Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells
by
 ,
,
Chuan Chen
1,† ,
,
Guanzhao Wen
1,†,
Zijie Xiao
1,†,
Jun Peng
1,
Rong Hu
2,
Zhifeng Chen
1,
Chengyun Zhang
1 and
Wei Zhang
1,3,4,* 1
School of Physics and Materials Science, Guangzhou University, Guangzhou 510006, China
2
School of Materials Science and Engineering, Chongqing University of Arts and Sciences, Chongqing 402160, China
3
Research Center for Advanced Information Materials (CAIM), Huangpu Research and Graduate School of Guangzhou University, Guangzhou 510006, China
4
Guangzhou University-Linköping University Research Center on Urban Sustainable Development, Guangzhou University, Guangzhou 510006, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Photonics 2022, 9(12), 892; https://doi.org/10.3390/photonics9120892
Submission received: 23 October 2022
/
Revised: 15 November 2022
/
Accepted: 20 November 2022
/
Published: 23 November 2022
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:In this work, charge photogeneration dynamics in PTQ10:Y6 solar cells were studied by steady-state and time-resolved spectroscopies. For neat donor and acceptor films, we determined the exciton diffusion coefficients of PTQ10 and Y6 as 1.3 × 10−3 cm2·s−1 and 6.8 × 10−3 cm2·s−1, respectively. Furthermore, we find the LUMO and HOMO level offsets of 0.14 eV and 0.11 eV are sufficient for the dissociation of donor and acceptor excitons, respectively. For PTQ10:Y6 blend films, we find DIO additive could increase the scales of acceptor and donor phases. The acceptor phase increased slightly from 17.2 nm to 20.0 nm, while the donor phase increased from 2.3 nm to 5.8 nm. In addition, we find bimolecular recombination is a critical form for carrier recombination and DIO additive can significantly suppress the carrier recombination rate of PTQ10:Y6 active layer in an ultrafast time scale. This work is helpful for understanding the charge photogeneration processes in non-fullerene polymer solar cells.
1. Introduction
With the advantages of lightweight, low cost, and easy preparation, polymer solar cells (PSCs) have attracted the attention of academia and industry [1,2,3,4,5]. Historically, fullerene-based PSCs have dominated the development of organic solar cells (OSCs), and the power conversion efficiency (PCE) of such solar cells is usually less than 15% [6]. After 2019, the emergence of high-efficient non-fullerene acceptors, represented by Y6 [7], greatly promoted the improvement of the device performance of PSCs. At present, the PCE of non-fullerene PSCs has exceeded 19% [8], showing a broad commercial prospect.
Morphology optimization is one of the essential steps for PSCs to achieve high performance. Among the methods for morphology optimization, adding 1,8-diiodooctane (DIO) into the blend solution is a common one [9,10,11]. In fullerene-based OSCs, the relationship between DIO additive and morphology, photoelectric conversion process, and device performance has been studied widely. For PTB7:PC71BM [12], PTB7-Th:PC71BM [13], PBDTTT-C:PC71BM [14], PDPP3T:PC71BM [15], and PBDTTT-E:PC71BM [16], the use of DIO additive has been found to form a network interpenetrating structure and reduce the phase separation scale, especially for the fullerene phase. This kind of morphology can greatly enhance the dissociation efficiency of excitons, and thus improve the performance of devices. Moreover, it also reported that DIO treatment can suppress carrier recombination processes in PTB7:PC71BM [12] and PTB7-Th:PC71BM [13] devices. For non-fullerene solar cells, there are also some reports that DIO can improve the PCE of devices. For example, Ding et al. found that DIO additive promoted the ordered packing of TTC8-O1-4F in PBDB-T:TTC8-O1-4F solar cells, and the device performance can be thus enhanced from 10.75% to 12.05% [17]. Choi et al. found the DIO additive led to a uniform surface morphology of photoactive layer and well-distributed interpenetrating networks in PBDB-T:ITIC solar cells, which is a critical reason for the increased PCE [18].
PTQ10 is a promising polymer donor with the advantages of low cost, high efficiency, and thickness insensitivity [19]. When it is blended with Y6 small molecule acceptor, the PCE of solar cells can reach 15.68%. With the addition of DIO, the PCE can further reach 16.28% [20]. To understand the hole transfer process in PTQ10/Y6 solar cells, Li et al. studied the dissociation dynamics of Y6 excitons in PTQ10/Y6 blend films by time-resolved absorption spectroscopy. They observe efficient hole transfer processes in PTQ10/Y6 blend films after the photoexcitation of Y6 selectively [21]. Up to now, the detailed electron transfer and carrier recombination processes of PTQ10/Y6 solar cells in ultrafast timescale are still not very clear. Moreover, the role of DIO additive on charge photogeneration and recombination dynamics of PTQ10/Y6 solar cells needs to be explored.
In this paper, we studied carrier photogeneration and recombination dynamics in the PTQ10:Y6 system by various steady-state and time-resolved spectroscopies. First, the exciton properties of neat PTQ10 and Y6 films, including exciton diffusion coefficients (D) and exciton diffusion length (Ld), were studied by excitation-fluency-dependent transient absorption (TA) spectroscopies. Subsequently, the effect of DIO additive on the surface morphology, exciton dissociation, and carrier recombination processes was examined and discussed. This work is helpful for understanding the charge photogeneration processes in non-fullerene PSCs.
2. Materials and Methods
2.1. Sample Information and Film Preparation
The donor material PTQ10 was purchased from Shenzhen Derthon optoelectronics Materials Science Technology Co LTD. (Shenzhen, China). Y6 material was purchased from Nanjing Zhiyan Technology Inc. (Nanjing, China). The materials are used directly without further processing.
For film preparation, the neat PTQ10, Y6, and PTQ10/Y6 (1:1.2, wt) were dissolved into chloroform solvent to obtain the 7 mg/mL, 8.4 mg/mL, and 15.4 mg/mL solutions, respectively. To obtain the DIO-treated blend solution, 0.25% (by volume) DIO was added as an additive into the PTQ10:Y6 (1:1.2, wt) solution, which was then stirred for 0.5 h at room temperature before the spin coating process. The quartz substrates were first washed with detergent, and then were ultrasonically cleaned in ionic water, acetone, isopropanol, and ethanol for 10 min, respectively. The washed quartz substrates were treated with plasma (Cpc-a-13.56, Beijing Huayihang company, Beijing, China) for 20 min, on which the neat and blend films were spin-coated (2500 rpm, 40 s). The spin-coated films were annealed at 100 °C for 10 min by a thermal plate. All preparation processes were carried out in a glove box filled with nitrogen.
2.2. Steady-State Spectra Measurements
The steady-state absorption spectra for neat and blend films were measured by a UV-visible spectrometer (UV-3600i Plus, Shimadzu company, Kyoto, Japan). The photoluminescence (PL) spectrum for neat and blend films at room temperature was measured by a home-built system. A continuous wave laser (532 nm and 780 nm) was focused on the sample at an incident angle of 45°. The emitted photons were collected by two lenses with a focal length of 10 cm and were then guided into a fiber-optic spectrometer (Avaspec-ULS2048CL-RS-EVO-UA, Avantes company, Apeldoorn, The Netherlands) with a detection wavelength range of 300–1050 nm. The measured spectrum was finally calibrated by a reference light source (AvaLight-DH-S-BAL, Avantes company, Apeldoorn, The Netherlands). The steady-state absorption and PL measurements were performed at room temperature in air.
Temperature-dependent PL of PTQ10 film was measured by a home-built PL detection system equipped with a cryostat (NCV15-2W, East Changing Technologies, Beijing, China) and a fiber-optic spectrometer (Avaspec-ULS2048CL-RS-EVO-UA, Avantes company, Apeldoorn, The Netherlands) with a detection wavelength range of 300–1050 nm. The measurement of temperature-dependent PL of Y6 film was conducted on a fluorescence spectrometer (FS5, Edinburgh Instruments, Livingston, UK) with a detection wavelength range of 600–2000 nm.
The surface morphology of blend films was measured by atomic force microscopy (AFM, Agilent, AFM-5500, Santa Clara, CA, USA).
