
Self-Emulsifying Formulations to Increase the Oral Bioavailability of 4,6,4′-Trimethylangelicin as a Possible Treatment for Cystic Fibrosis

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Metabolic Stability with Human Liver Microsomes
2.3. Solubility Studies
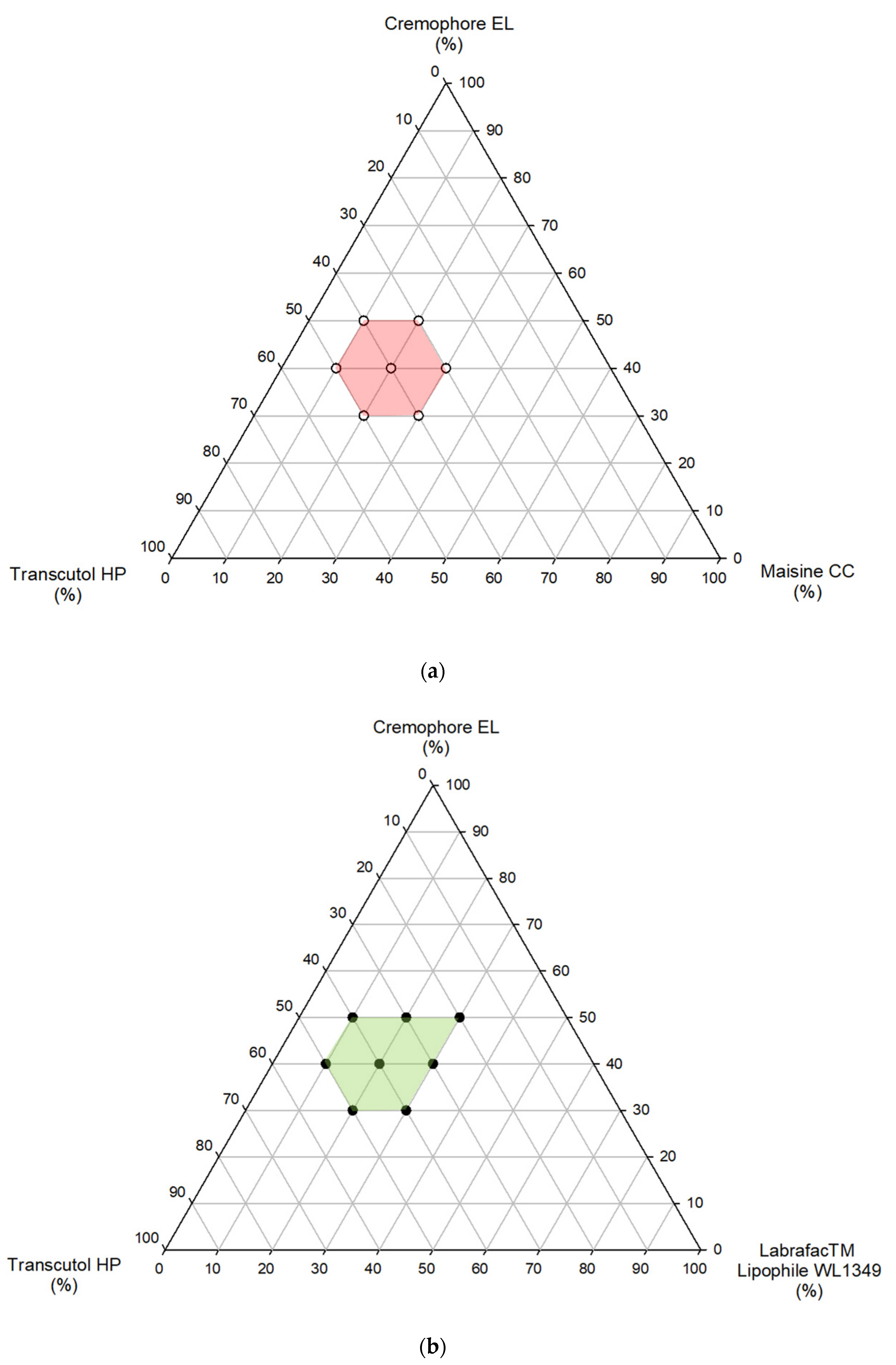
2.4. Phase Diagram Study and Determination of Self-Emulsification Time
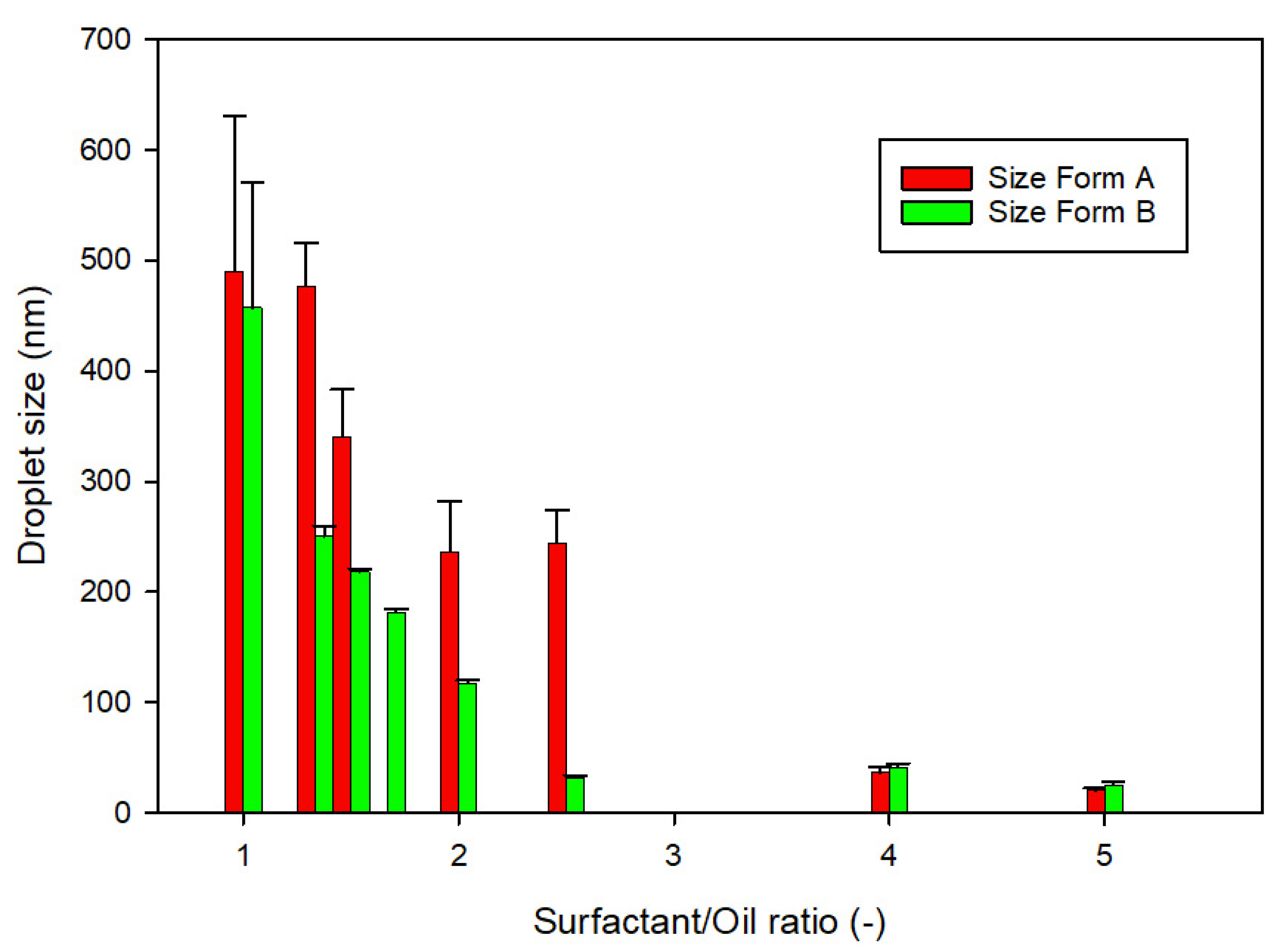
2.5. Droplet Size of Microemulsion and Zeta Potential
2.6. In Vitro Dissolution Test
