
Nano Differential Scanning Fluorimetry as a Rapid Stability Assessment Tool in the Nanoformulation of Proteins
 , , and
, , and 
Abstract
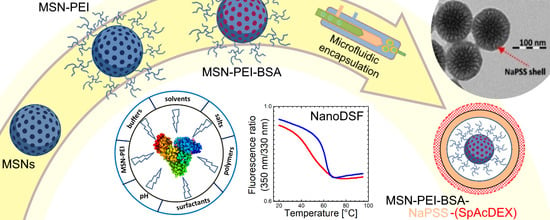
:
1. Introduction
2. Materials
3. Methods
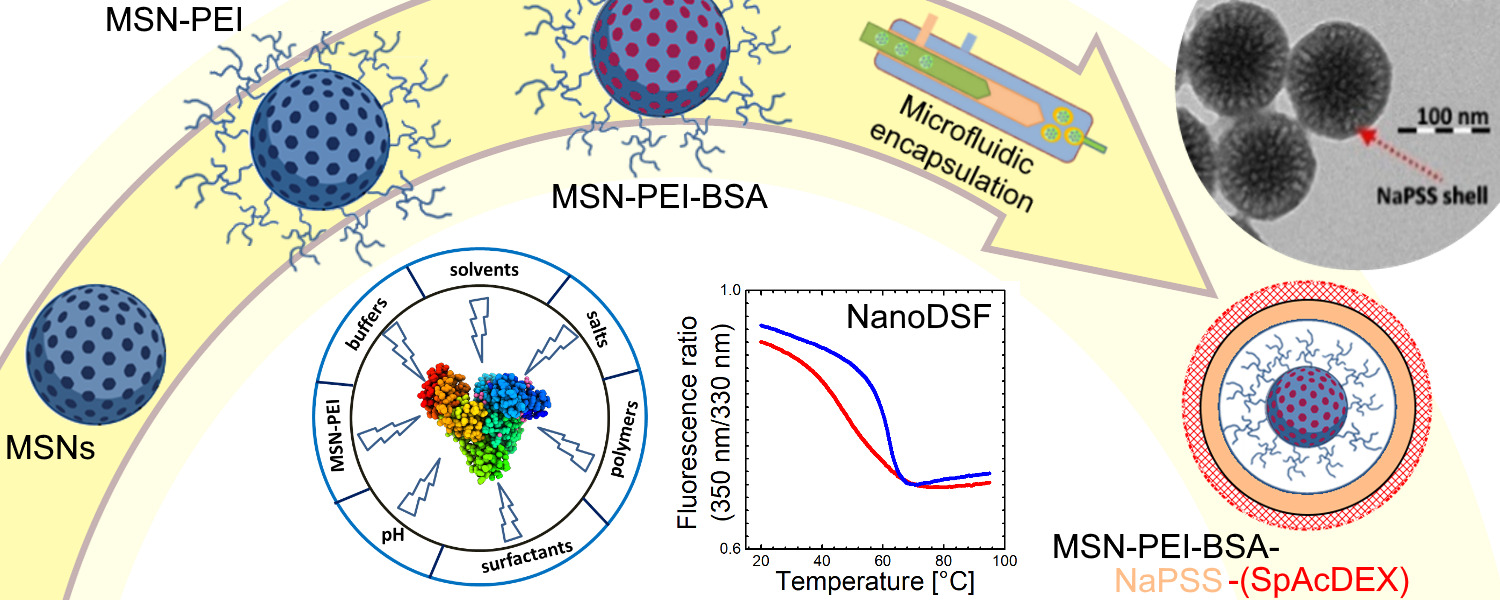
3.1. Synthesis of Large Pore Size 3D Dendritic MSNs
3.2. Preparation of Hyperbranched PEI—Functionalized MSNs (MSN-PEI)
3.3. Protein Loading into MSN-PEI and Loading Capacity Measurement
3.4. Synthesis of SpAcDEX Polymer
3.5. Microfluidic Encapsulation Studies
3.5.1. Fabrication of the 3D Microfluidic Coflow Glass Capillary Device
3.5.2. Preparation of the MSN-PEI–BSA–NaPSS Nanoparticles
3.5.3. Preparation of the MSN-PEI–BSA–NaPSS–SpAcDEX Nanoparticles