2.3. Transient Absorption Measurements
Femtosecond transient absorption measurements were carried out using a home-built TA system. The light source of the TA system is a femtosecond laser out from a regenerative amplifier (Legend Elite F 1K HE+II, Coherent, CA, USA), operating at 800 nm with a repletion rate of 1 kHz. The fundamental light is divided into two parts. One part of the light is frequency doubled (400 nm) or used directly as the pump light. The other part is used to excite a sapphire plate for the generation of supercontinuum white light, which is further used as the probing light for differential absorption measurement. The probing light was guided into a monochromator (Omni-λ200i, Zolix, Beijing, China) and detected by a CCD detector (Pascher Instruments, Lund, Sweden). The pump “on” and “off” for a pair of sequential actinic pulses was regulated by a mechanical chopper (MC2000B-EC, Thorlabs, Newton, New Jersey) in the pump light. With the help of a mechanical delay line, the probe is time-delayed relative to the pump. The samples were tested in an optical chamber filled with nitrogen.
3. Results and Discussion
3.1. Molecular Structures and Steady-State Spectra
The chemical structures of PTQ10, Y6, and DIO additive are shown in Figure 1a. PTQ10 has a simple thiophene ring and the difluorinated quinoline, which is regarded as the donor and receptor unit, respectively [19]. For the small molecule acceptor Y6, it shows an A-D-A’-D-A structure (D represents the electron-donor unit and A represents the electron-withdrawing unit), which is composed of a central fusion ring dithienothiophene [3,2-b]-pyrrolobenzothiadiazole (TPBT, D-A’-D unit) based on a trapezoidal electron-deficient-core and two 2(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (2FIC) termini (A unit) [7]. In terms of the energy levels of the donor and acceptor materials, as shown in Figure 1b, the HOMO energy levels of PTQ10 and Y6 films are −5.54 eV and −5.65 eV, respectively [7,19], while the LUMO energy levels are −3.52 eV and −3.66 eV [7,19]. The HOMO energy offset (ΔEHOMO = EHOMO(D) − EHOMO(A)) and LUMO energy offset (ΔELUMO = ELUMO(D) − ELUMO(A)) between PTQ10 and Y6 are 0.11 eV and 0.14 eV, respectively.
The absorption spectra of neat and blended films are shown in Figure 1c. The neat PTQ10 film exhibits a maximum absorption peak at ~600 nm and an extra shoulder peak at ~560 nm, which can be assigned to the 0–0 and 0–1 vibronic bands of the transition from the ground state S0 to the lowest excited state S1, respectively. Moreover, it can be found that the peak intensity of 0–0 peak is higher than 0–1 peak, indicating that PTQ10 neat film shows the J-aggregate characteristics [22]. For the Y6 film, it shows a maximum absorption peak at ~840 nm and a shoulder peak at 750 nm. The shoulder peak at 750 nm originates from the excitation from the ground state to higher electronic or vibrational levels of the lowest excited state [23]. When PTQ10 is blended with Y6, the blend films show two strong absorption bands at 400~650 nm and 700~900 nm, which mainly originate from the absorption of PTQ10 donor and Y6 acceptor in the blend film, respectively. Moreover, we find the ~810 nm absorption peak of PTQ10:Y6 blend film redshift to ~830 nm after the treatment by DIO additive, suggesting the DIO additive can influence the absorption properties of PTQ10:Y6 blend film.
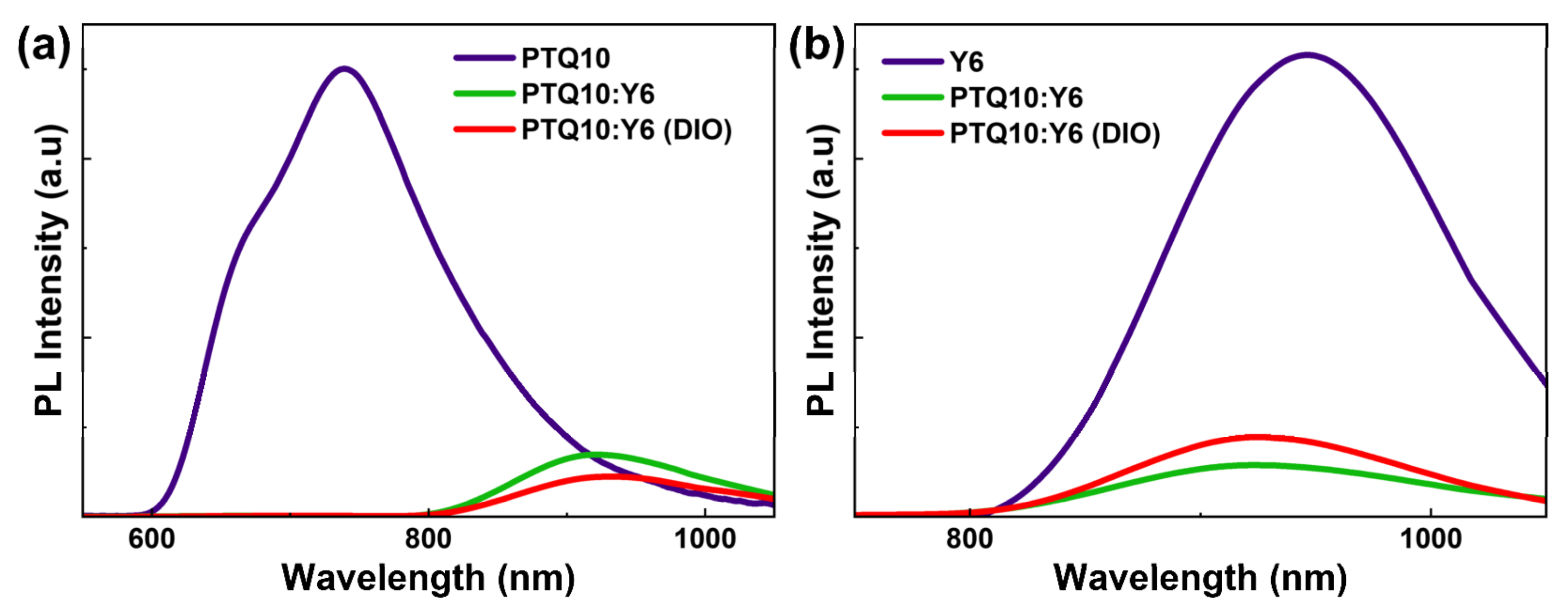
To investigate the influence of DIO additive on the exciton dissociation process, we performed the steady-state PL spectra of neat and blended films, as shown in Figure 2. The PL spectrum of PTQ10 neat film shows a broad PL band of 600~1000 nm with a maximum PL peak at ~770 nm, which originates from the emission of PTQ10 exciton. For the blend films, we find an extra emission peak at ~920 nm, which can be attributed to the emission of Y6 acceptor. The PL quenching efficiency [24] has been calculated to estimate the exciton dissociation efficiency of neat donor (acceptor) exciton. In this work, the PL peak of ~720 nm was almost invisible for blend films after excitation at 532 nm, as shown in Figure 2a, which suggests the exciton dissociate efficiency for PTQ10 approaches unity. For the dissociation of Y6 excitons, we find that the PL intensity of DIO-treated PTQ10:Y6 blend film was slightly lower than that without DIO-treated blend film, and the quenched efficiency for the blend films without and with DIO treatments are 85.87% and 82.36%, respectively. This indicates the DIO additive has little influence on the donor exciton dissociation process but could slightly reduce the exciton dissociation efficiency of the acceptor.