2.7. In Vivo Pharmacokinetics
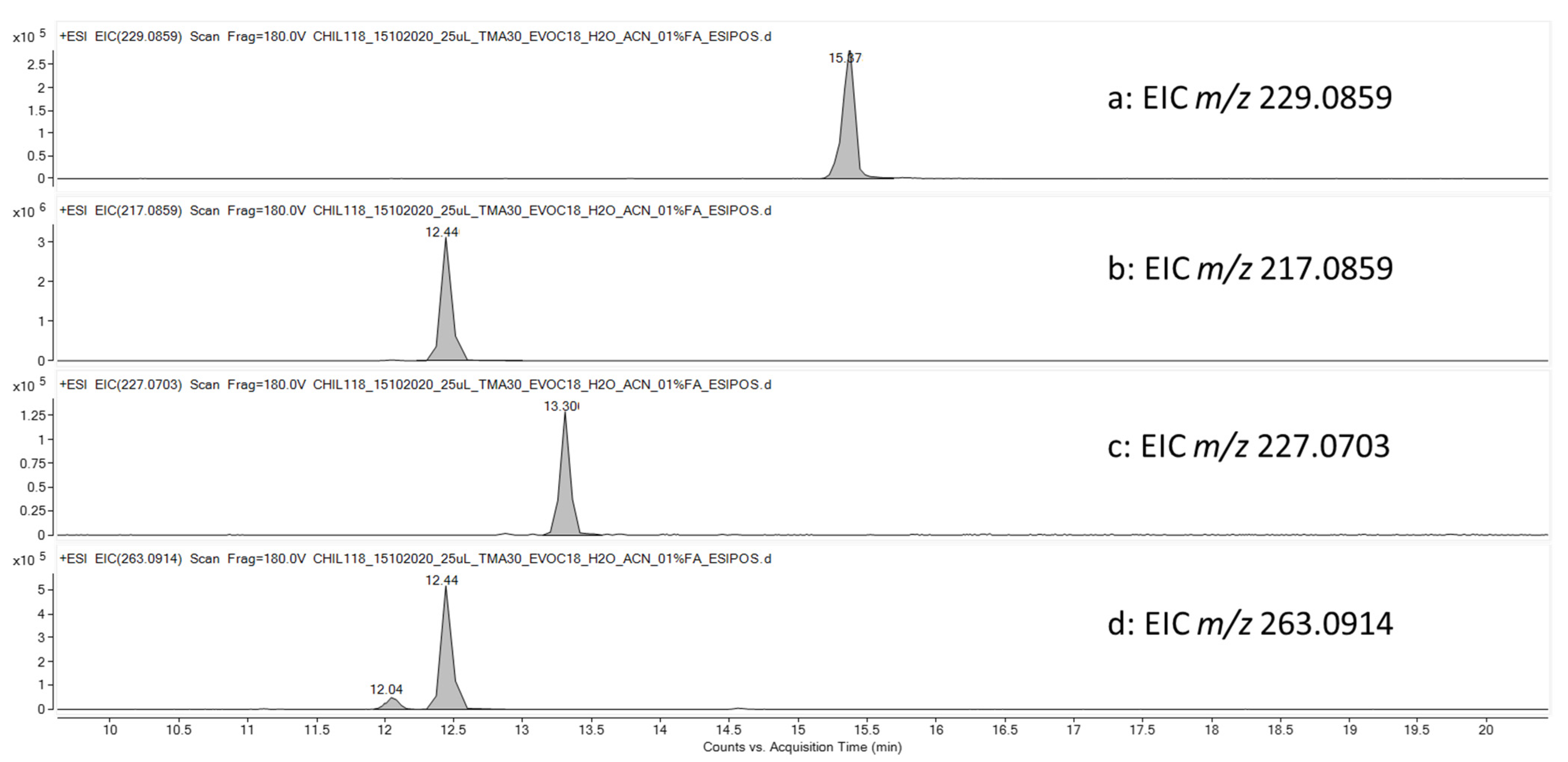
2.8. Identification of TMA-Related Metabolites in HLMs Mixture and Quantification of TMA in Plasma Samples by UHPLC-HRMS
2.8.1. Stability of TMA in HLMs
