
HMG-CoA Reductase Inhibitor Statins Activate the Transcriptional Activity of p53 by Regulating the Expression of TAZ

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
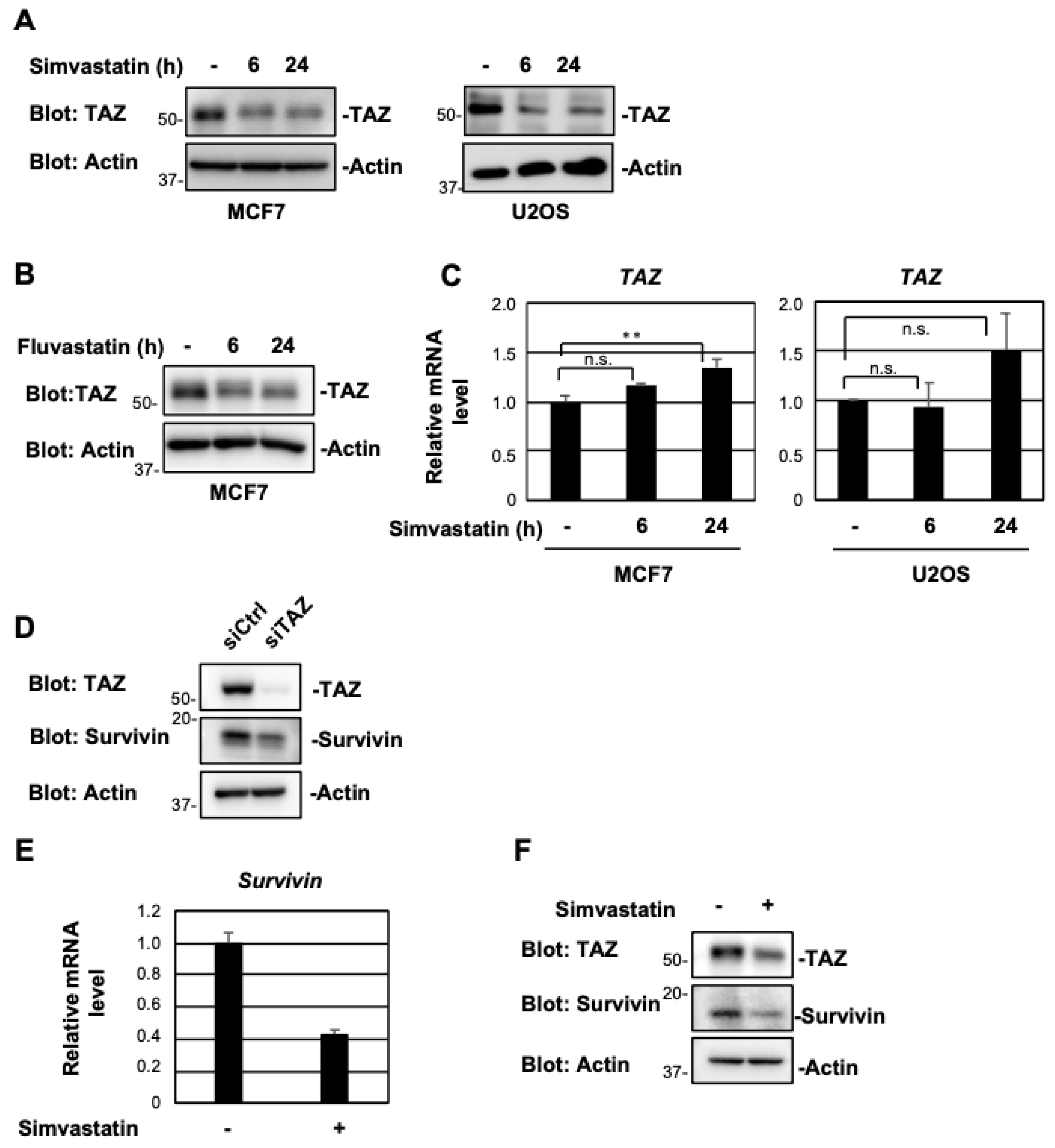
2.1. Simvastatin and Fluvastatin Suppress TAZ Protein Expression
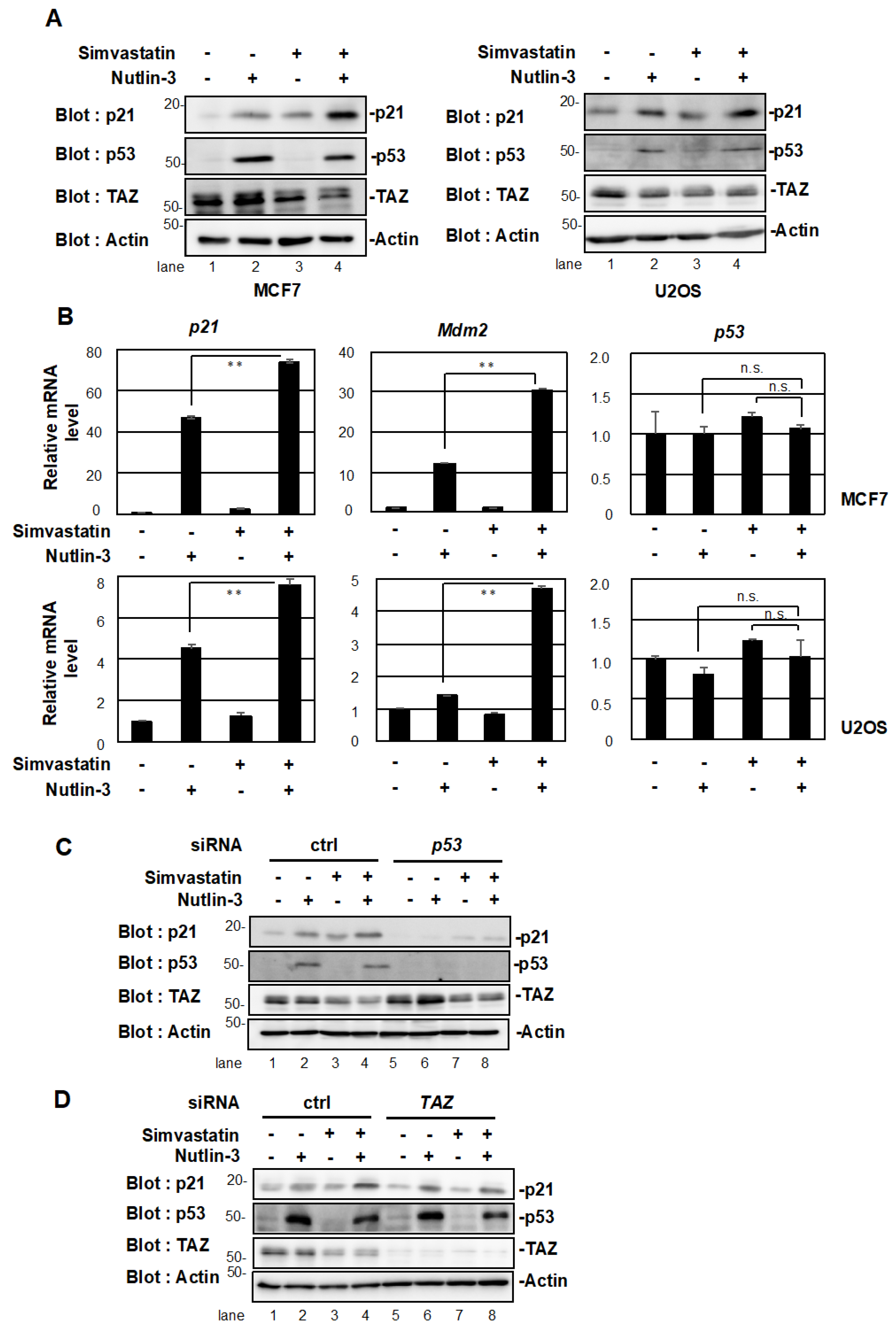
2.2. Simvastatin Enhances p53 Target Gene Expression
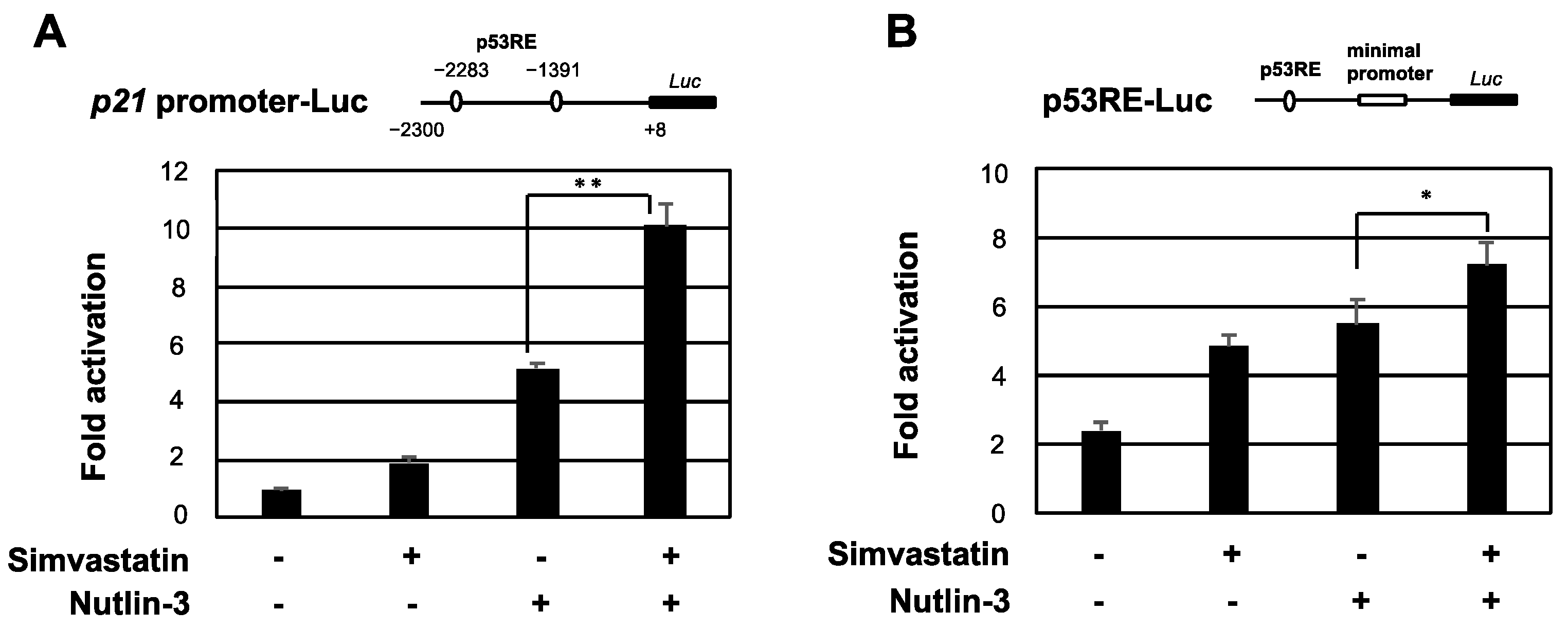
2.3. Simvastatin Enhances the Transcriptional Activation of p53
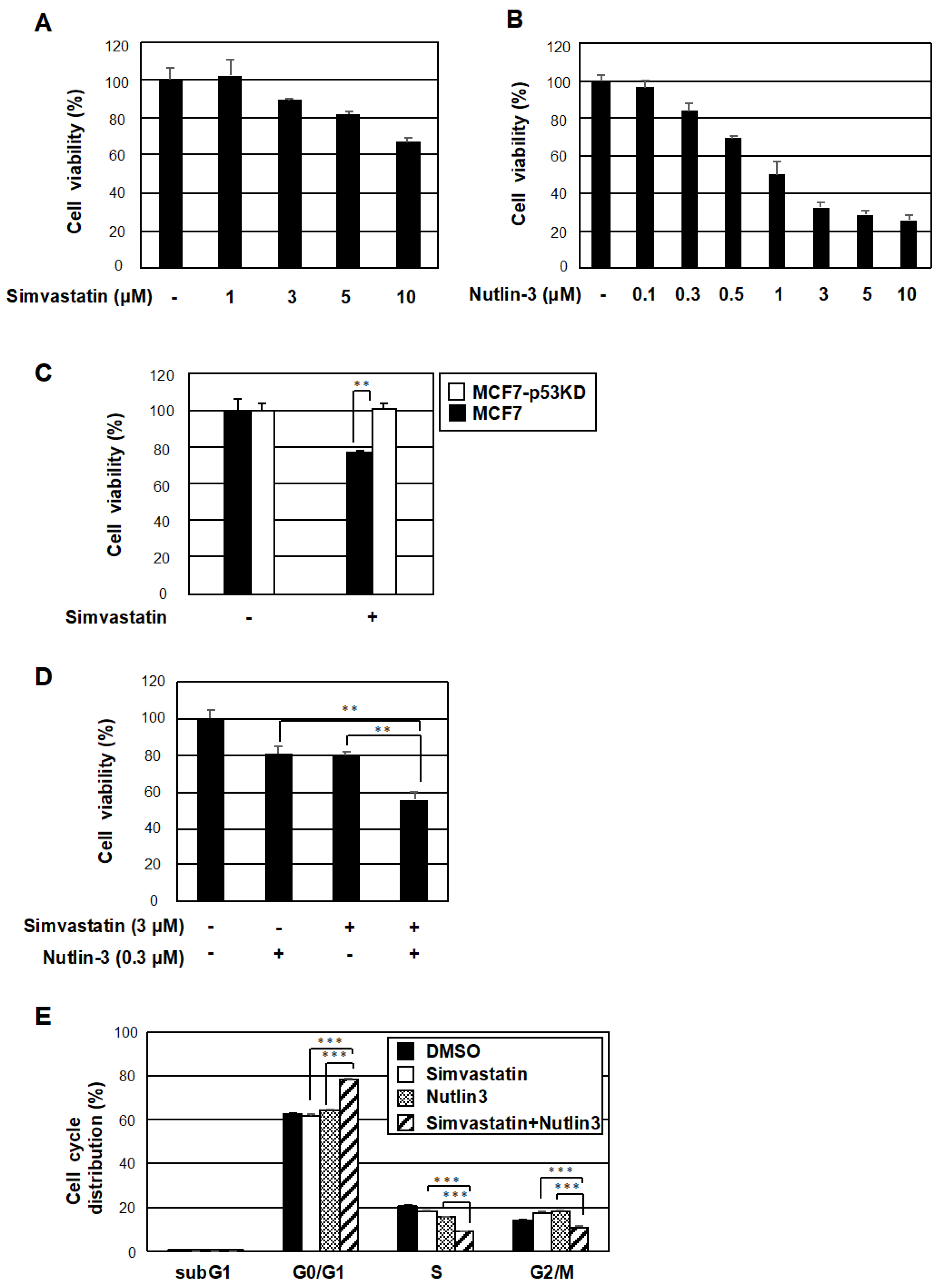
2.4. The Co-Treatment with Simvastatin and Nutlin-3 Efficiently Reduces Cancer Cell Survival
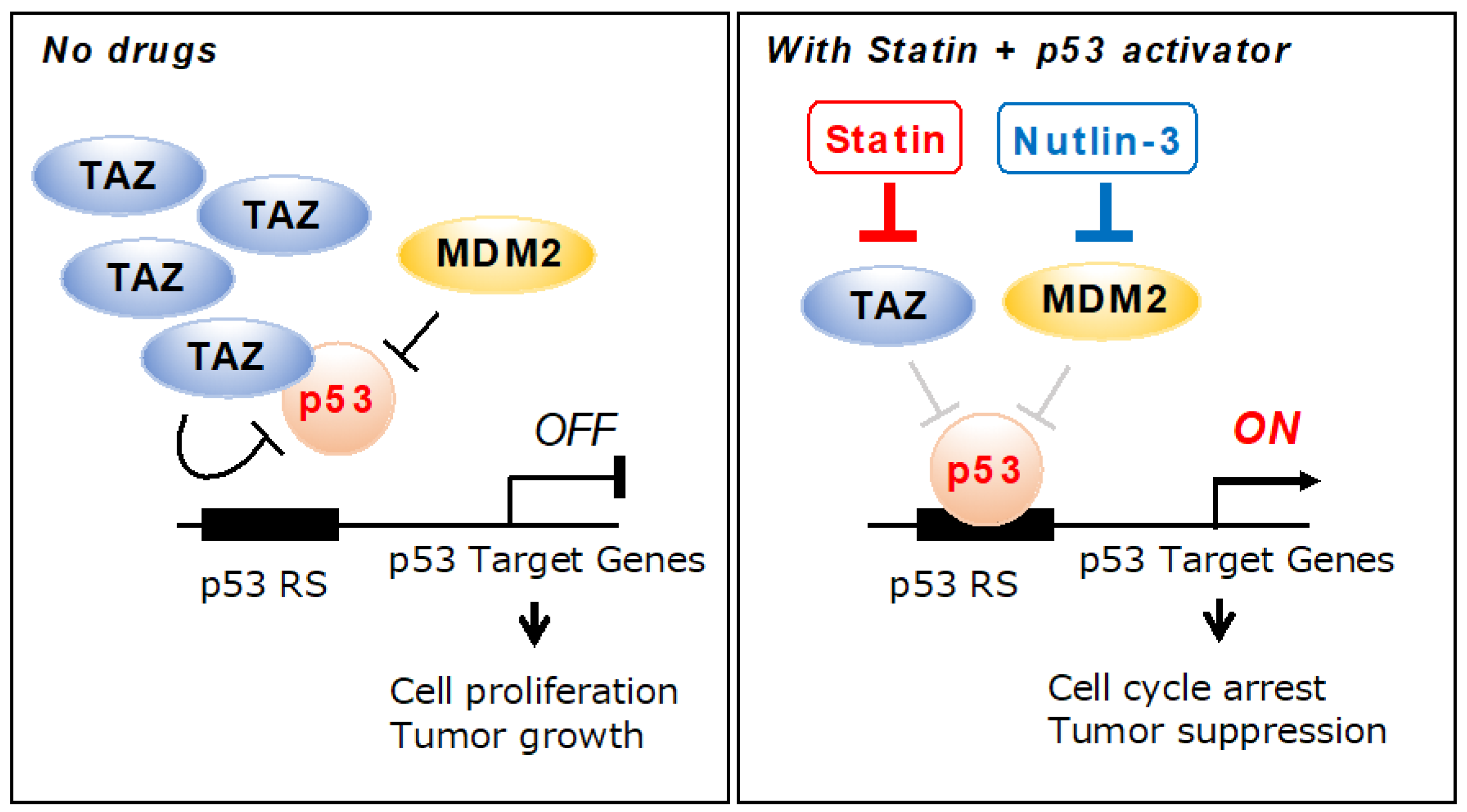
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines, Plasmids, and Transfection
4.3. RNA Extraction, Reverse Transcription, and Quantitative PCR (qPCR)
4.4. Immunochemical Methods and Antibodies
4.5. Reporter Assay
4.6. Cell Viability Assay