3.2. Exciton Properties of PTQ10 and Y6 Neat Films
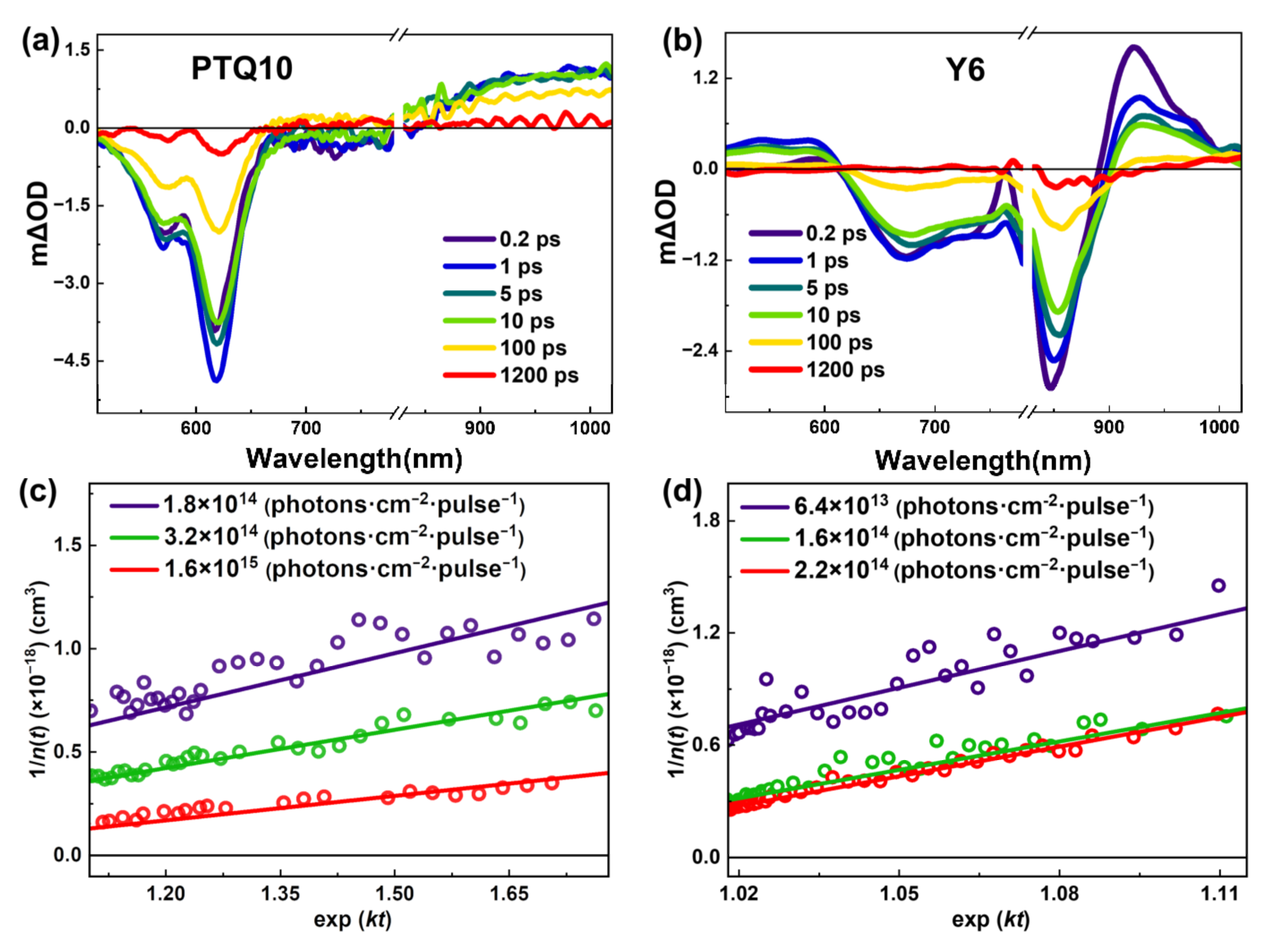
In PSCs, exciton dissociation efficiency is one of the key processes determining the PCE of devices. In blend film, the exciton dissociation depends on the relationship between the diffusion length of excitons and the phase separation scale. If the diffusion length of excitons is much longer than the phase separation scale, the excitons can be dissociated effectively at the interface. Otherwise, the excitons would recombine back to the ground state, leading to energy loss. To understand the diffusion characteristics of PTQ10 and Y6 excitons, we studied the exciton diffusion coefficient and exciton lifetime (τ), which determine the exciton diffusion length, by transient absorption spectroscopy [25]. Figure 3a,b show the TA spectra of neat PTQ10 and Y6 films at different delay times, respectively. For neat PTQ10 film, the transients in the wavelength range of 500~800 nm and 800~1050 nm appear immediately after photoexcitation, which can be attributed to the ground state bleaching (GSB) and exciton absorption, respectively. Similarly, the negative signal in 500~880 nm and the positive absorption signal in 900~1050 nm, are attributed to the signals induced by the ground state bleaching and exciton absorption for Y6 film, respectively. For neat PTQ10 and Y6 films, the exciton kinetics can be described as [26].
where n(t) is the concentration of the excitons, γ is exciton annihilation rate, k represents the exciton decay rate, which can be extracted by fitting the excited state absorption (ESA) kinetics of neat film. By fitting the kinetics of donor and acceptors, the exciton decay rates (lifetime) of PTQ10 and Y6 films are determined as 3.5 × 10−9 s−1 (~286 ps) and 1.4 × 10−9 s−1 (~693 ps), respectively, as shown in Supporting Information Figure S1 and Table S1. The solution of Equation (1) can be written in the form as [27]
By fitting the GSB kinetics of PTQ10 and Y6 films, as shown in Figure 3c,d, the exciton annihilation rate γ can be determined as 1.6 × 10−9 cm3·s−1 and 8.5 × 10−9 cm3·s−1 for PTQ10 and Y6 exciton, respectively. The exciton diffusion coefficient can be estimated by the γ in the form as [28]
where Ra represents the annihilation radius. By taking the literature value of annihilation radius (Ra = 1 nm) [29], D of PTQ10 and Y6 films is calculated as ~1.3 × 10−3 cm2·s−1 and ~6.8 × 10−3 cm2·s−1, respectively. Based on the exciton diffusion coefficient and exciton lifetime, the three-dimensional exciton diffusion length can be determined by
[30], which are ~14.8 nm and ~53.1 nm for PTQ10 and Y6 film, respectively. We find that the exciton diffusion length of PTQ10 is much longer than that of classical polymer donor materials, such as P3HT (~8 nm), PTB7-Th (~5.4 nm), and PM6 (~4.5 nm) [31]. Moreover, the exciton diffusion length of Y6 is much larger than that of classical non-fullerene small molecule acceptor materials, such as ITIC (~18.2 nm), IT-4F (~18.8 nm) [32], IDIC (~35 nm) [33], and L8-BO (~43.0 nm) [8]. In PSCs, exciton dissociation is an essential process for charge photogeneration processes, and the longer exciton diffusion length would enable the exciton to diffuse to the interface more efficiently; therefore, the long diffusion length of Y6 and PTQ10 excitons is one of the reasons for PTQ10:Y6 solar cells achieving high PCE.