2.8.2. TMA and TMA-Related Metabolites in Plasma Samples
3. Results
3.1. Development of TMA Formulations and In Vitro Characterizations
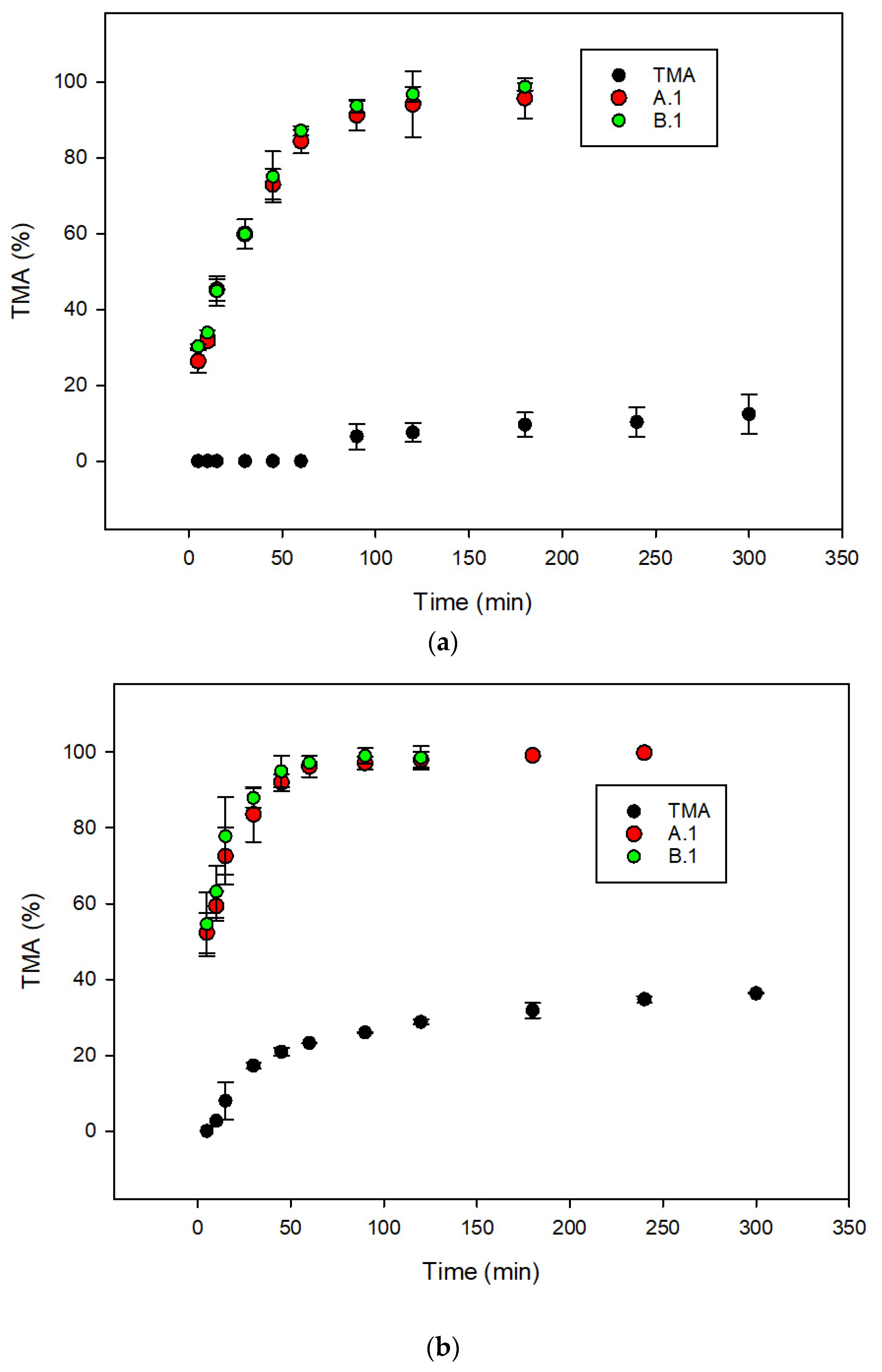
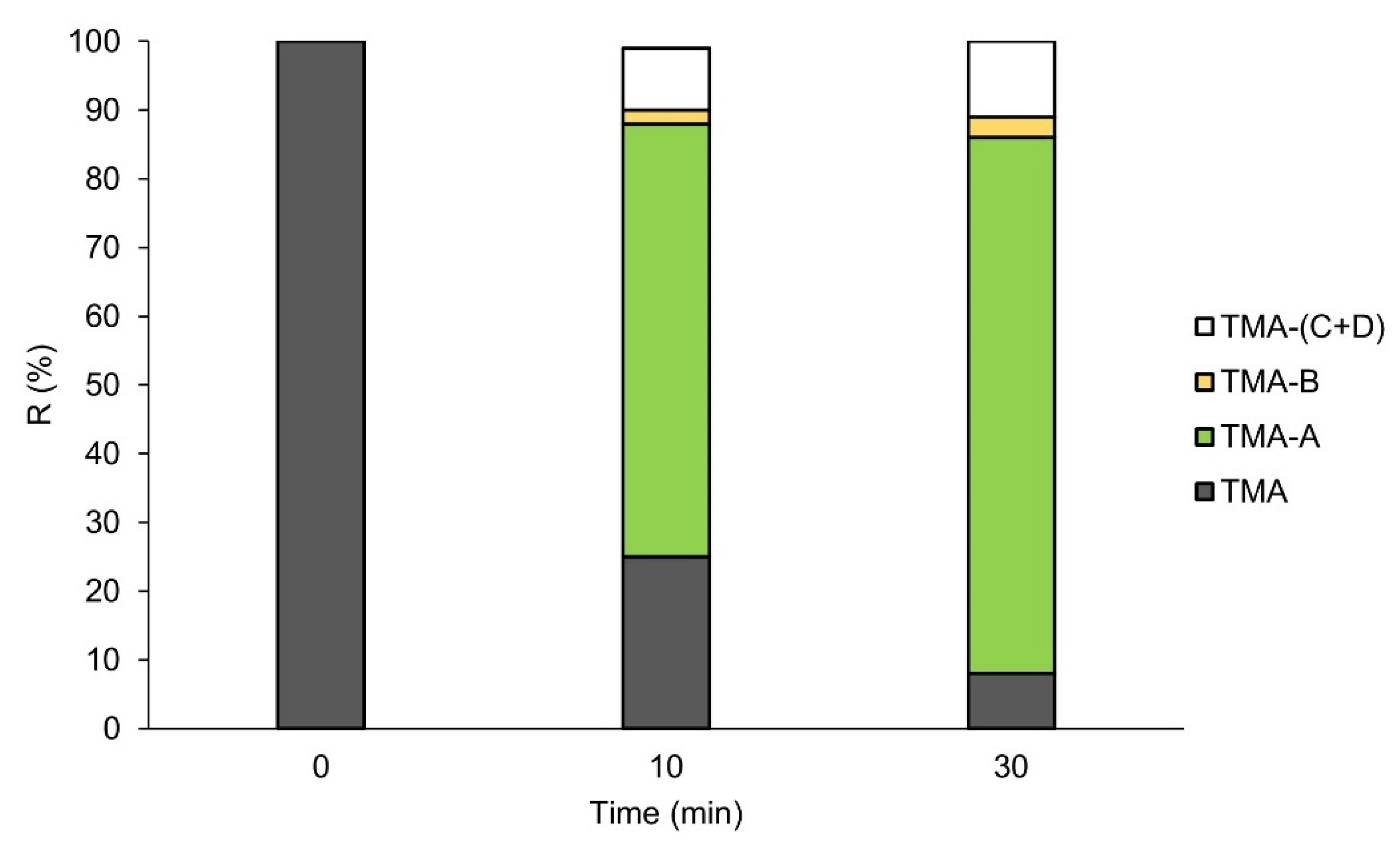
3.2. Metabolic Stability of TMA
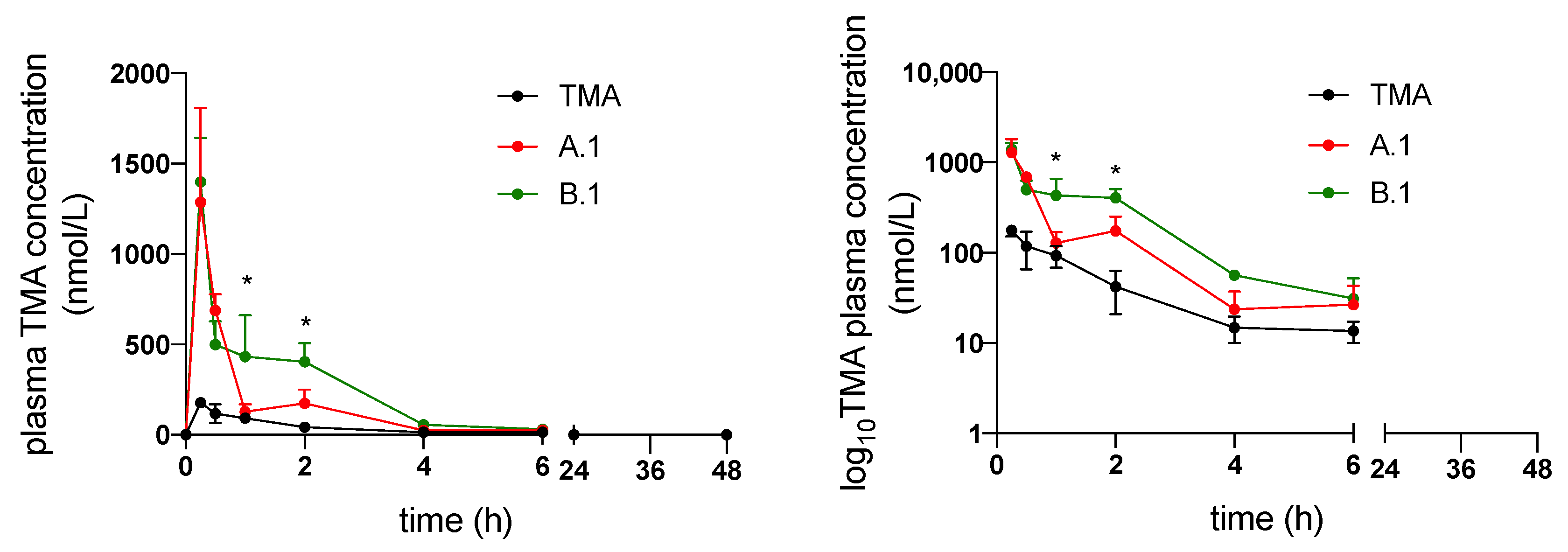
