
Paeoniflorin Alleviates Skeletal Muscle Atrophy in Ovariectomized Mice through the ERα/NRF1 Mitochondrial Biogenesis Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
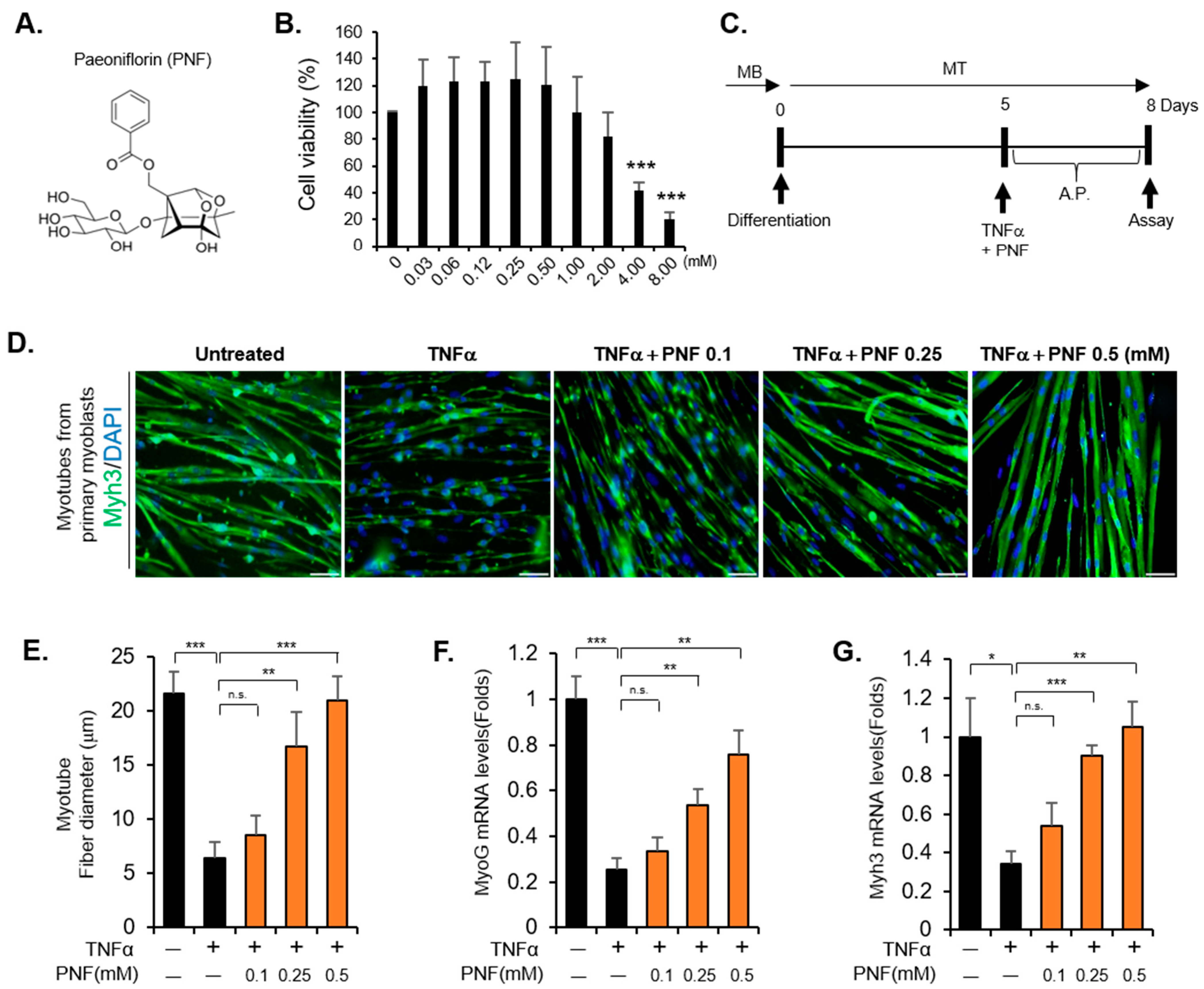
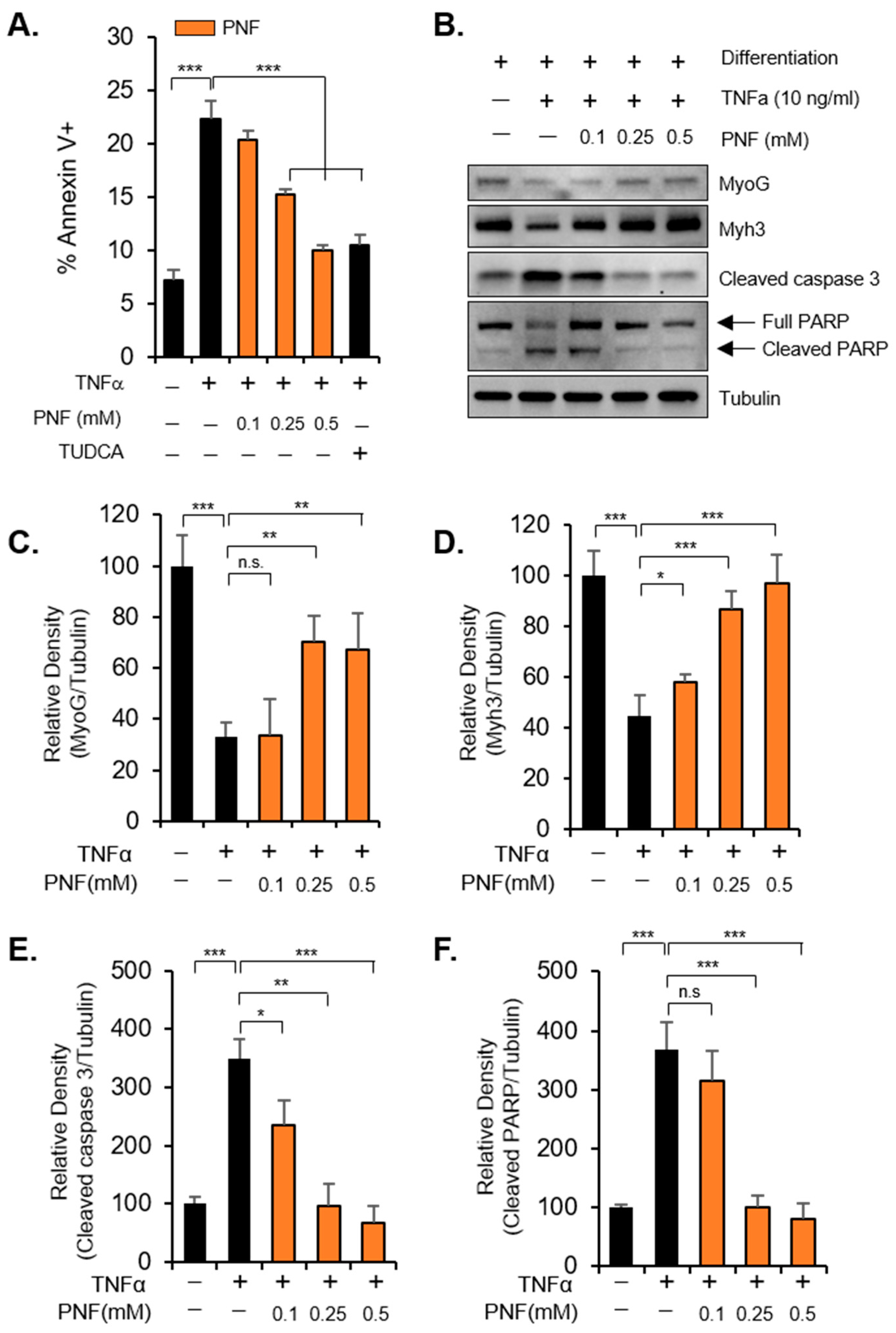