3.3. Temperature-Dependent PL Properties of PTQ10 and Y6 Film
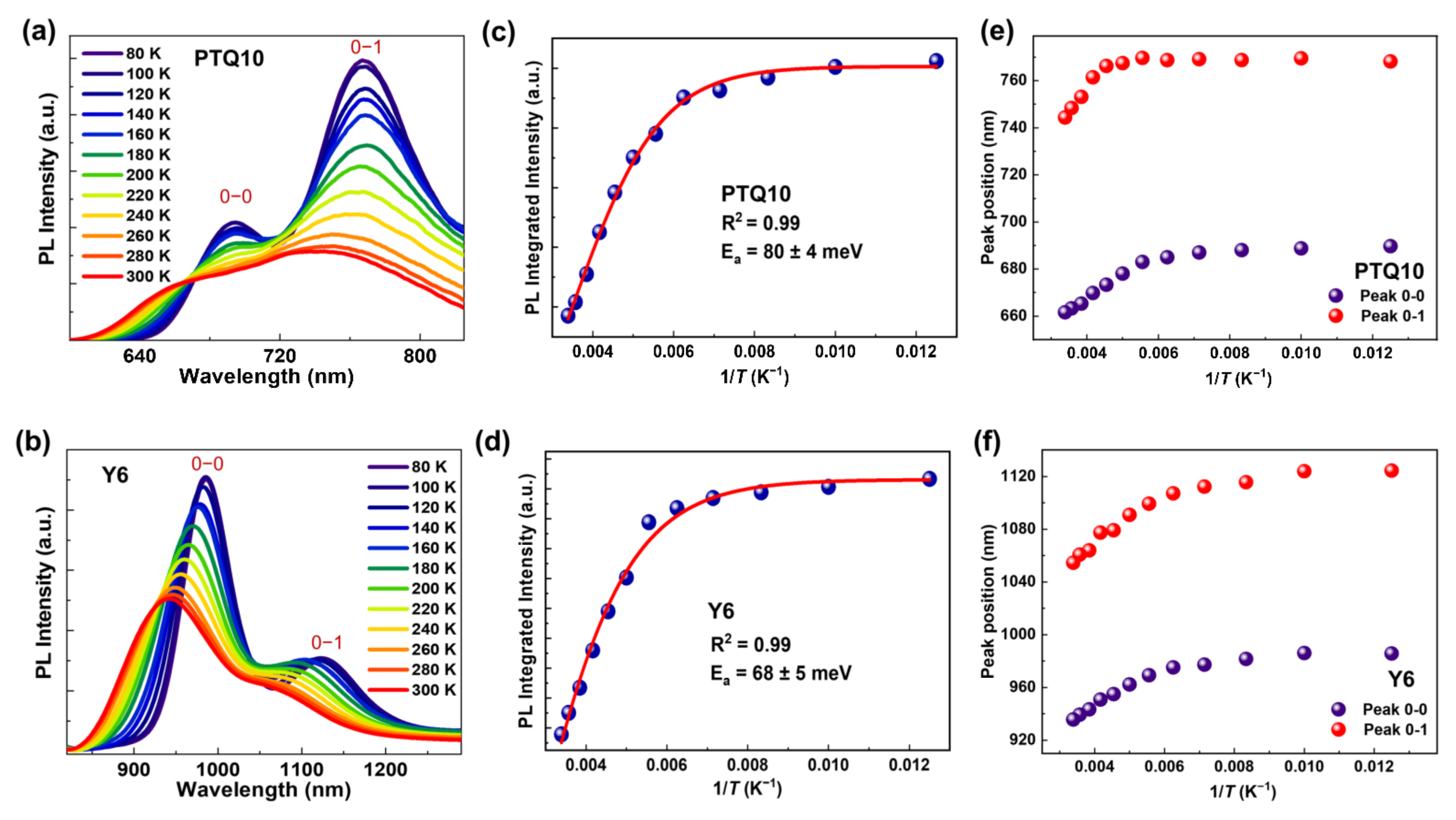
To examine the role of temperature on the exciton emission processes, we performed temperature-dependent PL of PTQ10 and Y6 films, as shown in Figure 4a. For the PTQ10 film, we find that the integrated PL intensity enhances with the decrease of temperature, as shown in Figure 4c. This suggests the PL emission loss of PTQ10 film increases with temperature. We noted that this integrated PL intensity could be explained by the Arrhenius equation, which can be expressed as [34]
where I(T) and I0 are the integrated PL intensity at the corresponding temperature and 0 K, respectively, Ea represents the activation energy for the exciton transit from emission state to non-radiative state, and kB is the Boltzmann constant. By fitting the integrated PL intensity, we extracted the activation energy of ~80 meV for PTQ10 exciton. A similar phenomenon was observed for Y6 film, and the activation energy of ~68 meV was estimated by using the temperature-dependent PL measurement, as shown in Figure 4b,d.
Aside from the variation of PL intensity with temperature, we also observe the blueshift of the PL emission peaks (0–0 and 0–1 peaks) of PTQ10 and Y6 films with the increase in temperature, as shown in Figure 4e,f. In fact, the blue shift of exciton emission peaks with temperature has been observed in many organic semiconductors, such as the conjugated polymer (PPV [35], MeLPPP [36], etc.) and small molecule semiconductors (Y6Se [37], Y6 [34], etc.). One explanation for this blueshift is that the excitons in PTQ10 and Y6 film cannot migrate to the low energy state easily at high temperatures. In detail, the photogenerated excitons will diffuse randomly through the density of states via energy transfer, and they will be trapped on a low-energy site, where the exciton emission occurs [36]. When the temperature increases, the excitons cannot easily diffuse to the low energy state, and they remain localized at a relatively high energy site that leads to a blueshift of the exciton emission compared to that at low temperature [35,36].
3.4. Charge Photogeneration and Recombination Processes in PTQ10:Y6 Blend Films
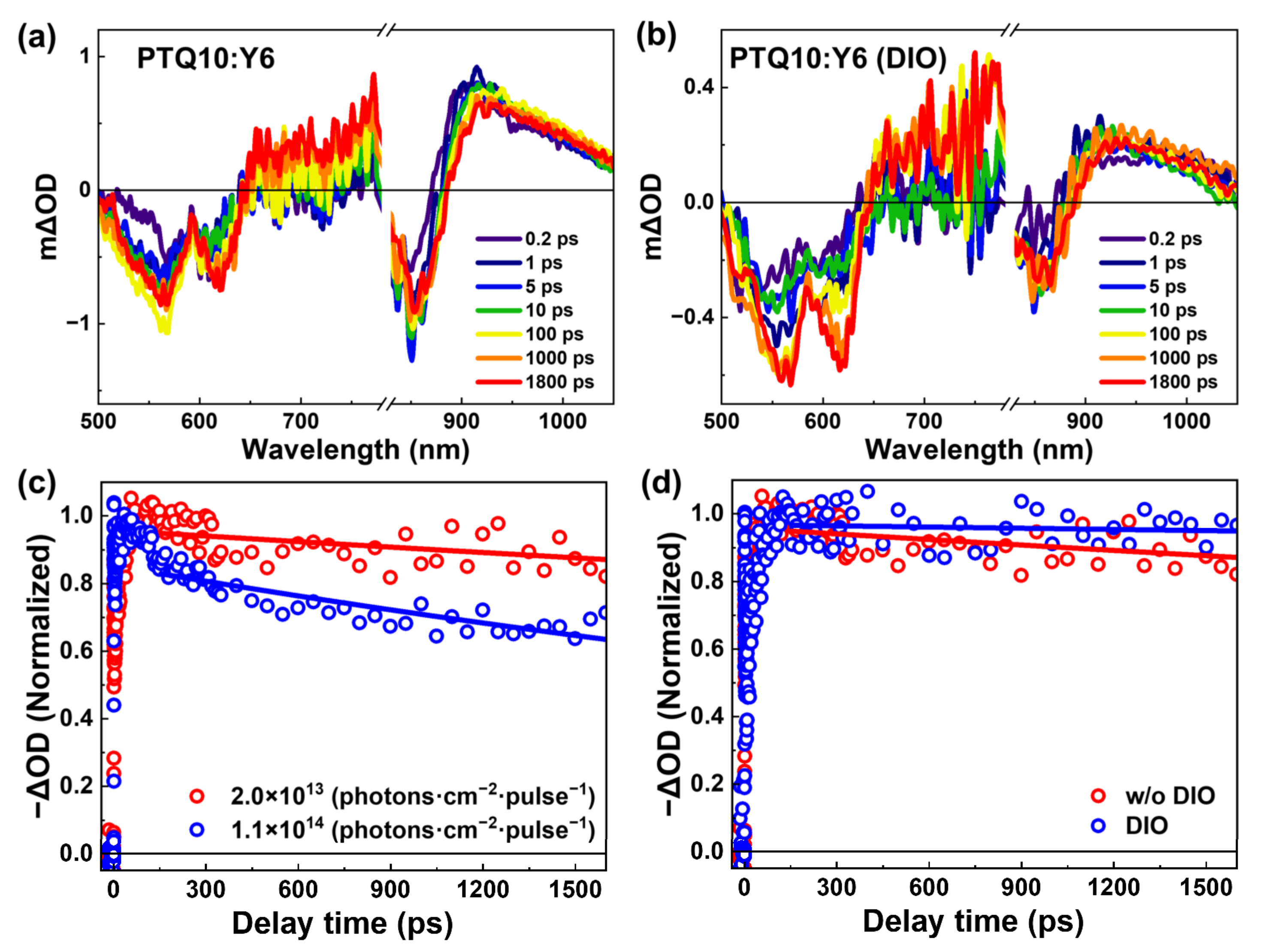
To understand the role of DIO additive on charge photogeneration and recombination processes, TA measurements of PTQ10:Y6 blend films were performed. Figure 5a,b, and Supporting Information Figure S2 show the TA spectra of PTQ10:Y6 blend films after excitation at 800 nm, which can selectively excite the Y6 in blend films. We find the TA spectral characteristics of DIO-treated PTQ10:Y6 film are similar to that of without DIO-treated blend film. In detail, the TA spectrum shows three negative peaks at ~570 nm, ~620 nm, and ~860 nm. The negative peaks of ~570 nm and ~620 nm can be attributed to the GSB peaks of PTQ10 material in blend film, while the negative peak of ~860 nm is GSB of Y6 in blend film. Both TA spectra of PTQ10:Y6 films without/with DIO treatment show a positive absorption band in the wavelength range of 900~1100 nm, which can be attributed to the absorption of excited-state populations in blend films. In Figure 5a, we observe the GSB peaks at ~570 nm built up, when the delay time increases to 100 ps. This phenomenon can be attributed to the hole transfer process from the Y6 acceptor to the PTQ10 donor after the selective excitation of the Y6 acceptor in the blend film. This indicates that the Y6 exciton can be dissociated under the HOMO energy offset of 0.11 eV. Similar properties can be found in DIO-treated blend film in Figure 5b.