3.5.4. Characterization of the Synthesized MSNs, MSN-PEI, MSN-PEI–BSA, MSN-PEI–BSA–NaPSS, and MSN-PEI–BSA–NaPSS–SpAcDEX Nanoparticles
3.6. Protein Thermal Stability Studies
3.7. Circular Dichroism (CD) Measurements
3.8. Data Analysis
4. Results and Discussion
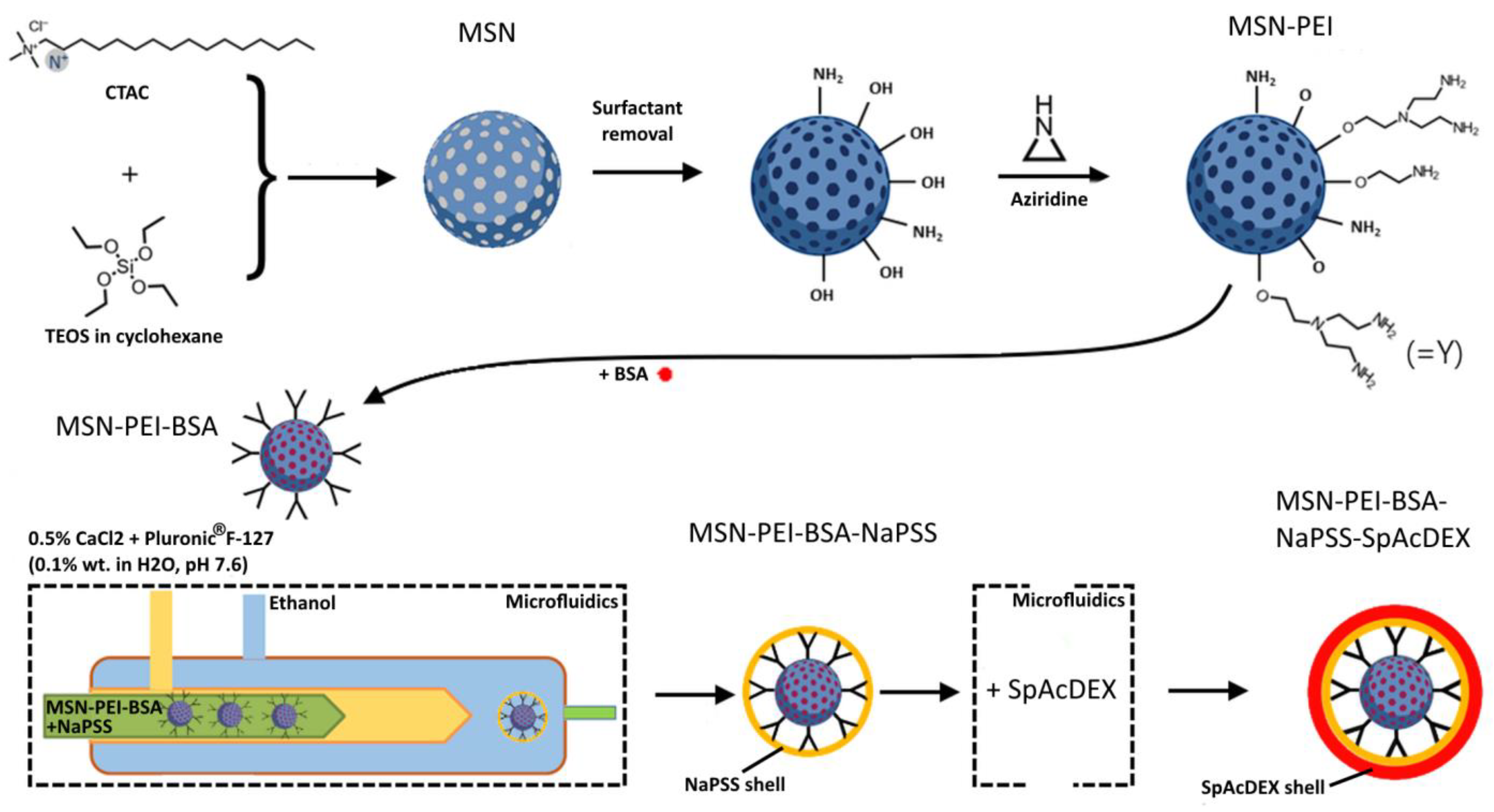
4.1. Synthesis and Characterization of Plain MSNs and MSN-PEI
4.2. Protein-Loaded MSN-PEI
4.3. Microfluidic Encapsulation Studies
4.3.1. Preparation of MSN-PEI–BSA–NaPSS Nanoparticles (First Coating)
4.3.2. Preparation of the MSN-PEI–BSA–NaPSS–SpAcDEX Nanoparticles (Second Coating)
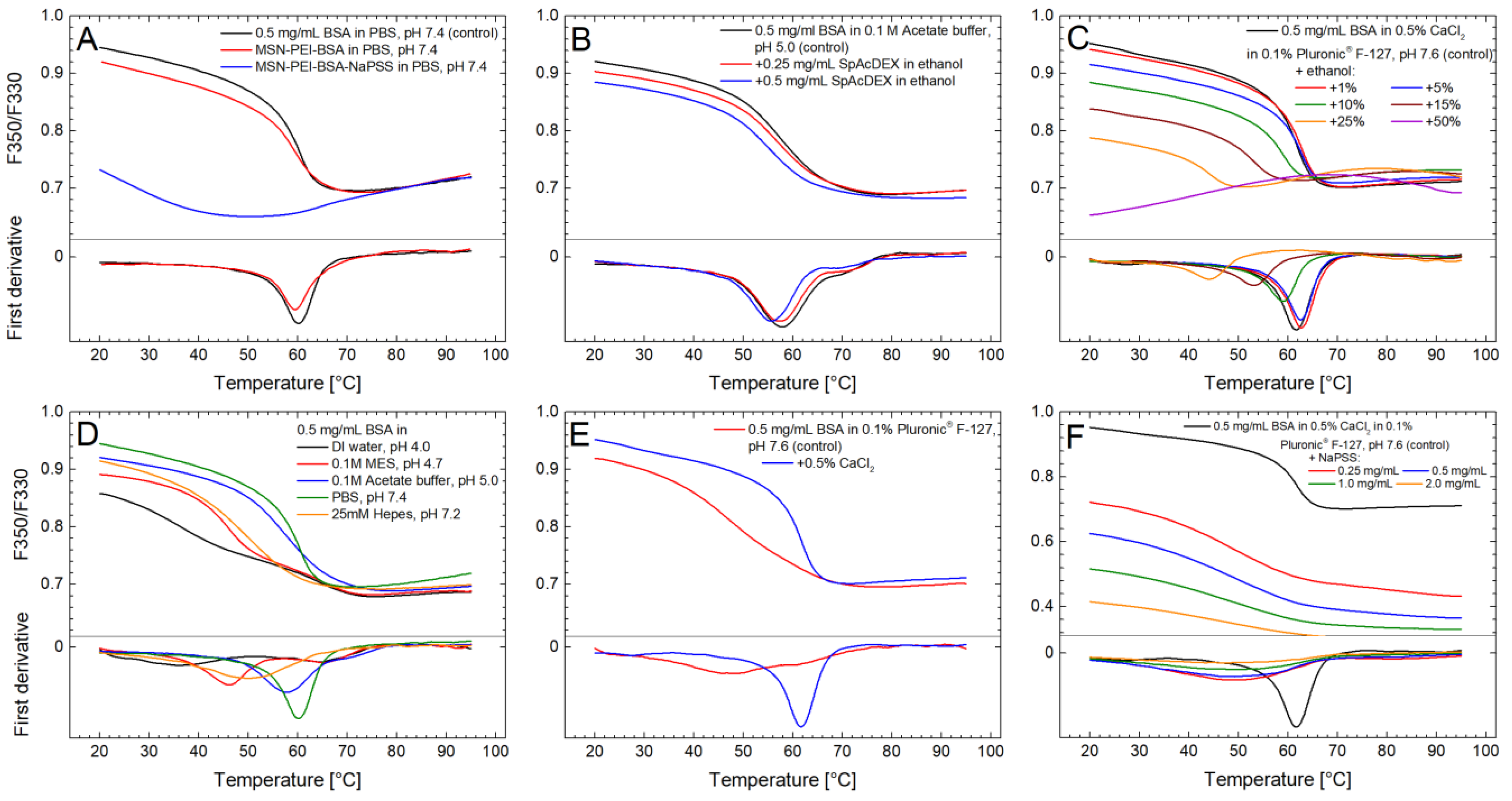
4.4. Protein Thermal Stability Studies