2.1. PNF Restores Atrophied Muscle by Inhibiting Apoptotic Signals Induced by TNFα
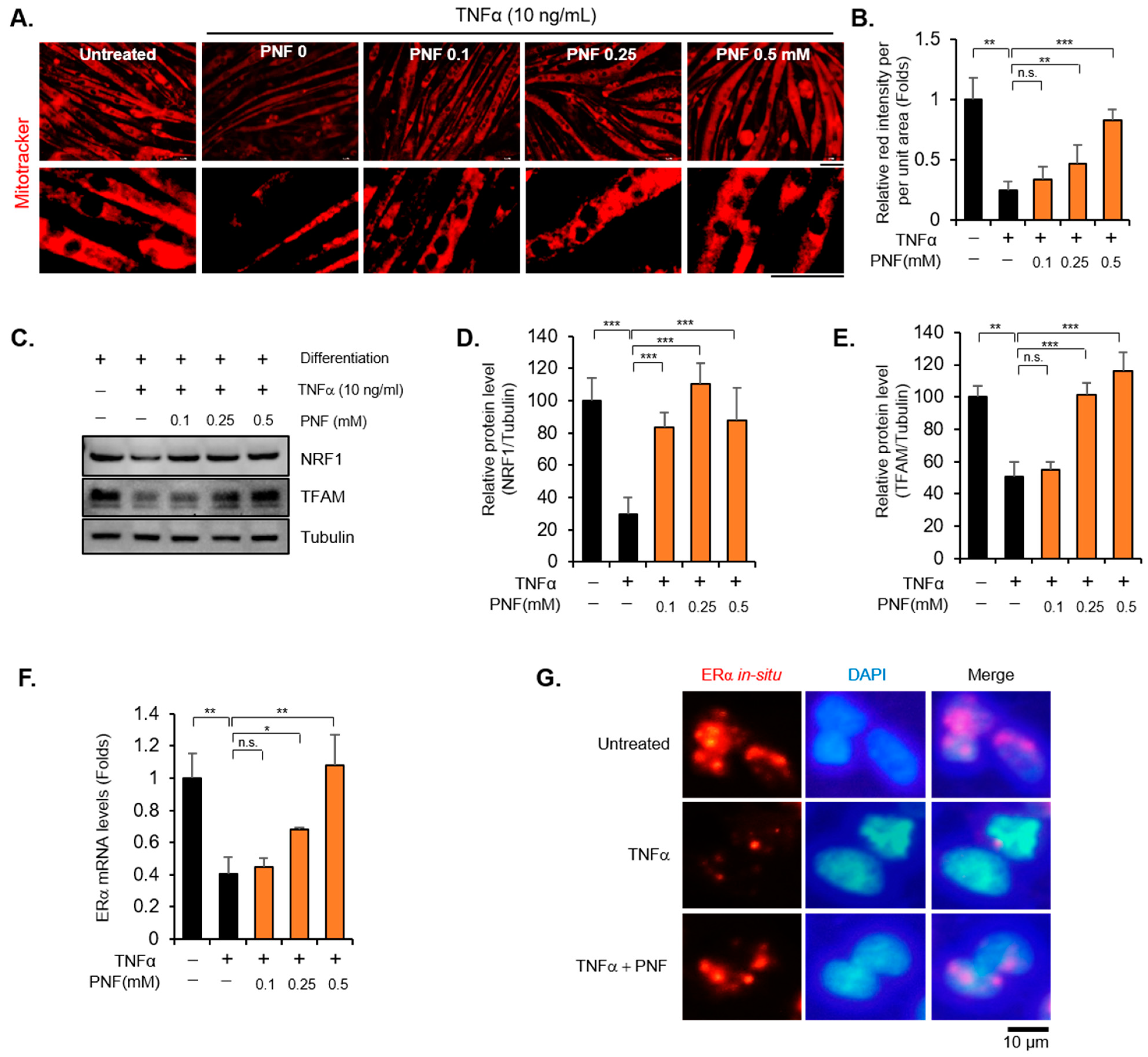
2.2. PNF Increases Mitochondrial Function by Regulating ERα and NRF1
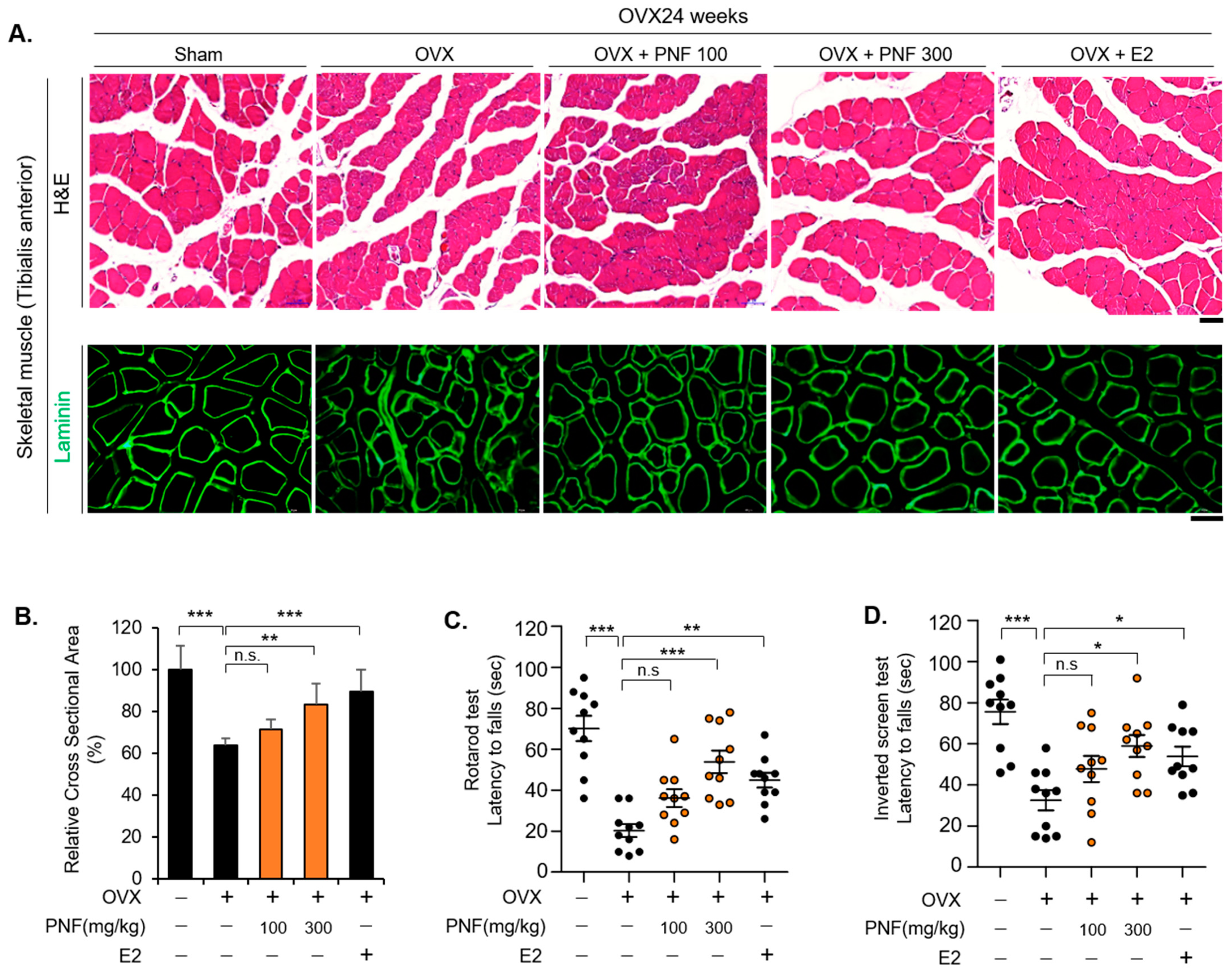