The charge transfer process is clearly shown in the TA kinetics at ~570 nm of PTQ10:Y6 blend films, which corresponds to the GSB kinetics of PTQ10, as shown in Figure 5d. We observe that the GSB kinetic shows an instantaneous generation, a slow rise and a slow decay process. The rise component of the bleaching kinetics can be fitted by double-exponential function: I = A1 × (1 − exp(-t/τ1)) + A2 × (1 − exp(-t/τ2)), where τ1 and τ2 represent the lifetimes for fast and slow hole transfer processes, respectively [38]. The detailed parameters can be found in Supporting Information Table S2.
The instantaneous generation and slow rise processes can be attributed to the instantaneous hole transfer for the excitons at the interface and exciton diffusion-mediated hole transfer processes, respectively [39]. In this work, the bi-exponential function fitting of the hole transfer kinetics show: τ1 = within instrumental response function (IRF), τ2 = 18.1 ps, and τ1 = within IRF, τ2 = 25.0 ps for PTQ10:Y6 blend films without and with DIO treatment, respectively. These lifetimes are much shorter than that of the singlet exciton in neat Y6 film (~693 ps), suggesting the efficient exciton dissociation efficiency of Y6 film via hole transfer processes. The observed highly efficient hole transfer of Y6 excitons in PTQ10/Y6 film is consistent with the study of PTQ10/Y6 film by Li et al. [21]. These fast and slow hole transfer processes were also observed in many PTQ10-based non-fullerene solar cells, such as PTQ10/ZITI-S and PTQ10/ZITI-N [40]. The various exciton diffusion times are determined by the phase-separate size of Y6 in blend films. Accordingly, the longer the exciton diffusion time in DIO-treated PTQ10:Y6 film, the larger the acceptor phase size.
From the AFM measurement (Supporting Information Figures S3 and S4), we find the blend film with DIO treatment exhibits a rather rough top surface with a root-mean-square roughness (RMS) of 2.65 nm, which is significantly higher than that of without DIO-treated PTQ10:Y6 blend film (1.16 nm). For polymer solar cells, the volatilization of solvent could affect the aggregation of donor and acceptor in blend film during the spin coating and film-drying processes. As PTQ10 and Y6 can be dissolved in DIO, the volatilization of DIO would also influence the film formation processes of PTQ10/Y6 film. Compared with the boiling point of the main solvent chloroform (61.2 ℃ at 760 mmHg), DIO additive has a higher boiling point (332.5 ℃ at 760 mmHg). Consequently, the DIO additive would volatilize more slowly during the aggregation process of donor and acceptor in the blend films, which could facilitate phase separation and result in larger sizes of donor and acceptor phases. In fact, it is a common method to control the phase separation of the blend film by controlling the volatilization rate of the solvent. For example, for the classical system P3HT/PCBM, the use of solvents with a slower volatilization rate can facilitate phase separation of donor and acceptor in the blend film [41,42,43]. The surface roughness is also closely related to the phase separation of the film. In many cases, a better phase separation would increase the surface roughness [42,43]; therefore, the larger phase size and roughness (RMS) of the film with DIO additive, could be related to the slower volatilization of DIO additive during film formation.
Based on the exciton diffusion coefficient of neat Y6 film (6.8 × 10−3 cm2·s−1) and the exciton-diffusion mediated lifetime (τ2), the average phase size Ls(A) can be determined as Ls(A) = 2(6Dτ2)1/2, which are ~17.2 nm and ~20.0 nm for PTQ10:Y6 blend films without/with DIO-treatment respectively. In PSCs, a larger phase size would increase the time for exciton diffusion to the D/A interface for dissociation, and reduce the exciton dissociation efficiency; however, we note that the PCE of DIO-treated PTQ10:Y6 is higher than that without DIO treatment [20], suggesting the exciton diffusion and dissociation processes are not the critical factors affecting the performance of PTQ10:Y6 devices in these two conditions.
For the charge recombination process in PSCs, there are two kinds of carrier recombination: one is geminate recombination and the other is bimolecular recombination [44]. The geminate recombination typically involves the recombination of charge carriers that were generated from the same exciton, and would thus be independent of the excitation fluency. Bimolecular recombination happens for a pair of electron and hole originates from different photons, which decays faster with the increase in excitation power. For the blend films, decay of ~570 nm kinetics can be attributed to charge recombination processes, as the exciton dissociation has been achieved when the decay time is longer than 100 ps. We find the kinetics under excitation fluence of 1.4 × 1013 photons·cm−2·pulse−1 decays slower than that of 1.4 × 1014 photons·cm−2·pulse−1 in both types of the blend films, as shown in Figure 5c. This indicates bimolecular type recombination is a critical form for the recombination process in these two blend films in ultrafast timescale. This bimolecular type was also reported in some PTQ10 based non-fullerene solar cells, such as PTQ10/IDIC [24]. In general, the exciton dissociates into the charge with the help of energy offsets and the charge states can be divided into the charge transfer state and free carrier in PSCs. The recombination of the charge transfer state is geminate recombination, while the recombination of the free carrier is more like bimolecular recombination when the carrier concentration is high. The observed biomolecule carrier recombination also suggests free carriers already exist in large quantities in ultrafast timescales. Moreover, by comparing the GSB kinetics of ~570 nm (as shown in Figure 5c), the charge recombination lifetime with DIO-treated PTQ10:Y6 blend film is ~40 ns, which is significantly longer than that of without DIO-treated blend film (~19 ns), as shown in Figure 5d, Supporting Information Figure S5 and Table S3. This indicates the DIO additive treatment can obviously suppress the charge recombination in PTQ10:Y6 blend film.
Moreover, we performed the TA measurements of the blend films after photoexcitation at 520 nm, to investigate the effect of DIO additive on donor exciton dissociation, electron transfer, and charge recombination processes. Figure 6 and Supporting Information Figure S6 show the representative TA spectra of the blend films with and without DIO treatment. For the GSB peaks of the acceptor at ~860 nm, we observed the signal built up within the time delays from 0.2 ps to ~10 ps, originating from the electron transfer from PTQ10 to Y6 material. Hence, we can conclude that the donor exciton can be dissociated efficiently under the ΔELUMO of 0.14 eV.
Then, we extracted the GSB kinetics of ~860 nm for without/with DIO-treated PTQ10:Y6 blend films after excitation at 520 nm with an excitation fluence of 2.0 × 1013 photons·cm−2·pulse−1, as shown in Supporting Information Figure S7. For the blend films, it shows three processes, including an ultrafast generation (<0.2 ps), a slow rising process (0.2~100 ps), and a long-lived decay process (>100 ps). The ultrafast generation and slow-rising processes can be attributed to the generation of Y6 exciton and the ultrafast electron transfer process, and the exciton-diffusion-mediate electron transfer after excitation, respectively. We find the electron transfer processes can be well fit by the bi-exponential function; the detailed parameters are shown in Supporting Information Table S4. For electron transfer, the lifetimes of exciton-diffusion-mediate electron transfer are ~1.7 ps and ~10.8 ps for PTQ10:Y6 blend films without and with DIO treatment, respectively. Obviously, the lifetime of exciton-diffusion-mediate electron transfer for the DIO-treated film is longer than that of without DIO-treated blend film, indicating the DIO additive slightly increases the phase separate size of donor materials. Furthermore, based on the exciton diffusion coefficient of donor exciton (1.3 × 10−3 cm2·s−1) and the lifetime of exciton-diffusion mediate, we quantitatively estimated the size of donor domain (LS(D)) as ~2.3 nm and ~5.8 nm for without/with DIO-treaded blend films.