4.4.1. Effect of PEI-Functionalized MSNs
4.4.2. Effect of Polymers
4.4.3. Effect of Solvents
4.4.4. Effect of Buffers and pH
4.4.5. Effect of Surfactants and Salts
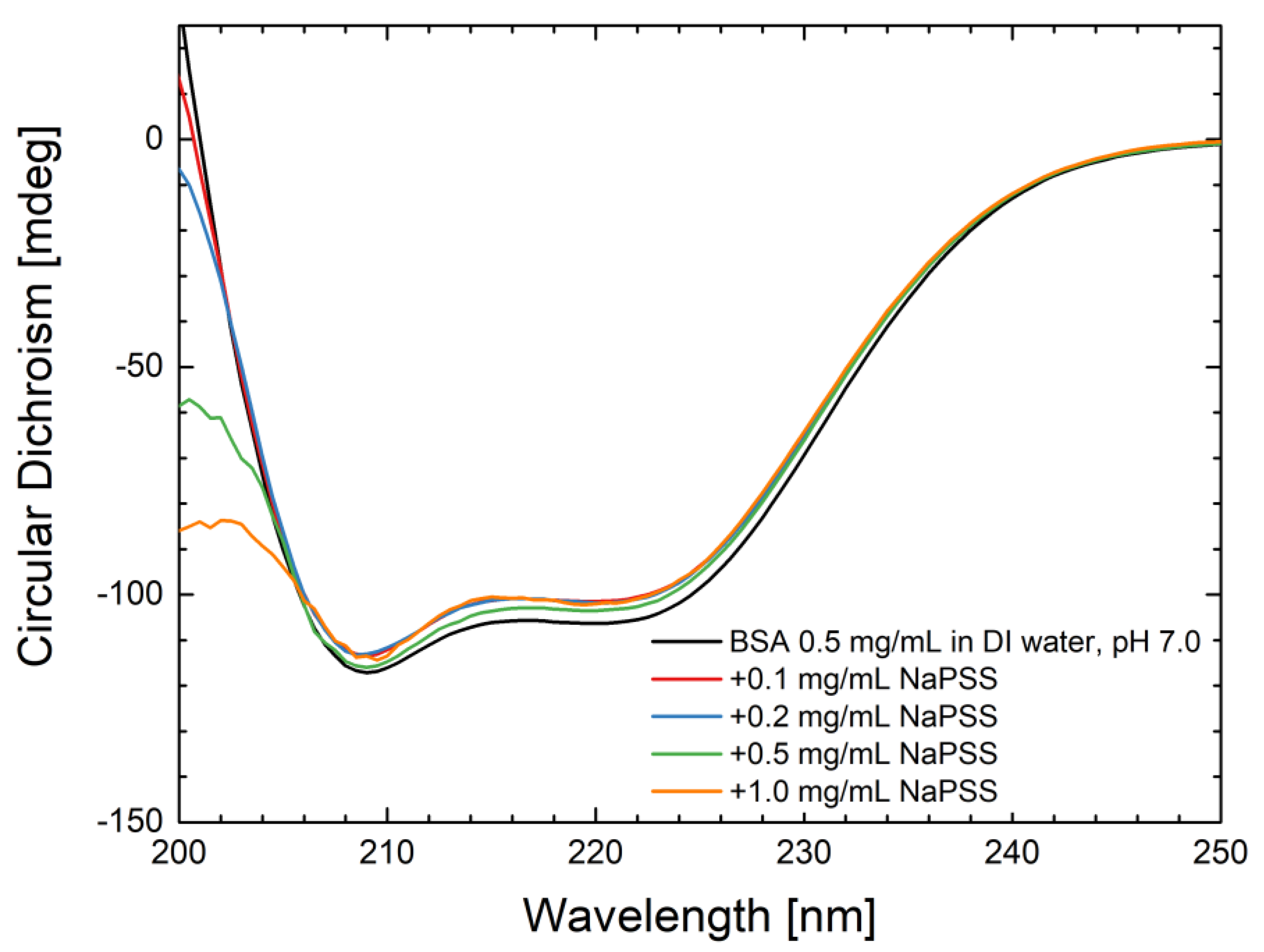
4.5. CD Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, L.; Barnes, T.J.; Prestidge, C.A. Oral Delivery of Protein-Based Therapeutics: Gastroprotective Strategies, Physiological Barriers and in Vitro Permeability Prediction. Int. J. Pharm. 2020, 585, 119488. [Google Scholar] [CrossRef] [PubMed]
- Evaluate Total Global Biotech Drugs Sales from 2012 to 2021* (in Billion U.S. Dollars). Available online: https://www.statista.com/statistics/280578/global-biologics-spending/ (accessed on 11 March 2022).