4.7. Cell Cycle Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, X.; He, Q.; Gimple, R.C.; Liao, Y.; Wang, L.; Wu, R.; Xie, Q.; Rich, J.N.; Shen, K.; et al. Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. USA 2020, 117, 33295–33304. [Google Scholar] [CrossRef]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef]
- Feng, J.; Ren, P.; Gou, J.; Li, Z. Prognostic significance of TAZ expression in various cancers: A meta-analysis. OncoTargets Ther. 2016, 9, 5235–5244. [Google Scholar] [CrossRef]
- Bálint, E.E.; Vousden, K.H. Activation and activities of the p53 tumour suppressor protein. Br. J. Cancer 2001, 85, 1813–1823. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Onel, K.; Cordon-Cardo, C. MDM2 and prognosis. Mol. Cancer Res. 2004, 2, 1–8. [Google Scholar] [CrossRef]
- Miyajima, C.; Kawarada, Y.; Inoue, Y.; Suzuki, C.; Mitamura, K.; Morishita, D.; Ohoka, N.; Imamura, T.; Hayashi, H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells 2020, 9, 171. [Google Scholar] [CrossRef]
- Demierre, M.F.; Higgins, P.D.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and cancer prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef]
- Mundy, G.; Garrett, R.; Harris, S.; Chan, J.; Chen, D.; Rossini, G.; Boyce, B.; Zhao, M.; Gutierrez, G. Stimulation of bone formation in vitro and in rodents by statins. Science 1999, 286, 1946–1949. [Google Scholar] [CrossRef]
- Davignon, J.; Laaksonen, R. Low-density lipoprotein-independent effects of statins. Curr. Opin. Lipidol. 1999, 10, 543–559. [Google Scholar] [CrossRef]
- Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. [Google Scholar] [CrossRef]
- Hindler, K.; Cleeland, C.S.; Rivera, E.; Collard, C.D. The role of statins in cancer therapy. Oncologist 2006, 11, 306–315. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588. [Google Scholar] [CrossRef]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The potential use of simvastatin for cancer treatment: A review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Di Bello, E.; Zwergel, C.; Mai, A.; Valente, S. The Innovative Potential of Statins in Cancer: New Targets for New Therapies. Front. Chem. 2020, 8, 516. [Google Scholar] [CrossRef]