To investigate the charge recombination process in PTQ10:Y6 films, we also performed the TA spectra with different excitation fluencies and extracted the TA kinetics of ~570 nm as shown in Figure 6d, Supporting Information Figure S8 and Table S5. We find that the charge recombination process in PTQ10:Y6 blend film is dominated by bimolecular recombination. Moreover, we find that the lifetime of charge recombination without DIO treatment PTQ10:Y6 film is ~17 ns, which is significantly faster than that of DIO-treated film (~80 ns). This long-lived recombination process suggests that DIO additive treatment can efficiently suppress the bimolecular recombination in PTQ10:Y6 blend film.
Based on the above discussion, the addition of DIO increases the phase size of donor and acceptor phases, which is consistent with the results of steady-state PL measurements. This is very different from the case of fullerene solar cells. In fullerene solar cells, the use of DIO can reduce the phase separation scale and form an interpenetrating network structure [45,46], which can suppress the reduction of power conversion efficiency caused by excessive phase separation in fullerene solar cells. Although the addition of DIO reduces the phase size and exciton dissociation efficiency slightly, the exciton dissociation efficiency is still very high in PTQ10:Y6 solar cells with DIO treatment. In addition, DIO treatment can significantly inhibit carrier recombination, and ultimately improve the power conversion efficiency.
4. Conclusions
In this work, the charge photogeneration and recombination processes of PTQ10:Y6 blend films have been systemically investigated by steady-state and time-resolved spectroscopies. For the neat film, we find the lifetime of singlet exciton for PTQ10 and Y6 materials can be found as ~286 ps and ~693 ps, respectively. The exciton diffusion length is estimated as ~14.8 nm and ~53.1 nm for PTQ10 and Y6 film, respectively. For PTQ10:Y6 blend films, we find the PTQ10 and Y6 excitons can be dissociated into charge under the LUMO energy offset of 140 meV and HOMO energy offset of 110 meV, respectively. Moreover, we find that the DIO additive can increase the phase separate sizes of PTQ10 and Y6 phase in blend film, resulting in a slight reduction of dissociation efficiency of Y6 excitons. Meanwhile, the bimolecular recombination is a critical form for carrier recombination and DIO additive can significantly suppress the carrier recombination rate of the PTQ10:Y6 active layer in an ultrafast time scale. This work is helpful for understanding the charge photogeneration processes in non-fullerene polymer solar cells.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/photonics9120892/s1, Table S1: The detailed fitting parameters for TA kinetics of neat PTQ10 and Y6 films; Table S2: The detailed fitting parameters for the rising kinetics of blend films at ~570 nm after excitation at 800 nm; Table S3: The detailed fitting parameters for charge recombination kinetics of blend films after excitation at 800 nm with different excitation fluencies; Table S4: The detailed fitting parameters for rising kinetics of blend films at ~860 nm after excitation at 520 nm; Table S5: The detailed fitting parameters for charge recombination kinetics of blend films after excitation at 520 nm with different excitation fluencies; Figure S1: Normalized TA kinetics of PTQ10 and Y6 films at ~940 nm; Figure S2: Color plot of TA spectra for blend films after photoexcitation at 800 nm; Figure S3: AFM height and phase images of blend films; Figure S4: AFM 3D model diagram of blend films; Figure S5: The comparison of TA bleaching kinetics of PTQ10:Y6 films with and without DIO-treatment after photoexcitation at 800 nm; Figure S6: Color plot of TA spectra for the blend films after photoexcitation at 520 nm; Figure S7: TA kinetics of PTQ10:Y6 blend films without and with DIO-treatment at the probe wavelength of 860 nm; Figure S8: The comparison of TA bleaching kinetics of PTQ10:Y6 films with and without DIO-treatment after photoexcitation at 520 nm; Figure S9: Thickness measurements of PTQ10 and Y6 films by AFM lower step method; Figure S10: Reciprocal of exciton density vs exp (kt) of neat PTQ10 and Y6 films under various excitation fluencies; Figure S11: TA kinetics of neat Y6 and PTQ10 films.; Figure S12: TA spectra of Y6 film after excitation at 520 nm.
Author Contributions
Conceptualization, W.Z.; methodology, Z.C., C.Z. and W.Z.; software, W.Z. and G.W.; validation, C.C., Z.X. and R.H.; formal analysis, G.W., C.C., W.Z., Z.X. and J.P.; investigation, C.C., Z.X. and R.H.; resources, W.Z., Z.C., J.P. and R.H.; data curation, C.C., R.H. and Z.X.; writing—original draft preparation, C.C.; writing—review and editing, Z.X., G.W., R.H. and W.Z.; visualization, C.C. and G.W.; supervision, W.Z., R.H. and G.W.; project administration, W.Z.; funding acquisition, W.Z., R.H. and Z.C.; All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 21903017, 21603020), Natural Science Foundation of Guangdong Province (Grant No. 2019A1515010783, 2020A1515010411), Guangzhou Science and Technology Planning Project (Grant No. 202102010443), and Young Talents Program of Guangzhou University (Grant No. RQ2020080).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the funding support from the National Natural Science Foundation of China, Department of Science and Technology of Guangdong Province, Guangzhou Science and Technology Bureau, and Guangzhou University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gillett, A.J.; Privitera, A.; Dilmurat, R.; Karki, A.; Qian, D.; Pershin, A.; Londi, G.; Myers, W.K.; Lee, J.; Yuan, J.; et al. The Role of Charge Recombination to Triplet Excitons in Organic Solar Cells. Nature 2021, 597, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.W. Two-layer Organic Photovoltaic Cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Wadsworth, A.; Moser, M.; Marks, A.; Little, M.S.; Gasparini, N.; Brabec, C.J.; Baran, D.; McCulloch, I. Critical Review of the Molecular Design Progress in Non-Fullerene Electron Acceptors towards Commercially Viable Organic Solar Cells. Chem. Soc. Rev. 2019, 48, 1596–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Inganäs, O.; Friend, R.H.; Gao, F. Organic Solar Cells Based on Non-Fullerene Acceptors. Nat. Mater. 2018, 17, 119–128. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, J.; Chow, P.C.Y.; Jiang, K.; Zhang, J.; Zhu, Z.; Zhang, J.; Huang, F.; Yan, H. Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells. Chem. Rev. 2018, 118, 3447–3507. [Google Scholar] [CrossRef]
- Wang, G.; Melkonyan, F.S.; Facchetti, A.; Marks, T.J. All-Polymer Solar Cells: Recent Progress, Challenges, and Prospects. Angew. Chem. Int. Ed. 2019, 58, 4129–4142. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, M.; Xu, J.; Li, C.; Yan, J.; Zhou, G.; Zhong, W.; Hao, T.; Song, J.; Xue, X.; et al. Single-Junction Organic Solar Cells with over 19% Efficiency Enabled by a Refined Double-Fibril Network Morphology. Nat. Mater. 2022, 21, 656–663. [Google Scholar] [CrossRef]
