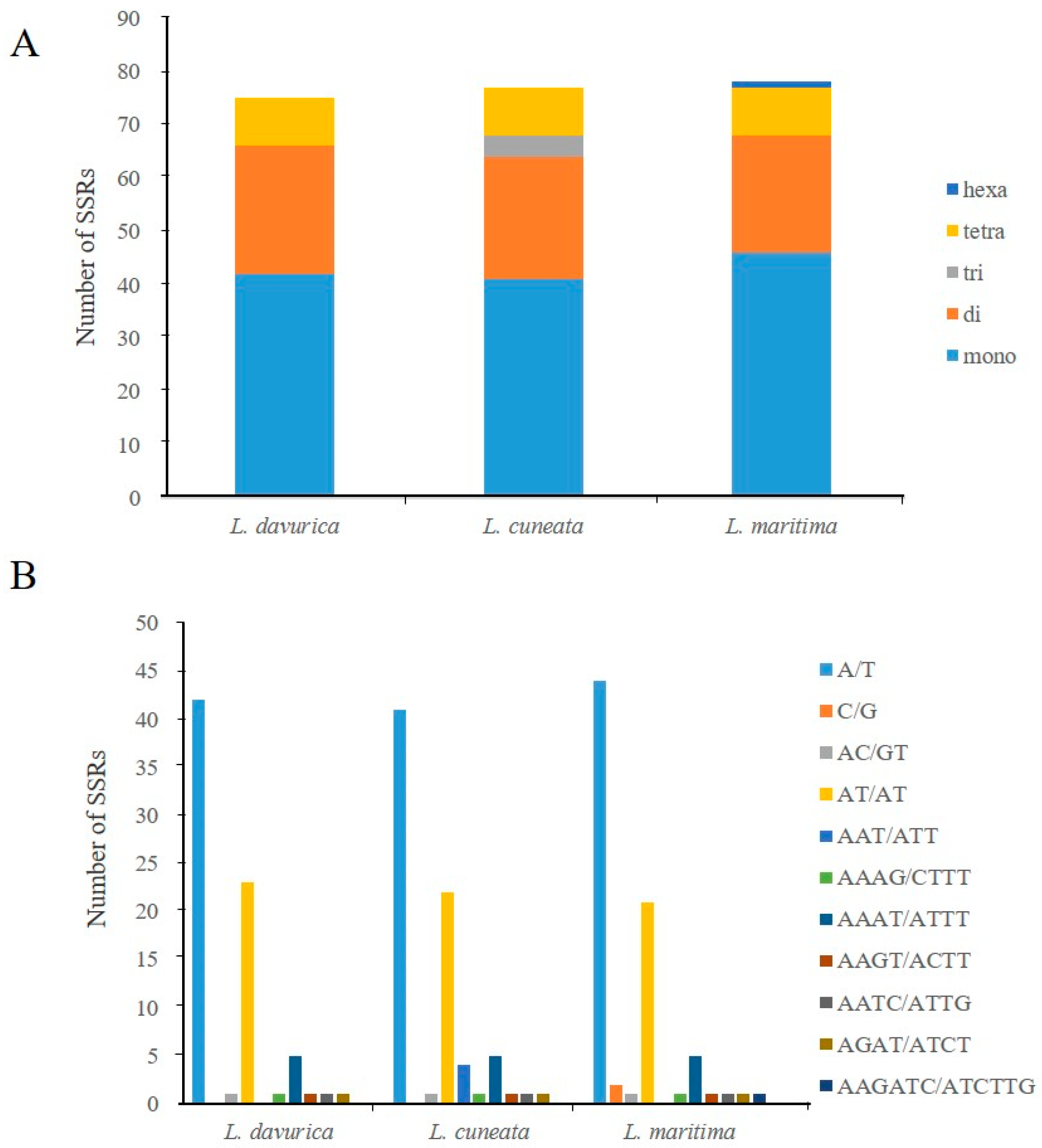
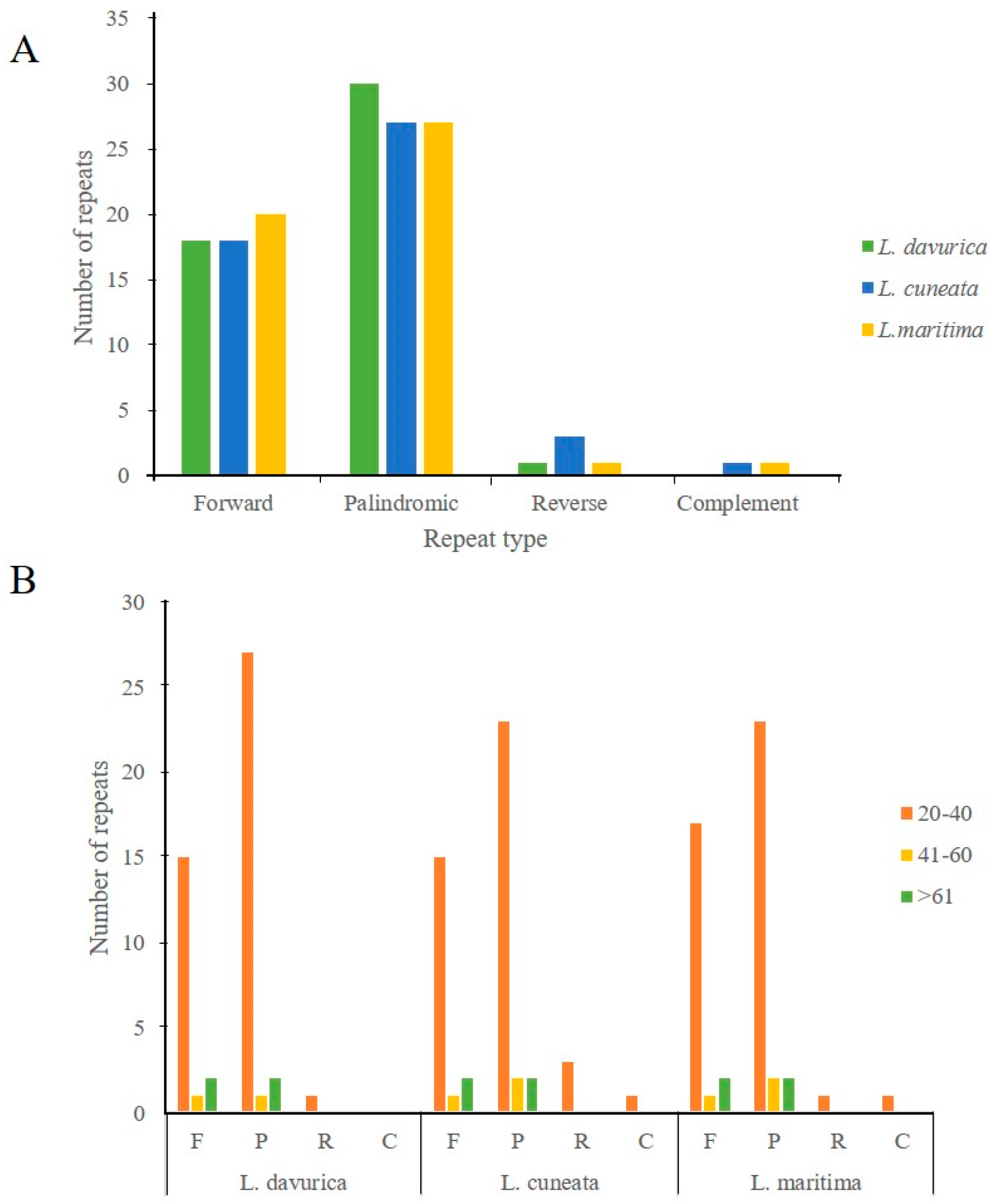
2.3. Simple Sequence Repeats and Repeat Structure Analysis
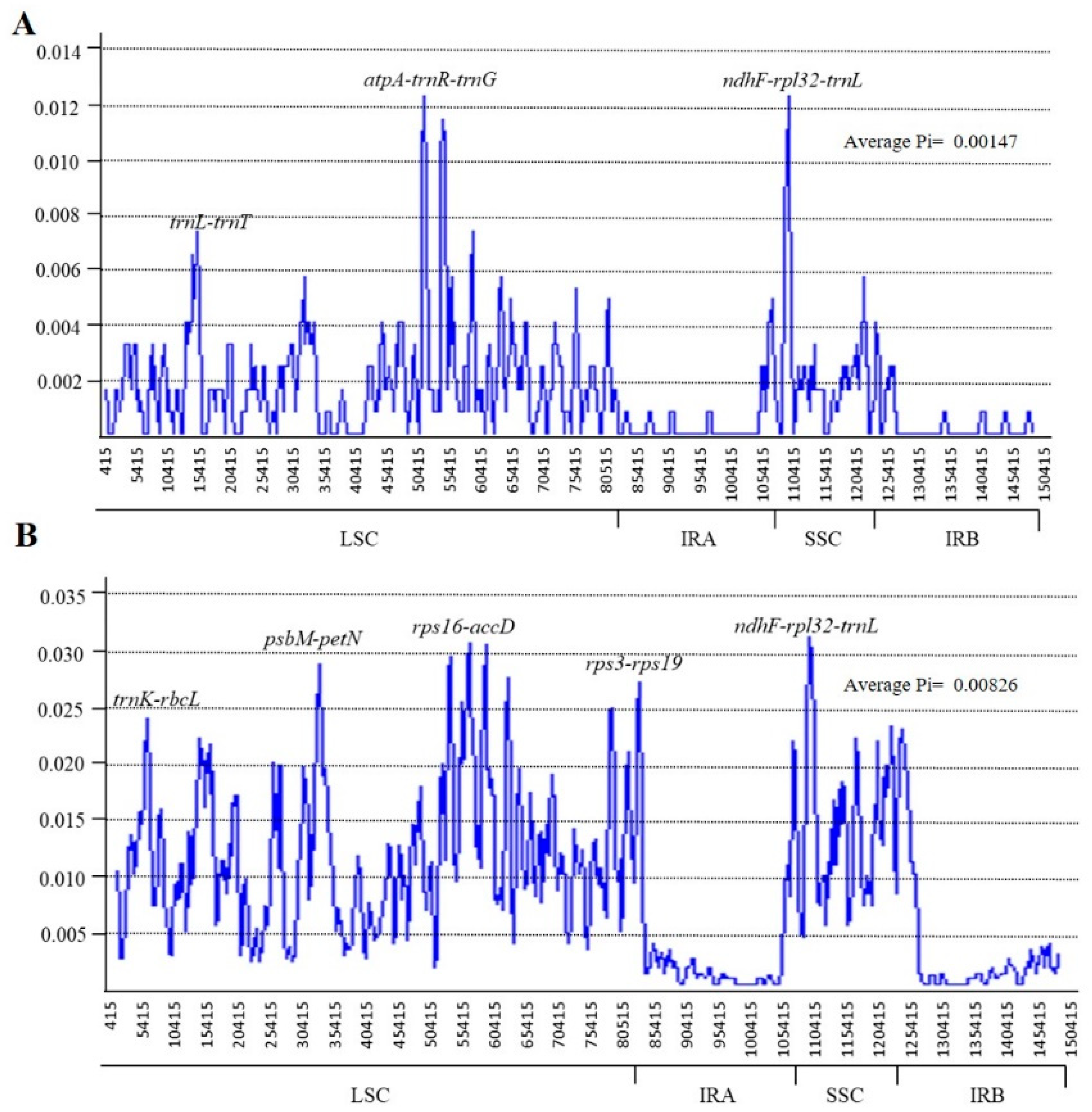
2.4. Sequence Divergence Analysis
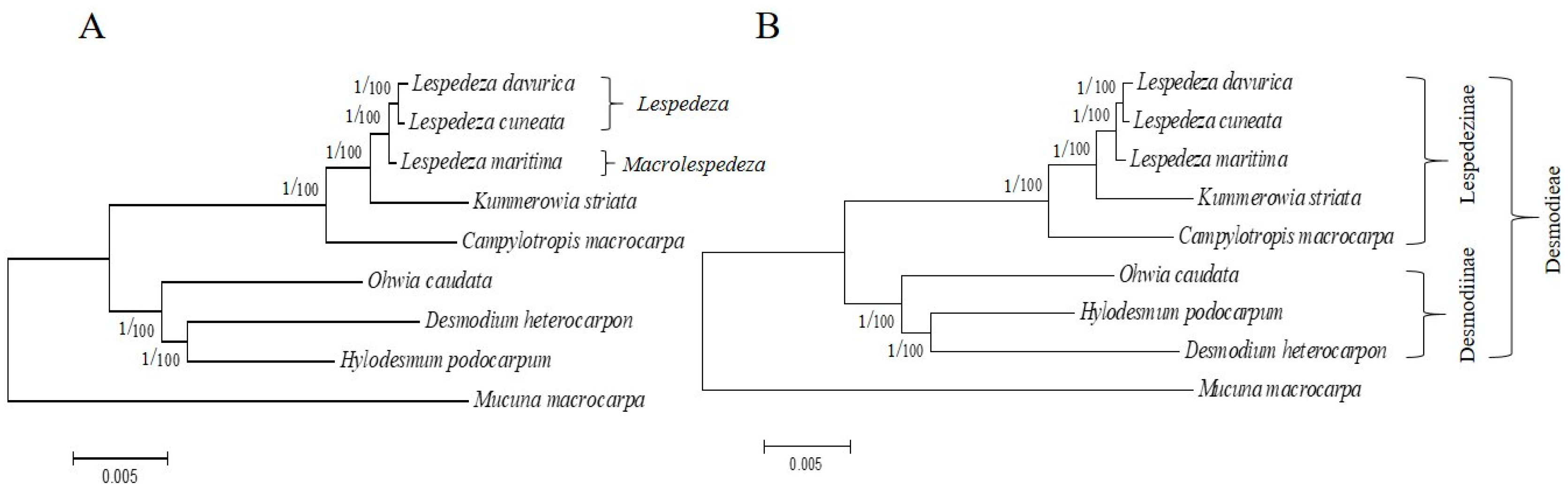
2.5. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and DNA Sequencing
4.2. Genome Assembly and Annotation
4.3. Codon Usage Bias, RNA Editing Sites, and Sequence Divergence
4.4. Repeat Sequences Analysis
4.5. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Han, J.E.; Chung, K.H.; Nemoto, T.; Choi, B.H. Phylogenetic analysis of eastern Asian and eastern North American disjunct Lespedeza (Fabaceae) inferred from nuclear ribosomal ITS and plastid region sequences. Bot. J. Linn. Soc. 2010, 164, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.H. Differences in leaf δ 13C among four dominant species in a secondary succession sere on the Loess Plateau of China. Photosynthetica 2003, 41, 525–531. [Google Scholar] [CrossRef]
- Guan, X.K.; Zhang, X.H.; Turner, N.C.; Xu, B.C.; Li, F.M. Two perennial legumes (Astragalus adsurgens Pall. and Lespedeza davurica S.) adapted to semiarid environments are not as productive as Lucerne (Medicago sativa L.) but use less water. Grass Forage Sci. 2013, 68, 469–478. [Google Scholar] [CrossRef]
- Chen, X.L.; An, S.; Liu, S.R.; Li, G.Q. Micro scale spatial heterogeneity and the loss of carbon, nitrogen and phosphorus in degraded grassland in Ordos Plateau, northwestern China. Plant Soil 2004, 259, 29–37. [Google Scholar] [CrossRef]