- Duskey, J.T.; da Ros, F.; Ottonelli, I.; Zambelli, B.; Vandelli, M.A.; Tosi, G.; Ruozi, B. Enzyme Stability in Nanoparticle Preparations Part 1: Bovine Serum Albumin Improves Enzyme Function. Molecules 2020, 25, 4593. [Google Scholar] [CrossRef] [PubMed]
- Matsarskaia, O.; Bühl, L.; Beck, C.; Grimaldo, M.; Schweins, R.; Zhang, F.; Seydel, T.; Schreiber, F.; Roosen-Runge, F. Evolution of the Structure and Dynamics of Bovine Serum Albumin Induced by Thermal Denaturation. Phys. Chem. Chem. Phys. 2020, 22, 18507–18517. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K. Pharmacokinetic and Pharmacodynamic Considerations for the next Generation Protein Therapeutics. J. Pharm. Pharm. 2015, 42, 553–571. [Google Scholar] [CrossRef]
- Singh, A.P.; Shin, Y.G.; Shah, D.K. Application of Pharmacokinetic-Pharmacodynamic Modeling and Simulation for Antibody-Drug Conjugate Development. Pharm. Res. 2015, 32, 3508–3525. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Kong, L.; Rupp, B. Protein Stability: A Crystallographer’s Perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and Applications of Differential Scanning Fluorimetry in Early-Stage Drug Discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Svilenov, H.; Markoja, U.; Winter, G. Isothermal Chemical Denaturation as a Complementary Tool to Overcome Limitations of Thermal Differential Scanning Fluorimetry in Predicting Physical Stability of Protein Formulations. Eur. J. Pharm. Biopharm. 2018, 125, 106–113. [Google Scholar] [CrossRef]
- BPI Contributor. Exploring Protein Stability by NanoDSF. Available online: https://bioprocessintl.com/sponsored-content/exploring-protein-stability-by-nanodsf/ (accessed on 12 March 2022).
- Magnusson, A.O.; Szekrenyi, A.; Joosten, H.J.; Finnigan, J.; Charnock, S.; Fessner, W.D. NanoDSF as Screening Tool for Enzyme Libraries and Biotechnology Development. FEBS J. 2019, 286, 184–204. [Google Scholar] [CrossRef]
- Kim, S.H.; Yoo, H.J.; Park, E.J.; Na, D.H. Nano Differential Scanning Fluorimetry-Based Thermal Stability Screening and Optimal Buffer Selection for Immunoglobulin G. Pharmaceuticals 2022, 15, 29. [Google Scholar] [CrossRef]
- Cecchetti, C.; Strauss, J.; Stohrer, C.; Naylor, C.; Pryor, E.; Hobbs, J.; Tanley, S.; Goldman, A.; Byrne, B. A Novel High-Throughput Screen for Identifying Lipids That Stabilise Membrane Proteins in Detergent Based Solution. PLoS ONE 2021, 16, e0254118. [Google Scholar] [CrossRef] [PubMed]
- Küçüktürkmen, B.; Rosenholm, J.M. Mesoporous Silica Nanoparticles as Carriers for Biomolecules in Cancer Therapy. Bio-Nanomed. Cancer Ther. 2021, 1295, 99–120. [Google Scholar]
- Desai, D.; Zhang, J.; Sandholm, J.; Lehtimäki, J.; Grönroos, T.; Tuomela, J.; Rosenholm, J.M. Lipid Bilayer-Gated Mesoporous Silica Nanocarriers for Tumor-Targeted Delivery of Zoledronic Acid in Vivo. Mol. Pharm. 2017, 14, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Åkerfelt, M.; Prabhakar, N.; Toriseva, M.; Näreoja, T.; Zhang, J.; Nees, M.; Rosenholm, J.M. Factors Affecting Intracellular Delivery and Release of Hydrophilic versus Hydrophobic Cargo from Mesoporous Silica Nanoparticles on 2D and 3D Cell Cultures. Pharmaceutics 2018, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Köper, I. Polyelectrolyte-Coated Gold Nanoparticles: The Effect of Salt and Polyelectrolyte Concentration on Colloidal Stability. Polymers 2018, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhou, X.; He, C.; Qiu, K.; Nie, W.; Chen, L.; Wang, H.; Mo, X.; Zhang, Y. Polyelectrolyte Multilayer Functionalized Mesoporous Silica Nanoparticles for PH-Responsive Drug Delivery: Layer Thickness-Dependent Release Profiles and Biocompatibility. J. Mater. Chem. B 2013, 1, 5886–5898. [Google Scholar] [CrossRef]
- Harris, C.M.; Miller, S.G.; Andresen, K.; Thompson, L.B. Quantitative Measurement of Sodium Polystyrene Sulfonate Adsorption onto CTAB Capped Gold Nanoparticles Reveals Hard and Soft Coronas. J. Colloid Interface Sci. 2018, 510, 39–44. [Google Scholar] [CrossRef]