- Ma, R.; Yan, C.; Fong, P.W.-K.; Yu, J.; Liu, H.; Yin, J.; Huang, J.; Lu, X.; Yan, H.; Li, G. In Situ and Ex Situ Investigations on Ternary Strategy and Co-Solvent Effects towards High-Efficiency Organic Solar Cells. Energy Environ. Sci. 2022, 15, 2479–2488. [Google Scholar] [CrossRef]
- He, Q.; Sheng, W.; Zhang, M.; Xu, G.; Zhu, P.; Zhang, H.; Yao, Z.; Gao, F.; Liu, F.; Liao, X.; et al. Revealing Morphology Evolution in Highly Efficient Bulk Heterojunction and Pseudo-Planar Heterojunction Solar Cells by Additives Treatment. Adv. Energy Mater. 2021, 11, 2003390. [Google Scholar] [CrossRef]
- Song, J.; Zhu, L.; Li, C.; Xu, J.; Wu, H.; Zhang, X.; Zhang, Y.; Tang, Z.; Liu, F.; Sun, Y. High-Efficiency Organic Solar Cells with Low Voltage Loss Induced by Solvent Additive Strategy. Matter 2021, 4, 2542–2552. [Google Scholar] [CrossRef]
- Su, X.; Hu, R.; Wen, G.; Zou, X.; Qing, M.; Peng, J.; He, X.; Zhang, W. Understanding of Photophysical Processes in DIO Additive-Treated PTB7:PC71BM Solar Cells. Crystals 2021, 11, 1139. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, S.; Xu, Z.; Qiao, B.; Huang, D.; Xu, X. Two Effects of 1,8-Diiodooctane on PTB7-Th:PC71BM Polymer Solar Cells. Org. Electron. 2016, 34, 188–192. [Google Scholar] [CrossRef]
- Zusan, A.; Gieseking, B.; Zerson, M.; Dyakonov, V.; Magerle, R.; Deibel, C. The Effect of Diiodooctane on the Charge Carrier Generation in Organic Solar Cells Based on the Copolymer PBDTTT-C. Sci. Rep. 2015, 5, 8286. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiao, C.; Huang, H.; Zhang, F. The Effect of DIO Additive on Performance Improvement of Polymer Solar Cells. Chin. Sci. Bull. 2014, 59, 3227–3231. [Google Scholar] [CrossRef]
- Huo, M.-M.; Hu, R.; Zhang, Q.-S.; Chen, S.; Gao, X.; Zhang, Y.; Yan, W.; Wang, Y. Morphology and Carrier Non-Geminate Recombination Dynamics Regulated by Solvent Additive in Polymer/Fullerene Solar Cells. RSC Adv. 2020, 10, 23128–23135. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, X.; Feng, H.; Ke, X.; Meng, L.; Sun, Y.; Guo, Z.; Cai, Y.; Jiao, C.; Wan, X.; et al. Subtle Morphology Control with Binary Additives for High-Efficiency Non-Fullerene Acceptor Organic Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 27425–27432. [Google Scholar] [CrossRef]
- Choi, J.Y.; Han, Y.W.; Jeon, S.J.; Ko, E.J.; Moon, D.K. Introduction of Co-Additives to Form Well Dispersed Photoactive Layer to Improve Performance and Stability of Organic Solar Cells. Sol. Energy 2019, 185, 1–12. [Google Scholar] [CrossRef]
- Sun, C.; Pan, F.; Bin, H.; Zhang, J.; Xue, L.; Qiu, B.; Wei, Z.; Zhang, Z.-G.; Li, Y. A Low Cost and High Performance Polymer Donor Material for Polymer Solar Cells. Nat. Commun. 2018, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Yang, H.; Wu, Y.; Yildiz, O.; Zhu, X.; Marszalek, T.; Blom, P.W.M.; Cui, C.; Li, Y. Anthracene-Assisted Morphology Optimization in Photoactive Layer for High-Efficiency Polymer Solar Cells. Adv. Funct. Mater. 2021, 31, 2103944. [Google Scholar] [CrossRef]
- Sun, C.; Pan, F.; Chen, S.; Wang, R.; Sun, R.; Shang, Z.; Qiu, B.; Min, J.; Lv, M.; Meng, L.; et al. Achieving Fast Charge Separation and Low Nonradiative Recombination Loss by Rational Fluorination for High-Efficiency Polymer Solar Cells. Adv. Mater. 2019, 31, 1905480. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Wen, G.; Hu, R.; Dong, G.; Zhang, C.; Zhang, W.; Huang, H.; Dang, W. An Insight into the Excitation States of Small Molecular Semiconductor Y6. Molecules 2020, 25, 4118. [Google Scholar] [CrossRef]
- Cha, H.; Zheng, Y.; Dong, Y.; Lee, H.H.; Wu, J.; Bristow, H.; Zhang, J.; Lee, H.K.H.; Tsoi, W.C.; Bakulin, A.A.; et al. Exciton and Charge Carrier Dynamics in Highly Crystalline PTQ10:IDIC Organic Solar Cells. Adv. Energy Mater. 2020, 10, 2001149. [Google Scholar] [CrossRef]
- Shin, H.-Y.; Woo, J.H.; Gwon, M.J.; Barthelemy, M.; Vomir, M.; Muto, T.; Takaishi, K.; Uchiyama, M.; Hashizume, D.; Aoyama, T.; et al. Exciton Diffusion in Near-Infrared Absorbing Solution-Processed Organic Thin Films. Phys. Chem. Chem. Phys. 2013, 15, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; Ruseckas, A.; Gaudin, O.P.M.; Webster, G.R.; Burn, P.L.; Samuel, I.D.W. Singlet Exciton Diffusion in MEH-PPV Films Studied by Exciton–Exciton Annihilation. Org. Electron. 2006, 7, 452–456. [Google Scholar] [CrossRef]
- Shaw, P.E.; Ruseckas, A.; Samuel, I.D.W. Exciton Diffusion Measurements in Poly(3-Hexylthiophene). Adv. Mater. 2008, 20, 3516–3520. [Google Scholar] [CrossRef]
- Hedley, G.J.; Ruseckas, A.; Samuel, I.D.W. Light Harvesting for Organic Photovoltaics. Chem. Rev. 2017, 117, 796–837. [Google Scholar] [CrossRef] [Green Version]
- Wen, G.; Zou, X.; Hu, R.; Peng, J.; Chen, Z.; He, X.; Dong, G.; Zhang, W. Ground- and Excited-State Characteristics in Photovoltaic Polymer N2200. RSC Adv. 2021, 11, 20191–20199. [Google Scholar] [CrossRef]
- Long, Y.; Hedley, G.J.; Ruseckas, A.; Chowdhury, M.; Roland, T.; Serrano, L.A.; Cooke, G.; Samuel, I.D.W. Effect of Annealing on Exciton Diffusion in a High Performance Small Molecule Organic Photovoltaic Material. ACS Appl. Mater. Interfaces 2017, 9, 14945–14952. [Google Scholar] [CrossRef]
- Riley, D.B.; Sandberg, O.J.; Li, W.; Meredith, P.; Armin, A. Quasi-Steady-State Measurement of Exciton Diffusion Lengths in Organic Semiconductors. Phys. Rev. Appl. 2022, 17, 024076. [Google Scholar] [CrossRef]
- Sajjad, M.T.; Ruseckas, A.; Jagadamma, L.K.; Zhang, Y.; Samuel, I.D.W. Long-Range Exciton Diffusion in Non-Fullerene Acceptors and Coarse Bulk Heterojunctions Enable Highly Efficient Organic Photovoltaics. J. Mater. Chem. A 2020, 8, 15687–15694. [Google Scholar] [CrossRef]
- Chandrabose, S.; Chen, K.; Barker, A.J.; Sutton, J.J.; Prasad, S.K.K.; Zhu, J.; Zhou, J.; Gordon, K.C.; Xie, Z.; Zhan, X.; et al. High Exciton Diffusion Coefficients in Fused Ring Electron Acceptor Films. J. Am. Chem. Soc. 2019, 141, 6922–6929. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, J.; Guo, Y.; Yang, C.; Yi, Y.; Wei, Z. Small Exciton Binding Energies Enabling Direct Charge Photogeneration Towards Low-Driving-Force Organic Solar Cells. Angew. Chem. Int. Ed. 2021, 60, 15348–15353. [Google Scholar] [CrossRef]
- Lim, S.-H.; Bjorklund, T.G.; Bardeen, C.J. Temperature-Dependent Exciton Dynamics in Poly(p-Phenylene Vinylene) Measured by Femtosecond Transient Spectroscopy. Chem. Phys. Lett. 2001, 342, 555–562. [Google Scholar] [CrossRef]
- Guha, S.; Rice, J.D.; Yau, Y.T.; Martin, C.M.; Chandrasekhar, M.; Chandrasekhar, H.R.; Guentner, R.; Scanduicci de Freitas, P.; Scherf, U. Temperature-Dependent Photoluminescence of Organic Semiconductors with Varying Backbone Conformation. Phys. Rev. B 2003, 67, 125204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, L.; Xu, C.; Jin, P.; Huang, M.; Li, Y.; Wang, H.; Yi, Y.; Zhang, C.; Yang, Y.; et al. Single Photovoltaic Material Solar Cells with Enhanced Exciton Dissociation and Extended Electron Diffusion. Cell Rep. Phys. Sci. 2022, 3, 100895. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, H. Photoinduced Charge Transfer and Recombination Dynamics in Star Nonfullerene Organic Solar Cells. J. Phys. Chem. Lett. 2022, 13, 1123–1130. [Google Scholar] [CrossRef]