- Hu, L.; Busby, R.R.; Gebhart, D.L.; Yannarell, A.C. Invasive Lespedeza cuneata and native Lespedeza virginica experience asymmetrical benefits from rhizobial symbionts. Plant Soil 2014, 384, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.R.; Rhyu, D.Y. Lespedeza davurica (Lax.) Schindl. extract protects against cytokine-induced β-cell damage and streptozotocin-induced diabetes. BioMed Res. Int. 2015, 2015, 169256. [Google Scholar] [CrossRef]
- Ohashi, H. Nomenclatural changes in Leguminosae of Japan. J. Jpn. Bot. 1982, 57, 29–30. [Google Scholar]
- Xu, B.; Gao, X.F.; Wu, N.; Zhang, L.B. Pollen diversity and its systematic implications in Lespedeza (Fabaceae). Syst. Bot. 2011, 36, 352–361. [Google Scholar] [CrossRef]
- Nemoto, T.; Ohashi, H. Seedling morphology in Lespedeza (Leguminosae). J. Plant Res. 1993, 106, 121–128. [Google Scholar] [CrossRef]
- Ohashia, H.; Nemoto, T. A New System of Lespedeza (Leguminosae Tribe Desmodieae). J. Jpn. Bot. 2014, 89, 1–11. [Google Scholar]
- Kim, Y.M.; Lee, J.; Park, S.H.; Lee, C.; Lee, J.W.; Lee, D.; Kim, N.; Lee, D.; Kim, H.Y.; Lee, C.H. LC-MS-based chemotaxonomic classification of wild-type Lespedeza sp. and its correlation with genotype. Plant Cell Rep. 2012, 31, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, D.F.; Jia, X.; Mei, W.L.; Dai, H.F.; Chen, X.T.; Peng, S.Q. Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the Malvales order. Front. Plant Sci. 2016, 7, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Cui, Y.; Chen, X.; Li, Y.; Xu, Z.; Duan, B.; Li, Y.; Song, J.; Yao, H. Complete Chloroplast Genomes of Papaver rhoeas and Papaver orientale: Molecular Structures, Comparative Analysis, and Phylogenetic Analysis. Molecules 2018, 23, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, D.P.; Choi, I.S.; Choi, B.H. Plastid