- Mehta, S.; Campbell, H.; Drummond, C.J.; Li, K.; Murray, K.; Slatter, T.; Bourdon, J.C.; Braithwaite, A.W. Adaptive homeostasis and the p53 isoform network. EMBO Rep. 2021, 22, e53085. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef]
- Scoumanne, A.; Chen, X. Protein methylation: A new mechanism of p53 tumor suppressor regulation. Histol. Histopathol. 2008, 23, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef]
- Shanzer, M.; Adler, J.; Ricardo-Lax, I.; Reuven, N.; Shaul, Y. The nonreceptor tyrosine kinase c-Src attenuates SCF(β-TrCP) E3-ligase activity abrogating Taz proteasomal degradation. Proc. Natl. Acad. Sci. USA 2017, 114, 1678–1683. [Google Scholar] [CrossRef]
- Arya, A.K.; El-Fert, A.; Devling, T.; Eccles, R.M.; Aslam, M.A.; Rubbi, C.P.; Vlatković, N.; Fenwick, J.; Lloyd, B.H.; Sibson, D.R.; et al. Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br. J. Cancer 2010, 103, 186–195. [Google Scholar] [CrossRef]
- Fukuura, K.; Inoue, Y.; Miyajima, C.; Watanabe, S.; Tokugawa, M.; Morishita, D.; Ohoka, N.; Komada, M.; Hayashi, H. The ubiquitin-specific protease USP17 prevents cellular senescence by stabilizing the methyltransferase SET8 and transcriptionally repressing p21. J. Biol. Chem. 2019, 294, 16429–16439. [Google Scholar] [CrossRef]