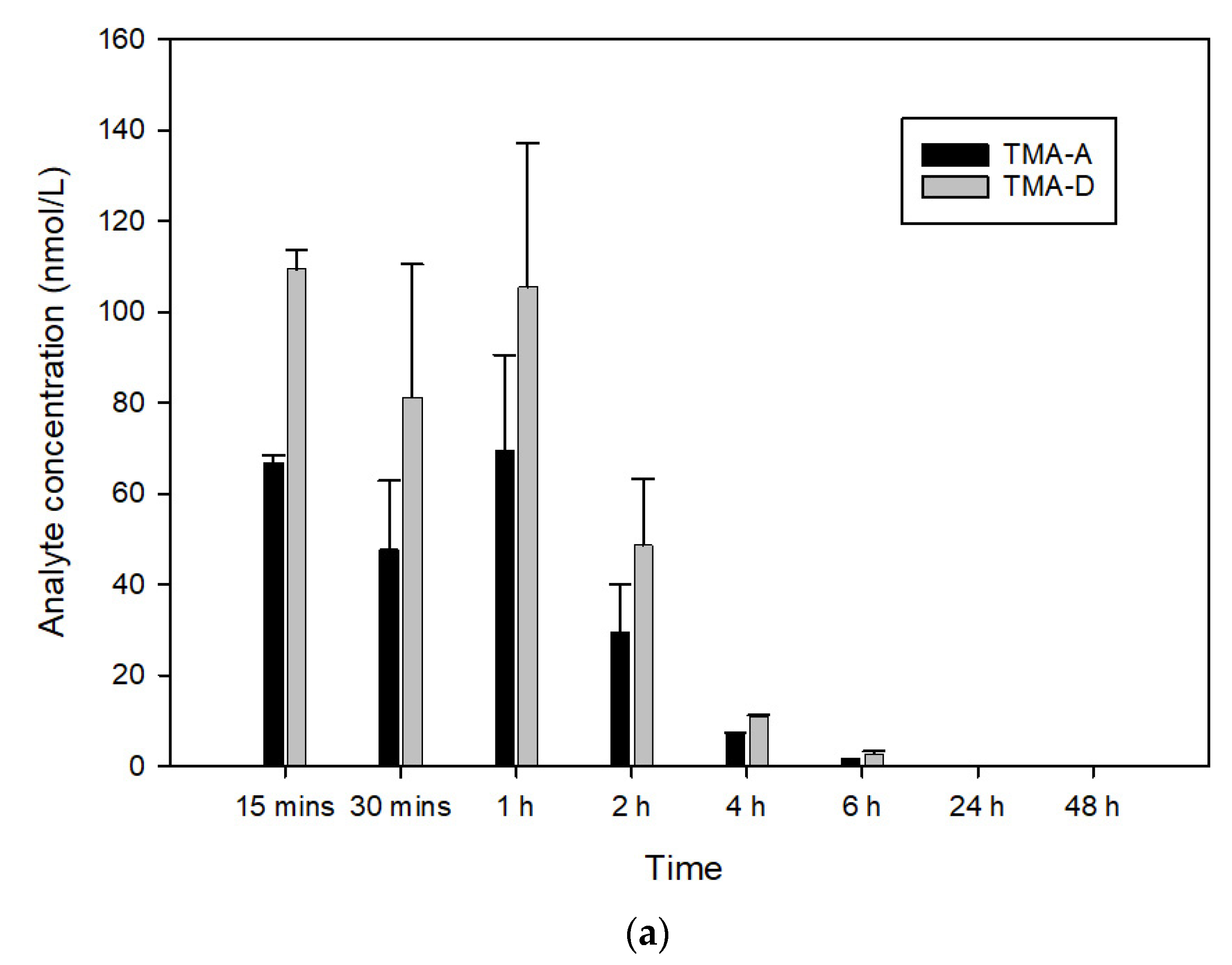
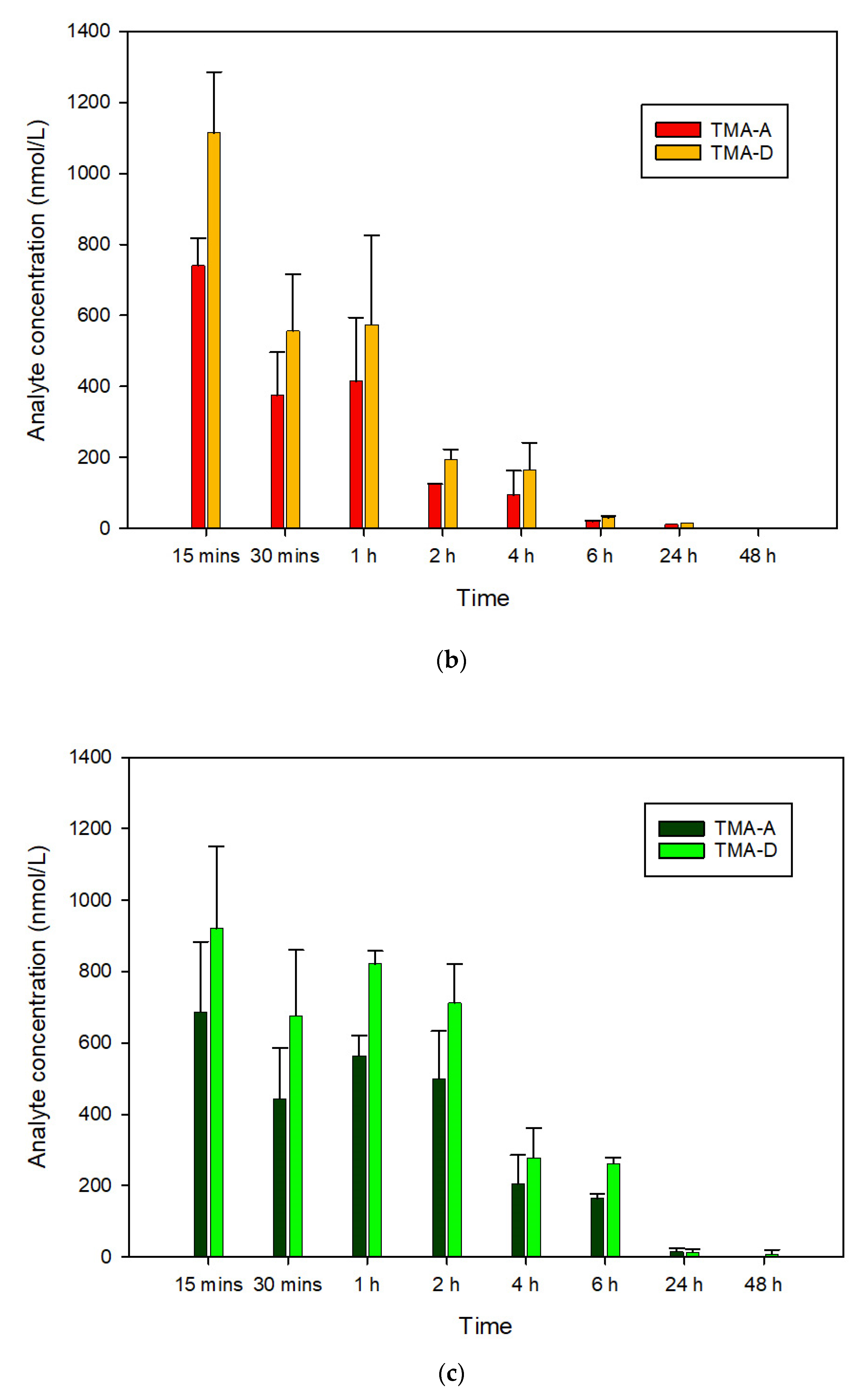
3.3. In Vivo Pharmacokinetics of TMA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharm. 2016, 8, 127–140. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2010, 10, 1662. [Google Scholar] [CrossRef]
- Europe Medicinal Agency. EU/3/13/1137: Orphan Designation for the Treatment of Cystic Fibrosis. 2020. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3131137 (accessed on 10 April 2022).