- Cohen, J.L.; Schubert, S.; Wich, P.R.; Cui, L.; Cohen, J.A.; Mynar, J.L.; Fréchet, J.M.J. Acid-Degradable Cationic Dextran Particles for the Delivery of SiRNA Therapeutics. Bioconjug. Chem. 2011, 22, 1056–1065. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Wang, L.; Liu, Z.; Wu, R.; Janoniene, A.; Ma, M.; Pan, G.; Baranauskiene, L.; Zhang, L.; et al. Microfluidic Encapsulation of Prickly Zinc-Doped Copper Oxide Nanoparticles with VD1142 Modified Spermine Acetalated Dextran for Efficient Cancer Therapy. Adv. Healthc. Mater. 2017, 6, 1601406. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.; Ma, Y.; Sun, J. Microfluidic Methods for Fabrication and Engineering of Nanoparticle Drug Delivery Systems. ACS Appl. Bio Mater. 2020, 3, 107–120. [Google Scholar] [CrossRef]
- Karnik, R.; Gu, F.; Basto, P.; Cannizzaro, C.; Dean, L.; Kyei-Manu, W.; Langer, R.; Farokhzad, O.C. Microfluidic Platform for Controlled Synthesis of Polymeric Nanoparticles. Nano Lett. 2008, 8, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.M.; Swami, A.; Gilson, L.M.; Chopra, S.; Choi, S.; Wu, J.; Langer, R.; Karnik, R.; Farokhzad, O.C. Ultra-High Throughput Synthesis of Nanoparticles with Homogeneous Size Distribution Using a Coaxial Turbulent Jet Mixer. ACS Nano 2014, 8, 6056–6065. [Google Scholar] [CrossRef]
- Wang, S.; Fontana, F.; Shahbazi, M.A.; Santos, H.A. Acetalated Dextran Based Nano- and Microparticles: Synthesis, Fabrication, and Therapeutic Applications. Chem. Commun. 2021, 57, 4212–4229. [Google Scholar] [CrossRef] [PubMed]
- Rosenholm, J.M.; Penninkangas, A.; Lindén, M. Amino-Functionalization of Large-Pore Mesoscopically Ordered Silica by a One-Step Hyperbranching Polymerization of a Surface-Grown Polyethyleneimine. Chem. Commun. 2006, 37, 3909–3911. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Yang, J.; Li, X.; Zhou, L.; Zhang, R.; Li, W.; Chen, L.; Wang, R.; Zhang, F.; Zhao, D. Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres. Nano Lett. 2014, 14, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Zampini, G.; Matino, D.; Quaglia, G.; Tarpani, L.; Gargaro, M.; Cecchetti, F.; Iorio, A.; Fallarino, F.; Latterini, L. Experimental Evidences on the Role of Silica Nanoparticles Surface Morphology on the Loading, Release and Activity of Three Proteins. Microporous Mesoporous Mater. 2019, 287, 220–227. [Google Scholar] [CrossRef]
- Shi, H.; Liu, S.; Cheng, J.; Yuan, S.; Yang, Y.; Fang, T.; Cao, K.; Wei, K.; Zhang, Q.; Liu, Y. Charge-Selective Delivery of Proteins Using Mesoporous Silica Nanoparticles Fused with Lipid Bilayers. ACS Appl. Mater. Interfaces 2019, 11, 3645–3653. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Shahbazi, M.A.; Mäkilä, E.; Herranz-Blanco, B.; Salonen, J.; Hirvonen, J.; Santos, H.A. Fabrication of a Multifunctional Nano-in-Micro Drug Delivery Platform by Microfluidic Templated Encapsulation of Porous Silicon in Polymer Matrix. Adv. Mater. 2014, 26, 4497–4503. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cito, S.; Zhang, Y.; Wang, C.F.; Sikanen, T.M.; Santos, H.A. A Versatile and Robust Microfluidic Platform toward High Throughput Synthesis of Homogeneous Nanoparticles with Tunable Properties. Adv. Mater. 2015, 27, 2298–2304. [Google Scholar] [CrossRef]
- Herranz-Blanco, B.; Ginestar, E.; Zhang, H.; Hirvonen, J.; Santos, H.A. Microfluidics Platform for Glass Capillaries and Its Application in Droplet and Nanoparticle Fabrication. Int. J. Pharm. 2017, 516, 100–105. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Microfluidic-Assisted Fabrication of Carriers for Controlled Drug Delivery. Lab Chip 2017, 17, 1856–1883. [Google Scholar] [CrossRef]