genome evolution in tribe Desmodieae (Fabaceae: Papilionoideae). PLoS ONE 2019, 14, e0218743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Zhang, X.W.; Liu, G.M.; Yin, Y.X.; Chen, K.F.; Yun, Q.Z.; Zhao, D.J.; Almssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.F.; Li, Z.H. Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes 2017, 8, 124. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.S.; Ming, Y.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Cheng, Y.F.; Deng, C.; Ma, Y.; Wang, Z.W.; Chen, X.H.; Xue, L.B. Comparative transcriptome analysis of eggplant (Solanum melongena L.) and turkey berry (Solanum torvum Sw.): Phylogenomics and disease resistance analysis. BMC Genom. 2014, 15, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Yang, C.; Liu, J.; Jin, S.; Harijati, N.; Hu, Z.; Diao, Y.; Zhao, L. Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 2019, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, J.; Yao, X.; Huang, H. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, e123–e126. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, J.; Cui, Y.; Wang, Y.; Duan, B.; Yao, H. Identification of Ligularia Herbs Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, J.; Song, Q.; Zhang, Q.; Tian, P.; Nan, Z. Comprehensive Analysis of Codon Usage Bias in Seven Epichloë Species and Their Peramine-Coding Genes. Front. Microbiol. 2017, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid Genome Comparative and Phylogenetic Analyses of the Key Genera in Fagaceae: Highlighting the Effect of Codon Composition Bias in Phylogenetic Inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Wu, Y.; Maliga, P.; Messing, J. RNA editing in chloroplasts of Spirodela polyrhiza, an aquatic monocotelydonous species. PLoS ONE 2015, 10, e0140285. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Ge, L.; Xing, G.; Wang, M.; Weining, S.; Nie, X. Identification and Analysis of RNA Editing Sites in the Chloroplast Transcripts of Aegilops tauschii L. Genes 2017, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Mcdevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Du, L.; Ao, L.; Chen, J.; Li, W.; Hu, W.; Wei, Z.; Kim, K.; Lee, S.C.; Yang, T.J. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Yang, S.; Choi, G.; Kim, W.J.; Moon, B.C. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum subgenus Lycoctonum. Molecules 2017, 22, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judd, W.S.; Campbell, C.S.; Kellog, E.A.; Stevens, P.F.; Donoghue, M.J. Plant Systematics: A Phylogenetic Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2002; pp. 287–292. ISBN 0-87893-403-0. [Google Scholar]
- Rahman, A.H.M.M.; Parvin, M.I.A. Study of Medicinal Uses on Fabaceae Family at Rajshahi, Bangladesh. Res. Plant Sci. 2014, 2, 6–8. [Google Scholar]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Magalion, S.A.; Sanderson, K.R. Absolute diversification rates in angiosperm clades. Evolution 2001, 55, 1762–1780. [Google Scholar] [CrossRef]
- Ohashi, H.; Polhill, R.H.; Schubert, B.G. Desmodieae. In Advances in Legume Systematics. Part 1; Polhill, R.M., Raven, P.H., Eds.; Royal Botanic Gardens, Kew: Richmond, VA, USA, 1981; pp. 292–300. [Google Scholar]