- Nagasaka, M.; Hashimoto, R.; Inoue, Y.; Ishiuchi, K.; Matsuno, M.; Itoh, Y.; Tokugawa, M.; Ohoka, N.; Morishita, D.; Mizukami, H.; et al. Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway. Molecules 2018, 23, 1394. [Google Scholar] [CrossRef]
- Inoue, Y.; Kawachi, S.; Ohkubo, T.; Nagasaka, M.; Ito, S.; Fukuura, K.; Itoh, Y.; Ohoka, N.; Morishita, D.; Hayashi, H. The CDK inhibitor p21 is a novel target gene of ATF4 and contributes to cell survival under ER stress. FEBS Lett. 2017, 591, 3682–3691. [Google Scholar] [CrossRef]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-β induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef]
- Miyajima, C.; Itoh, Y.; Inoue, Y.; Hayashi, H. Positive Regulation of Interleukin-2 Expression by a Pseudokinase, Tribbles 1, in Activated T Cells. Biol. Pharm. Bull. 2015, 38, 1126–1133. [Google Scholar] [CrossRef]
- Tokugawa, M.; Inoue, Y.; Ishiuchi, K.; Kujirai, C.; Matsuno, M.; Ri, M.; Itoh, Y.; Miyajima, C.; Morishita, D.; Ohoka, N.; et al. Periplocin and cardiac glycosides suppress the unfolded protein response. Sci. Rep. 2021, 11, 9528. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyajima, C.; Hayakawa, Y.; Inoue, Y.; Nagasaka, M.; Hayashi, H. HMG-CoA Reductase Inhibitor Statins Activate the Transcriptional Activity of p53 by Regulating the Expression of TAZ. Pharmaceuticals 2022, 15, 1015. https://doi.org/10.3390/ph15081015
Miyajima C, Hayakawa Y, Inoue Y, Nagasaka M, Hayashi H. HMG-CoA Reductase Inhibitor Statins Activate the Transcriptional Activity of p53 by Regulating the Expression of TAZ. Pharmaceuticals. 2022; 15(8):1015. https://doi.org/10.3390/ph15081015
Chicago/Turabian StyleMiyajima, Chiharu, Yurika Hayakawa, Yasumichi Inoue, Mai Nagasaka, and Hidetoshi Hayashi. 2022. "HMG-CoA Reductase Inhibitor Statins Activate the Transcriptional Activity of p53 by Regulating the Expression of TAZ" Pharmaceuticals 15, no. 8: 1015. https://doi.org/10.3390/ph15081015