- Tamanini, A.; Borgatti, M.; Finotti, A.; Piccagli, L.; Bezzerri, V.; Favia, M.; Guerra, L.; Lampronti, I.; Bianchi, N.; Dall’Acqua, F.; et al. Trimethylangelicin reduces IL-8 transcription and potentiates CFTR function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, 380–390. [Google Scholar] [CrossRef]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; Laselva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 48–61. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. The investigational Cystic Fibrosis drug Trimethylangelicin directly modulates CFTR by stabilizing the first membrane-spanning domain. Biochem. Pharmacol. 2016, 119, 85–92. [Google Scholar]
- Laselva, O.; Marzaro, G.; Vaccarin, C.; Lampronti, I.; Tamanini, A.; Lippi, G.; Gambari, R.; Cabrini, G.; Bear, C.E.; Chilin, A.; et al. Molecular Mechanism of Action of Trimethylangelicin Derivatives as CFTR Modulators. Front. Pharmacol. 2018, 9, 719. [Google Scholar] [CrossRef]
- Caffieri, S. Reversed-phase high performance liquid chromatography (RP-HPLC) determination of lipophilicity of furocumarins: Relationship with DNA interaction. J. Pharm. Sci. 2001, 90, 732–739. [Google Scholar] [CrossRef]
- Poovi, G.; Damodharan, N. Lipid nanoparticles: A challenging approach for oral delivery of BCS Class-II drugs. Future J. Pharm. Sci. 2018, 4, 191–205. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and “self-microemulsifying” drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L. Lipid-Based Excipients for Oral Drug Delivery. In Oral Lipid-Based Formulations, Enhancing the Bioavailability of Poorly Water-Soluble Drugs; Hauss, D.J., Ed.; Informa Healthcare: New York, NY, USA, 2007; pp. 33–84. [Google Scholar] [CrossRef]
- Baloch, J.; Sohail, M.F.; Sarwar, H.S.; Kiani, M.H.; Khan, G.M.; Jahan, S.; Rafay, M.; Chaudhry, M.T.; Yasinzai, M.; Shahnaz, G. Self-nanoemulsifying drug delivery system (Snedds) for improved oral bioavailability of chlorpromazine: In vitro and in vivo evaluation. Medicina 2019, 55, 210. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, Q.; Chen, Z.; Wu, W.; Zhu, Q.; Lu, Y. In vitro and in correlation for lipid-bases formulations: Current status and future perspectives. Acta Pharm. Sin. B 2021, 11, 2469–2487. [Google Scholar] [PubMed]
- Holm, R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv. Drug Deliv. Rev. 2019, 142, 118–127. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: The ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur. J. Pharm. Biopharm. 2007, 67, 96–105. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilization using liquid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar]
- Hauss, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev. 2007, 59, 667–676. [Google Scholar] [CrossRef]
- Khoo, S.M.; Shackleford, D.M.; Porter, C.J.H.; Edwards, G.A.; Charman, W.N. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm. Res. 2003, 20, 1460–1465. [Google Scholar] [CrossRef]
- Guiotto, A.; Rodighiero, P.; Manzini, P.; Pastorini, G.; Bordin, F.; Baccichetti, F.; Carlassare, F.; Vedaldi, D.; Dall’Acqua, F.; Dall’Acqua, F.; et al. 6-Methylangelicins: A new Series of Potential Photochemotherapeutic Agents for the Treatment of Psoriasis. J. Med. Chem. 1984, 27, 959–967. [Google Scholar] [CrossRef]
- Salmaso, S.; Mastrotto, F.; Roverso, M.; Gandin, V.; De Martin, S.; Gabbia, D.; De Franco, M.; Vaccarin, C.; Verona, M.; Chilin, A.; et al. Tyrosine kinase inhibitor prodrug-loaded liposomes for controlled release at tumor microenvironment. J. Control. Release 2021, 340, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, T.; Klein, S. Simplified biorelevant media for screening dissolution performance of poorly soluble drugs. Dissolution Technol. 2007, 14, 8–13. [Google Scholar] [CrossRef]
- Orlando, R.; de Martin, S.; Andrighetto, L.; Floreani, M.; Palatini, P. Fluvoxamine pharmacokinetics in healthy elderly subjects and elderly patients with chronic heart failure. Br. J. Clin. Pharmacol. 2010, 69, 279–286. [Google Scholar] [CrossRef]
- Assi, R.A.; Abdulbaqi, I.M.; Ming, T.S.; Yee, C.S.; Wahab, H.A.; Asif, S.M.; Darwis, Y. Liquid and solid self-emulsifying drug delivery systems (Sedds) as carriers for the oral delivery of azithromycin: Optimization, in vitro characterization and stability assessment. Pharmaceutics 2020, 12, 1052. [Google Scholar] [CrossRef] [PubMed]
- Ditzinger, F.; Price, D.J.; Ilie, A.R.; Köhl, N.J.; Jankovic, S.; Tsakiridou, G.; Aleandri, S.; Kalantzi, L.; Holm, R.; Nair, A.; et al. Lipophilicity and hydrophobicity considerations in bio-enabling oral formulations approaches—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 464–482. [Google Scholar] [CrossRef] [PubMed]