- Ma, X.; Özliseli, E.; Zhang, Y.; Pan, G.; Wang, D.; Zhang, H. Fabrication of Redox-Responsive Doxorubicin and Paclitaxel Prodrug Nanoparticles with Microfluidics for Selective Cancer Therapy. Biomater. Sci. 2019, 7, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xu, X.; Zhou, J.; Liu, C.; Zhang, L.; Wang, D.; Yang, F.; Zhang, H. Fabrication of a PH/Redox-Triggered Mesoporous Silica-Based Nanoparticle with Microfluidics for Anticancer Drugs Doxorubicin and Paclitaxel Codelivery. ACS Appl. Bio Mater. 2020, 3, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Svilenov, H.; Winter, G. Rapid Sample-Saving Biophysical Characterisation and Long-Term Storage Stability of Liquid Interferon Alpha2a Formulations: Is There a Correlation? Int. J. Pharm. 2019, 562, 42–50. [Google Scholar] [CrossRef]
- Kulakova, A.; Indrakumar, S.; Sønderby, P.; Gentiluomo, L.; Streicher, W.; Roessner, D.; Frieß, W.; Peters, G.H.J.; Harris, P. Small Angle X-ray Scattering and Molecular Dynamic Simulations Provide Molecular Insight for Stability of Recombinant Human Transferrin. J. Struct. Biol. X 2020, 4, 100017. [Google Scholar] [CrossRef] [PubMed]
- Gentiluomo, L.; Roessner, D.; Streicher, W.; Mahapatra, S.; Harris, P.; Frieß, W. Characterization of Native Reversible Self-Association of a Monoclonal Antibody Mediated by Fab-Fab Interaction. J. Pharm. Sci. 2020, 109, 443–451. [Google Scholar] [CrossRef]
- Satish, L.; Millan, S.; Das, S.; Jena, S.; Sahoo, H. Thermal Aggregation of Bovine Serum Albumin in Conventional Buffers: An Insight into Molecular Level Interactions. J. Solut. Chem. 2017, 46, 831–848. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Lindén, M. Wet-Chemical Analysis of Surface Concentration of Accessible Groups on Different Amino-Functionalized Mesoporous SBA-15 Silicas. Chem. Mater. 2007, 19, 5023–5034. [Google Scholar] [CrossRef]
- Maguire, C.M.; Rösslein, M.; Wick, P.; Prina-Mello, A. Characterisation of Particles in Solution–a Perspective on Light Scattering and Comparative Technologies. Sci. Technol. Adv. Mater. 2018, 19, 732–745. [Google Scholar] [CrossRef]
- Kaasalainen, M.; Aseyev, V.; von Haartman, E.; Karaman, D.Ş.; Mäkilä, E.; Tenhu, H.; Rosenholm, J.; Salonen, J. Size, Stability, and Porosity of Mesoporous Nanoparticles Characterized with Light Scattering. Nanoscale Res. Lett. 2017, 12, 74. [Google Scholar] [CrossRef]
- Liu, H.-J.; Xu, P. Smart Mesoporous Silica Nanoparticles for Protein Delivery. Nanomaterials 2019, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Kubiak-Ossowska, K.; Tokarczyk, K.; Jachimska, B.; Mulheran, P.A. Bovine Serum Albumin Adsorption at a Silica Surface Explored by Simulation and Experiment. J. Phys. Chem. B 2017, 121, 3975–3986. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Farooq, M.A.; Li, T.; Geng, T.; Kutoka, P.T.; Wang, B. Cationic Chitosan-modified Silica Nanoparticles for Oral Delivery of Protein Vaccine. J. Biomed. Mater. Res. A 2021, 109, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Boyle, A.L.; Friedrich, H.; Bomans, P.H.H.; Bussmann, J.; Sommerdijk, N.A.J.M.; Jiskoot, W.; Kros, A. Mesoporous Silica Nanoparticles with Large Pores for the Encapsulation and Release of Proteins. ACS Appl. Mater. Interfaces 2016, 8, 32211–32219. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Eltohamy, M.; Kim, M.; Perez, R.A.; Kim, J.-H.; Yun, Y.-R.; Jang, J.-H.; Lee, E.-J.; Knowles, J.C.; Kim, H.-W. Therapeutic Foam Scaffolds Incorporating Biopolymer-Shelled Mesoporous Nanospheres with Growth Factors. Acta Biomater. 2014, 10, 2612–2621. [Google Scholar] [CrossRef]
- Sang, L.C.; Vinu, A.; Coppens, M.O. General Description of the Adsorption of Proteins at Their Iso-Electric Point in Nanoporous Materials. Langmuir 2011, 27, 13828–13837. [Google Scholar] [CrossRef]
- Shahabi, S.; Döscher, S.; Bollhorst, T.; Treccani, L.; Maas, M.; Dringen, R.; Rezwan, K. Enhancing Cellular Uptake and Doxorubicin Delivery of Mesoporous Silica Nanoparticles via Surface Functionalization: Effects of Serum. ACS Appl. Mater. Interfaces 2015, 7, 26880–26891. [Google Scholar] [CrossRef]
- Yola, A.M.; Campbell, J.; Volodkin, D. Microfluidics Meets Layer-by-Layer Assembly for the Build-up of Polymeric Scaffolds. Appl. Surf. Sci. Adv. 2021, 5, 100091. [Google Scholar] [CrossRef]