- Saski, C.; Lee, S.B.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.G.; Jansen, R.K. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [Green Version]
- Yi, D.K.; Kim, K.J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [Green Version]
- Diekmann, K.; Hodkinson, T.R.; Wolfe, K.H.; van den Bekerom, R.; Dix, P.J.; Barth, S. Complete chloroplast genome sequence of a major allogamous forage species, perennial ryegrass (Lolium perenne L.). DNA Res. 2009, 16, 165–176. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Long, H.; Zhang, L.; Liu, Z.; Cao, H.; Shi, M.; Tan, X. The complete chloroplast genome sequence of tung tree (Vernicia fordii): Organization and phylogenetic relationships with other angiosperms. Sci. Rep. 2017, 7, 1869. [Google Scholar] [CrossRef] [PubMed]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin-A-Woeng, T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Kusnetsov, V.V. Chloroplasts: Structure and Expression of the Plastid Genome. Russ. J. Plant Physiol. 2018, 65, 465. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Hirose, T.; Ideue, T.; Wakasugi, T.; Sugiura, M. The chloroplast infA gene with a functional UUG initiation codon. FEBS Lett. 1999, 445, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Bi, Y.; Yang, F.; Zhang, M.; Chen, X.; Xue, J.; Zhang, X. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. Arapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P. The PREP Suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cui, Y.; Nie, L.; Hu, H.; Xu, Z.; Sun, W.; Gao, T.; Song, J.; Yao, H. Identification and Phylogenetic Analysis of the Complete Chloroplast Genomes of Three Ephedra Herbs Containing Ephedrine. BioMed Res. Int. 2019, 2019, 5921725. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and Phylogenetic Analyses of Ginger (Zingiberofficinale) in the Family Zingiberaceae Based on the Complete Chloroplast Genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, X.; Yu, Y.; Yuan, S.; Jiang, D.; Zhang, Y.; Zhang, T.; Zhong, W.; Yuan, Q.; Huang, L. Complete chloroplast genome sequences of Dioscorea: Characterization, genomic resources, and phylogenetic analyses. PeerJ 2018, 6, e6032. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Lespedeza davurica | Lespedeza cuneata |
|---|---|---|
| Accession Number | MH800328 | MN268503 |
| Total cpDNA Size | 149,010 | 149,010 |
| LSC | 82,373 | 82,395 |
| IR | 23,831 | 23,832 |
| SSC | 18,975 | 18,951 |
| Total CDS Length | 77,868 | 77,868 |
| Total tRNA Length | 2794 | 2794 |
| Total rRNA Length | 9062 | 9062 |
| Total Number of Genes | 128 | 128 |
| Coding Genes | 83 | 83 |
| rRNA Genes | 8 | 8 |
| tRNA Genes | 37 | 37 |
| Total GC % | 35.0 | 35.0 |
| LSC | 32.5 | 32.5 |
| IR | 42.2 | 42.2 |
| SSC | 28.1 | 28.1 |
| CDS | 36.0 | 36.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somaratne, Y.; Guan, D.-L.; Wang, W.-Q.; Zhao, L.; Xu, S.-Q. The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2020, 9, 51. https://doi.org/10.3390/plants9010051
Somaratne Y, Guan D-L, Wang W-Q, Zhao L, Xu S-Q. The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae). Plants. 2020; 9(1):51. https://doi.org/10.3390/plants9010051
Chicago/Turabian StyleSomaratne, Yamuna, De-Long Guan, Wen-Qiang Wang, Liang Zhao, and Sheng-Quan Xu. 2020. "The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae)" Plants 9, no. 1: 51. https://doi.org/10.3390/plants9010051