- Chong, K.; Xu, X.; Meng, H.; Xue, J.; Yu, L.; Ma, W.; Peng, Q. Realizing 19.05% Efficiency Polymer Solar Cells by Progressively Improving Charge Extraction and Suppressing Charge Recombination. Adv. Mater. 2022, 34, 2109516. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Zhou, G.; Yi, Y.; Xu, S.; Liu, F.; Zhu, H.; Zhu, X. Accurate Determination of the Minimum HOMO Offset for Efficient Charge Generation Using Organic Semiconducting Alloys. Adv. Energy Mater. 2020, 10, 1903298. [Google Scholar] [CrossRef]
- Ruderer, M.A.; Guo, S.; Meier, R.; Chiang, H.-Y.; Körstgens, V.; Wiedersich, J.; Perlich, J.; Roth, S.V.; Müller-Buschbaum, P. Solvent-Induced Morphology in Polymer-Based Systems for Organic Photovoltaics. Adv. Funct. Mater. 2011, 21, 3382–3391. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, R.; Li, D.; Huo, M.-M.; Ai, X.-C.; Zhang, J.-P. Primary Dynamics of Exciton and Charge Photogeneration in Solvent Vapor Annealed P3HT/PCBM Films. J. Phys. Chem. C 2012, 116, 4298–4310. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, N.-J.; Huo, M.-M.; Fu, L.-M.; Ai, X.-C.; Zhang, J.-P. Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films. Molecules 2012, 17, 13923–13936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, T.M.; Durrant, J.R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, A.K.K.; Wang, D.H.; Luo, C.; Cao, Y.; Nguyen, T.-Q.; Bazan, G.C.; Heeger, A.J. Effects of Solvent Additives on Morphology, Charge Generation, Transport, and Recombination in Solution-Processed Small-Molecule Solar Cells. Adv. Energy Mater. 2014, 4, 1301469. [Google Scholar] [CrossRef]
- Peet, J.; Brocker, E.; Xu, Y.; Bazan, G.C. Controlledβ-Phase Formation in Poly(9,9-Di-n-Octylfluorene) by Processing with Alkyl Additives. Adv. Mater. 2008, 20, 1882–1885. [Google Scholar] [CrossRef]
Figure 1.
(a) The chemical structures of PTQ10, Y6, and DIO additive. (b) The HOMO and LUMO energy levels for PTQ10 and Y6 materials. (c) The steady-state absorption spectra of neat and blended films.
Figure 1.
(a) The chemical structures of PTQ10, Y6, and DIO additive. (b) The HOMO and LUMO energy levels for PTQ10 and Y6 materials. (c) The steady-state absorption spectra of neat and blended films.

Figure 2.
(a) PL spectra of neat PTQ10 and PTQ10:Y6 blend films with and without DIO treatment after photoexcitation at 532 nm. (b) PL spectra of neat Y6 and PTQ10:Y6 blend films with and without DIO treatment after photoexcitation at 780 nm.
Figure 2.
(a) PL spectra of neat PTQ10 and PTQ10:Y6 blend films with and without DIO treatment after photoexcitation at 532 nm. (b) PL spectra of neat Y6 and PTQ10:Y6 blend films with and without DIO treatment after photoexcitation at 780 nm.

Figure 3.
The representative TA spectra of neat (a) PTQ10 and (b) Y6 films at indicated delay time after photoexcitation at 400 nm and 800 nm, respectively. Reciprocal of exciton density vs. exp (kt) of (c) PTQ10 and (d) Y6 films under various excitation fluencies.
Figure 3.
The representative TA spectra of neat (a) PTQ10 and (b) Y6 films at indicated delay time after photoexcitation at 400 nm and 800 nm, respectively. Reciprocal of exciton density vs. exp (kt) of (c) PTQ10 and (d) Y6 films under various excitation fluencies.

Figure 4.
Temperature-dependent PL spectra of neat (a) PTQ10 and (b) Y6 film after excitation at 532 nm and 780 nm, respectively. The correlations between integrated PL intensity and 1/T for neat (c) PTQ10 and (d) Y6 films. The correlations between PL emission peak positions and 1/T for (e) PTQ 10 and (f) Y6 films.
Figure 4.
Temperature-dependent PL spectra of neat (a) PTQ10 and (b) Y6 film after excitation at 532 nm and 780 nm, respectively. The correlations between integrated PL intensity and 1/T for neat (c) PTQ10 and (d) Y6 films. The correlations between PL emission peak positions and 1/T for (e) PTQ 10 and (f) Y6 films.

Figure 5.
TA spectra of PTQ10:Y6 blend films (a) without and (b) with DIO treatment at indicated delay times after excitation at 800 nm with an excitation fluency of 1.4 × 1013 photons·cm−2·pulse−1. (c) The normalized TA bleaching signal (~570 nm) of PTQ10:Y6 after photoexcitation at 800 nm with various excitation fluencies (1.4 × 1013 and 1.4 × 1014 photons·cm−2·pulse−1) (d) TA kinetics of PTQ10:Y6 blend films without and with DIO treatment at the probe wavelength of 570 nm with an excitation fluency of 1.4 × 1013 photons·cm−2·pulse−1.
Figure 5.
TA spectra of PTQ10:Y6 blend films (a) without and (b) with DIO treatment at indicated delay times after excitation at 800 nm with an excitation fluency of 1.4 × 1013 photons·cm−2·pulse−1. (c) The normalized TA bleaching signal (~570 nm) of PTQ10:Y6 after photoexcitation at 800 nm with various excitation fluencies (1.4 × 1013 and 1.4 × 1014 photons·cm−2·pulse−1) (d) TA kinetics of PTQ10:Y6 blend films without and with DIO treatment at the probe wavelength of 570 nm with an excitation fluency of 1.4 × 1013 photons·cm−2·pulse−1.

Figure 6.
TA spectra of PTQ10:Y6 blend films (a) without and (b) with DIO-treatment at indicated delay times after excitation at 520 nm with an excitation fluency of 2.0 × 1013 photons·cm−2·pulse−1. (c) The normalized TA bleaching signal (~570 nm) of PTQ10:Y6 after photoexcitation at 520 nm with various excitation fluencies (2.0 × 1013 and 1.1 × 1014 photons·cm−2·pulse−1) (d) TA kinetics of PTQ10:Y6 blend films without and with DIO-treatment at the probe wavelengths of 570 nm with an excitation fluency of 2.0 × 1013 photons·cm−2·pulse−1.
Figure 6.
TA spectra of PTQ10:Y6 blend films (a) without and (b) with DIO-treatment at indicated delay times after excitation at 520 nm with an excitation fluency of 2.0 × 1013 photons·cm−2·pulse−1. (c) The normalized TA bleaching signal (~570 nm) of PTQ10:Y6 after photoexcitation at 520 nm with various excitation fluencies (2.0 × 1013 and 1.1 × 1014 photons·cm−2·pulse−1) (d) TA kinetics of PTQ10:Y6 blend films without and with DIO-treatment at the probe wavelengths of 570 nm with an excitation fluency of 2.0 × 1013 photons·cm−2·pulse−1.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.; Wen, G.; Xiao, Z.; Peng, J.; Hu, R.; Chen, Z.; Zhang, C.; Zhang, W. Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells. Photonics 2022, 9, 892. https://doi.org/10.3390/photonics9120892
AMA Style
Chen C, Wen G, Xiao Z, Peng J, Hu R, Chen Z, Zhang C, Zhang W. Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells. Photonics. 2022; 9(12):892. https://doi.org/10.3390/photonics9120892
Chicago/Turabian StyleChen, Chuan, Guanzhao Wen, Zijie Xiao, Jun Peng, Rong Hu, Zhifeng Chen, Chengyun Zhang, and Wei Zhang. 2022. "Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells" Photonics 9, no. 12: 892. https://doi.org/10.3390/photonics9120892
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.