- Basset, C.; Harder, C.; Vidaud, C.; Déjugnat, C. Design of Double Stimuli-Responsive Polyelectrolyte Microcontainers for Protein Soft Encapsulation. Biomacromolecules 2010, 11, 806–814. [Google Scholar] [CrossRef]
- Arduino, I.; Liu, Z.; Rahikkala, A.; Figueiredo, P.; Correia, A.; Cutrignelli, A.; Denora, N.; Santos, H.A. Preparation of Cetyl Palmitate-Based PEGylated Solid Lipid Nanoparticles by Microfluidic Technique. Acta Biomater. 2021, 121, 566–578. [Google Scholar] [CrossRef]
- Inam, W.; Bhadane, R.; Akpolat, R.N.; Taiseer, R.A.; Filippov, S.K.; Salo-Ahen, O.M.H.; Rosenholm, J.M.; Zhang, H. Interactions between Polymeric Nanoparticles and Different Buffers as Investigated by Zeta Potential Measurements and Molecular Dynamics Simulations. View 2022, 3, 20210009. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of Bovine, Equine and Leporine Serum Albumin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Lord, H.; Knutson, N.; Wikström, M. Nano Differential Scanning Fluorimetry for Comparability Studies of Therapeutic Proteins. Anal. Biochem. 2020, 593, 113581. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, M.; Dyawanapelly, S.; Kansara, B.; Dandekar, P.; Jain, R. Understanding the Stability of Nanoparticle–Protein Interactions: Effect of Particle Size on Adsorption, Conformation and Thermodynamic Properties of Serum Albumin Proteins. ACS Appl. Nano Mater. 2018, 1, 5524–5535. [Google Scholar] [CrossRef]
- Xu, C.; Lei, C.; Yu, C. Mesoporous Silica Nanoparticles for Protein Protection and Delivery. Front. Chem. 2019, 7, 290. [Google Scholar] [CrossRef]
- Ohtake, S.; Kita, Y.; Arakawa, T. Interactions of Formulation Excipients with Proteins in Solution and in the Dried State. Adv. Drug Deliv. Rev. 2011, 63, 1053–1073. [Google Scholar] [CrossRef]
- Küçüktürkmen, B.; Inam, W.; Howaili, F.; Gouda, M.; Prabhakar, N.; Zhang, H.; Rosenholm, J.M. Microfluidic-Assisted Fabrication of Dual-Coated PH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery. Biosensors 2022, 12, 181. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Hirano, A.; Arakawa, T.; Shiraki, K. Effects of Alcohol on the Solubility and Structure of Native and Disulfide-Modified Bovine Serum Albumin. Int. J. Biol. Macromol. 2012, 50, 1286–1291. [Google Scholar] [CrossRef]
- Gupta, B.S.; Taha, M.; Lee, M.J. Buffers More than Buffering Agent: Introducing a New Class of Stabilizers for the Protein BSA. Phys. Chem. Chem. Phys. 2015, 17, 1114–1133. [Google Scholar] [CrossRef]
- Wang, W. Instability, Stabilization, and Formulation of Liquid Protein Pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Giancola, C.; De Sena, C.; Fessas, D.; Graziano, G.; Barone, G. DSC Studies on Bovine Serum Albumin Denaturation Effects of Ionic Strength and SDS Concentration. Int. J. Biol. Macromol. 1997, 20, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kerstens, S.; Murray, B.S.; Dickinson, E. Confocal Microscopy of Heat-Induced Aggregation and Gelation of β-Lactoglobulin in Presence of Non-Ionic Surfactant. Food Hydrocoll. 2005, 19, 625–633. [Google Scholar] [CrossRef]
- Treuheit, M.J.; Kosky, A.A.; Brems, D.N. Inverse Relationship of Protein Concentration and Aggregation. Pharm. Res. 2002, 19, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Neacsu, M.V.; Matei, I.; Micutz, M.; Staicu, T.; Precupas, A.; Popa, V.T.; Salifoglou, A.; Ionita, G. Interaction between Albumin and Pluronic F127 Block Copolymer Revealed by Global and Local Physicochemical Profiling. J. Phys. Chem. B 2016, 120, 4258–4267. [Google Scholar] [CrossRef]
- Prasanthan, P.; Kishore, N. Self-Assemblies of Pluronic Micelles in Partitioning of Anticancer Drugs and Effectiveness of This System towards Target Protein. RSC Adv. 2021, 11, 22057–22069. [Google Scholar] [CrossRef]
- Kita, Y.; Arakawa, T. Salts and Glycine Increase Reversibility and Decrease Aggregation during Thermal Unfolding of Ribonuclease-A. Biosci. Biotechnol. Biochem. 2002, 66, 880–882. [Google Scholar] [CrossRef]