- Han, S.F.; Yao, T.; Zhang, X.; Gan, L.; Zhu, C.; Yu, H.Z.; Gan, Y. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: Effects of lipid composition and formulation. Int. J. Pharm. 2009, 379, 18–24. [Google Scholar] [CrossRef]
- Rege, B.D.; Kao, J.P.; Polli, J.E. Effects of nonionic surfactants on membrane transporters in Caco2-cell monolayers. Eur. J. Pharm. Sci. 2002, 16, 237–246. [Google Scholar] [CrossRef]
- Hong, J.Y.; Kim, J.K.; Song, Y.K.; Park, J.S.; Kim, C.K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control. Release 2006, 110, 332–338. [Google Scholar] [CrossRef]
- Thomas, N.; Müllertz, A.; Graf, A.; Rades, T. Influence of lipid composition and drug load on the In Vitro performance of self-nanoemulsifying drug delivery systems. J. Pharm. Sci. 2012, 101, 1721–1731. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Role | Chemical Name | HLB | TMA Solubility (µg/mL) |
|---|---|---|---|---|
| Maisine® CC | Oil | Glycerol/Glyceryl monolinoleate | 1 | 6340 ± 377 |
| LabrafacTM Lipophile WL1349 | Medium chain triglycerides | 1 | 4639 ± 170 | |
| Transcutol® HP | Co-solvent | Diethylene glycol-monoethyl ether | - | 9568 ± 131 |
| Ethanol | Ethyl alcohol | - | 2509 ± 52 | |
| Capryol® PGMC | Co-surfactant | Propylene glycol monocaprylate | 6 | 7346 ± 380 |
| LauroglycolTM FCC | Propylene glycol monolaurate | 5 | 6484 ± 92 | |
| Cremophor® EL | Surfactant | Polyoxyl-35-castor oil | 12–14 | 9091 ± 217 |
| Tween 80 | Polysorbate 80 | 15 | 7040 ± 120 |
| Formulation | Maisine® CC (% w/w) | Cremophor® EL (% w/w) | Transcutol® HP (% w/w) | Self-Emulsification Time (s) | Droplet Size (nm) | PDI (-) | |
|---|---|---|---|---|---|---|---|
| A | 1 | 10 | 50 | 40 | 7 ± 1 | 20.37 ± 1.76 | 0.19 |
| 2 | 10 | 40 | 50 | 7 ± 1 | 36.29 ± 5.30 | 0.20 | |
| 3 | 20 | 50 | 30 | 8 ± 1 | 243.96 ± 29.71 | 0.29 | |
| 4 | 20 | 40 | 40 | 9 ± 2 | 236.30 ± 45.31 | 0.51 | |
| 5 | 20 | 30 | 50 | 9 ± 1 | 341.03 ± 42.66 | 0.48 | |
| 6 | 30 | 50 | 20 | - | - | - | |
| 7 | 30 | 40 | 30 | 39 ± 6 | 477.30 ± 38.40 | 0.52 | |
| 8 | 30 | 30 | 40 | 17 ± 4 | 490.31 ± 140.52 | 0.82 | |
| 9 | 30 | 20 | 50 | - | - | - | |
| Formulation | LabrafacTM Lipophile WL1349 (% w/w) | Cremophor® EL (% w/w) | Transcutol® HP (% w/w) | Self-Emulsification Time (s) | Droplet Size (nm) | PDI (-) | |
|---|---|---|---|---|---|---|---|
| B | 1 | 10 | 50 | 40 | 8 ± 3 | 25.37 ± 2.71 | 0.16 |
| 2 | 10 | 40 | 50 | 6 ± 2 | 41.32 ± 2.90 | 0.17 | |
| 3 | 20 | 50 | 30 | 11 ± 2 | 31.98 ± 1.71 | 0.21 | |
| 4 | 20 | 40 | 40 | 8 ± 1 | 117.73 ± 2.06 | 0.18 | |
| 5 | 20 | 30 | 50 | 8 ± 1 | 218.57 ± 2.61 | 0.19 | |
| 6 | 30 | 50 | 20 | >90 | 181.47 ± 2.91 | 0.25 | |
| 7 | 30 | 40 | 30 | 8 ± 1 | 250.33 ± 9.29 | 0.10 | |
| 8 | 30 | 30 | 40 | 10 ± 3 | 457.67 ± 113.07 | 0.8 | |
| 9 | 30 | 20 | 50 | - | - | - | |
| Formulation | Oil (% w/w) | Cremophor® EL (% w/w) | Transcutol® HP (% w/w) | Droplet Size (nm) | PDI (-) | Zeta Potential (mV) |
|---|---|---|---|---|---|---|
| A.1 | 10 | 50 | 40 | 22.80 ± 1.80 | 0.25 | −8.82 ± 1.84 |
| B.1 | 10 | 50 | 40 | 23.38 ± 0.61 | 0.11 | −9.03 ± 3.66 |
| Parameter (Mean ± S.D.) | Formulation | ||
|---|---|---|---|
| TMA | A.1 | B.1 | |
| Cmax (nmol/L) | 186.05 ± 21.14 | 1285.85 ± 738.43 * | 1399.60 ± 342.81 * |
| Tmax (h) | 0.38 ± 0.18 | 0.25 ± 0.00 | 0.25 ± 0.00 |
| AUC0-inf (nmol/L * h) | 297.83 ± 119.13 | 1059.39 ± 90.80 * | 1686.53 ± 504.57 ** |
| t1/2 (h) | 1.71 ± 0.16 | 1.09 ± 0.39 | 1.27 ± 0.80 |
| Vz/F (mL/g) | 0.183 ± 0.089 | 0.029 ± 0.008 * | 0.025 ± 0.004 * |
| CL/F (mL/h/g) | 0.073 ± 0.029 | 0.019 ± 0.002 * | 0.012 ± 0.004 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceschinis, E.; Roverso, M.; Gabbia, D.; De Martin, S.; Brusegan, M.; Vaccarin, C.; Bogialli, S.; Chilin, A. Self-Emulsifying Formulations to Increase the Oral Bioavailability of 4,6,4′-Trimethylangelicin as a Possible Treatment for Cystic Fibrosis. Pharmaceutics 2022, 14, 1806. https://doi.org/10.3390/pharmaceutics14091806
Franceschinis E, Roverso M, Gabbia D, De Martin S, Brusegan M, Vaccarin C, Bogialli S, Chilin A. Self-Emulsifying Formulations to Increase the Oral Bioavailability of 4,6,4′-Trimethylangelicin as a Possible Treatment for Cystic Fibrosis. Pharmaceutics. 2022; 14(9):1806. https://doi.org/10.3390/pharmaceutics14091806
Chicago/Turabian StyleFranceschinis, Erica, Marco Roverso, Daniela Gabbia, Sara De Martin, Matteo Brusegan, Christian Vaccarin, Sara Bogialli, and Adriana Chilin. 2022. "Self-Emulsifying Formulations to Increase the Oral Bioavailability of 4,6,4′-Trimethylangelicin as a Possible Treatment for Cystic Fibrosis" Pharmaceutics 14, no. 9: 1806. https://doi.org/10.3390/pharmaceutics14091806