- Donato, L.; Garnier, C.; Doublier, J.L.; Nicolai, T. Influence of the NaCl or CaCl2 Concentration on the Structure of Heat-Set Bovine Serum Albumin Gels at PH 7. Biomacromolecules 2005, 6, 2157–2163. [Google Scholar] [CrossRef]
- Hsieh, S.R.; Reddy, P.M.; Chang, C.J.; Kumar, A.; Wu, W.C.; Lin, H.Y. Exploring the Behavior of Bovine Serum Albumin in Response to Changes in the Chemical Composition of Responsive Polymers: Experimental and Simulation Studies. Polymers 2016, 8, 238. [Google Scholar] [CrossRef]
- Woody, R.W.; Tinoco, I. Optical Rotation of Oriented Helices. III. Calculation of the Rotatory Dispersion and Circular Dichroism of the Alpha- and 3 10 -Helix. J. Chem. Phys. 1967, 46, 4927–4945. [Google Scholar] [CrossRef]
- Schwinté, P.; Ball, V.; Szalontai, B.; Haikel, Y.; Voegel, J.C.; Schaaf, P. Secondary Structure of Proteins Adsorbed onto or Embedded in Polyelectrolyte Multilayers. Biomacromolecules 2002, 3, 1135–1143. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of Protein Secondary Structure from Circular Dichroism Using Theoretically Derived Spectra. Proteins Struct. Funct. Bioinform. 2012, 80, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hauser, P. New characterization of layer-by-layer self-assembly deposition of polyelectrolytes on cotton fabric. Cellulose 2009, 16, 1123–1131. [Google Scholar] [CrossRef]
- Anand, U.; Mukherjee, S. Reversibility in protein folding: Effect of β-cyclodextrin on bovine serum albumin unfolded by sodium dodecyl sulphate. Phys. Chem. Chem. Phys. 2013, 15, 9375–9383. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Hydrodynamic Particle Size (nm), in DI Water | PDI | ζ-Potential (mV), in 25 mM HEPES Buffer, pH 7.2 |
|---|---|---|---|
| MSNs | 151.0 ± 1.69 | 0.054 ± 0.03 | −21.1 ± 0.49 |
| MSN-PEI | 154.9 ± 1.50 | 0.028 ± 0.03 | +33.5 ± 0.44 |
| MSN-PEI–BSA | 148.1 ± 1.16 | 0.039 ± 0.02 | −11.9 ± 0.46 |
| MSN-PEI–BSA–NaPSS | 274.8 ± 8.43 | 0.134 ± 0.06 | −15.1 ± 1.16 |
| MSN-PEI–BSA–NaPSS–SpAcDEX | 296.9 ± 8.98 | 0.208 ± 0.05 | −5.16 ± 0.48 |
| Samples | Tm (°C) |
|---|---|
| 0.5 mg/mL BSA in PBS pH 7.4 (control) | 60.0 ± 0.03 |
| MSN-PEI–BSA in PBS pH 7.4 | 59.4 ± 0.20 |
| MSN-PEI–BSA–NaPSS in PBS pH 7.4 | - |
| 0.5 mg/mL BSA in 0.5% CaCl2 in 0.1% Pluronic® F-127, pH 7.6 (control) | 61.6 ± 0.04 |
| +0.25 mg/mL NaPSS | 48.8 ± 0.10 |
| +0.5 mg/mL NaPSS | 48.5 ± 0.10 |
| +1 mg/mL NaPSS | 48.1 ± 0.05 |
| +2 mg/mL NaPSS | 47.7 ± 0.10 |
| 0.5 mg/mL BSA in 0.1 M acetate buffer pH 5.0 (control) | 57.9 ± 0.05 |
| +0.25 mg/mL SpAcDEX in ethanol | 57.4 ± 0.10 |
| +0.5 mg/mL SpAcDEX in ethanol | 55.9 ± 0.24 |
| 0.5 mg/mL BSA in 0.5% CaCl2 in 0.1% Pluronic® F-127 pH 7.6 (control) | 61.6 ± 0.04 |
| +1% ethanol | 62.6 ± 0.10 |
| +5% ethanol | 62.3 ± 0.08 |
| +10% ethanol | 58.7 ± 0.10 |
| +15% ethanol | 53.0 ± 0.10 |
| +25% ethanol | 43.9 ± 0.08 |
| +50% ethanol | - |
| 0.5 mg/mL BSA in DI water pH 4.0 | 36.3 ± 0.10 |
| 0.5 mg/mL BSA in 0.1 M MES pH 4.7 | 46.0 ± 0.10 |
| 0.5 mg/mL BSA in 0.1 M acetate buffer pH 5.0 | 57.9 ± 0.05 |
| 0.5 mg/mL BSA in PBS pH 7.4 | 60.0 ± 0.03 |
| 0.5 mg/mL BSA in 25 mM HEPES pH 7.2 | 49.7 ± 0.10 |
| 0.5 mg/mL BSA in 0.1% Pluronic® F-127, pH 7.6 (control) | 47.5 ± 0.10 |
| +0.5% CaCl2 | 61.6 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisina, S.; Inam, W.; Huhtala, M.; Howaili, F.; Zhang, H.; Rosenholm, J.M. Nano Differential Scanning Fluorimetry as a Rapid Stability Assessment Tool in the Nanoformulation of Proteins. Pharmaceutics 2023, 15, 1473. https://doi.org/10.3390/pharmaceutics15051473
Lisina S, Inam W, Huhtala M, Howaili F, Zhang H, Rosenholm JM. Nano Differential Scanning Fluorimetry as a Rapid Stability Assessment Tool in the Nanoformulation of Proteins. Pharmaceutics. 2023; 15(5):1473. https://doi.org/10.3390/pharmaceutics15051473
Chicago/Turabian StyleLisina, Sofia, Wali Inam, Mikko Huhtala, Fadak Howaili, Hongbo Zhang, and Jessica M. Rosenholm. 2023. "Nano Differential Scanning Fluorimetry as a Rapid Stability Assessment Tool in the Nanoformulation of Proteins" Pharmaceutics 15, no. 5: 1473. https://doi.org/10.3390/pharmaceutics15051473