2.3. PNF May Improve Muscle Function by Suppressing Muscle Atrophy of OVX
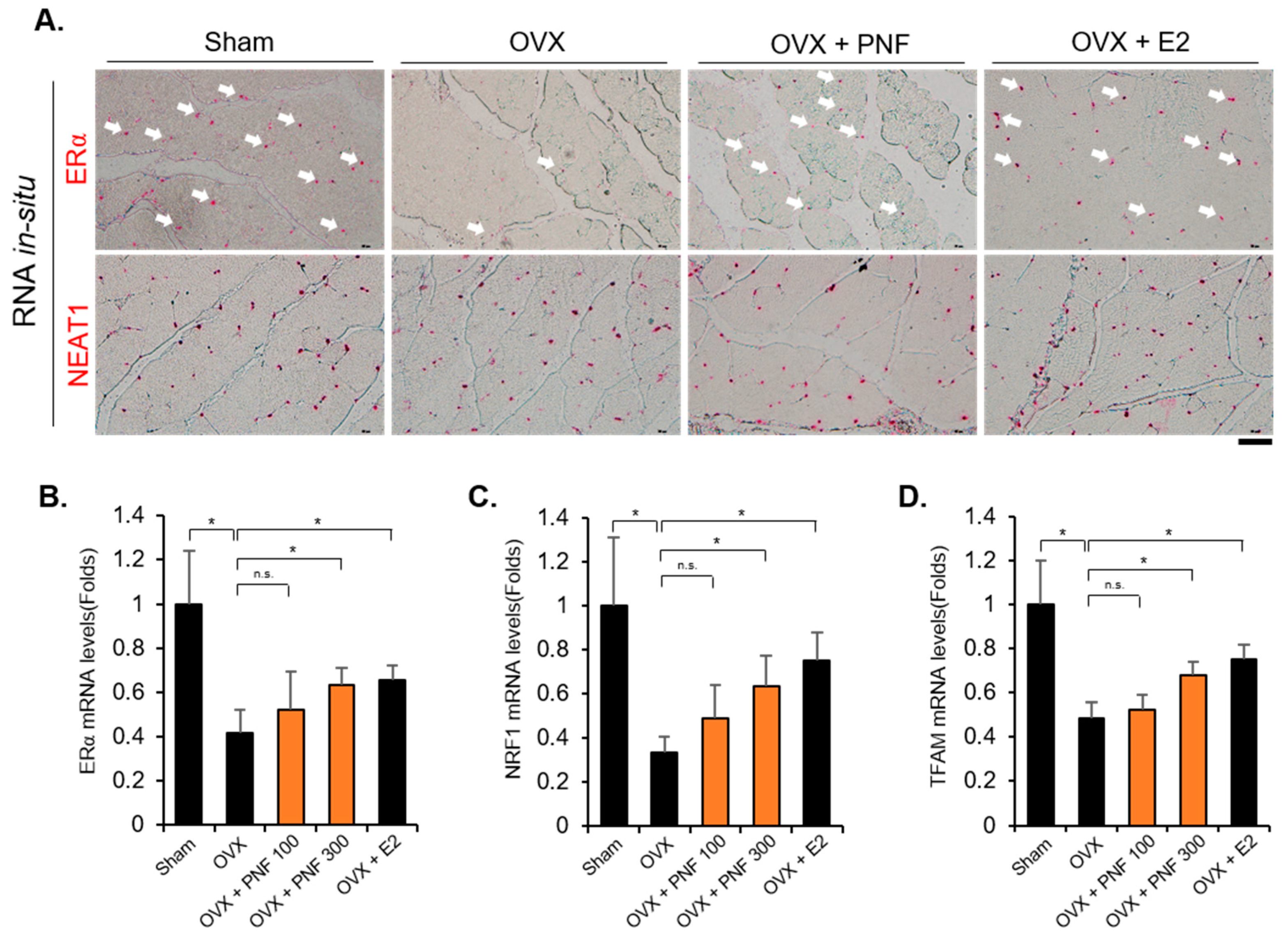
2.4. Reduced ERα in OVX May Be Maintained by PNF
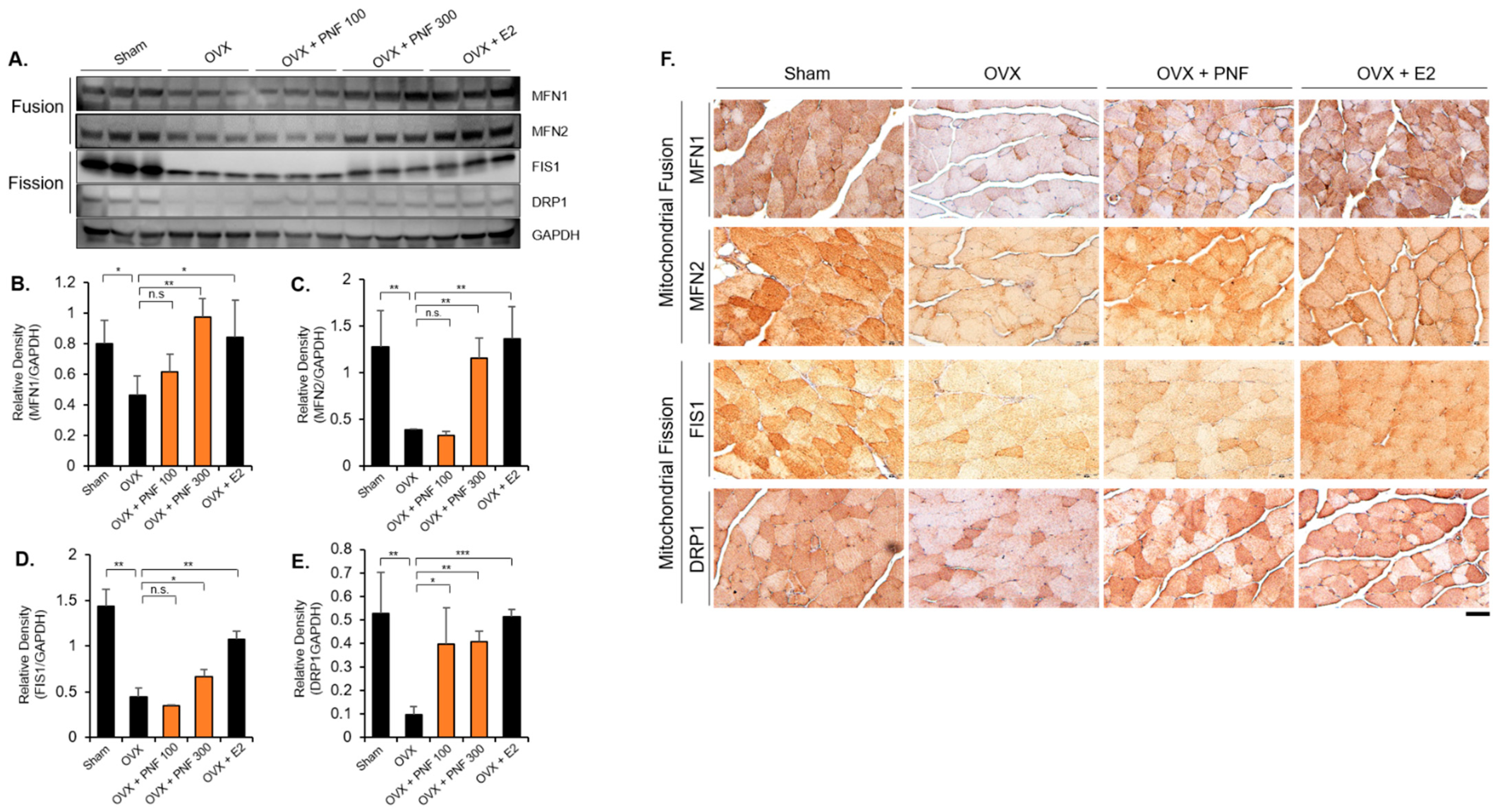
2.5. PNF Increases the Mitochondrial Fusion and Fission Markers of OVX
3. Discussion
4. Material and Methods
4.1. Animals Study Design
4.2. Cell Culture and Reagents
4.3. Cell-Viability Assay
4.4. Quantitative Real-Time PCR for RNA Samples
4.5. Annexin V Staining
4.6. Immunofluorescence
4.7. Immunohistochemistry
4.8. Immunoblotting
4.9. Rotarod and Inverted Screen Test
4.10. RNA In Situ Hybridization
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Campos, N.; Lopez-Pedrosa, J.M.; Rueda, R.; Rodriguez-Mañas, L. Skeletal muscle regulates metabolism via interorgan crosstalk: Roles in health and disease. J. Am. Med. Dir. Assoc. 2016, 17, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, K.L.; Shubert, T.; Doyle, J.; Soher, B.; Sakkas, G.K.; Kent-Braun, J.A. Muscle atrophy in patients receiving hemodialysis: Effects on muscle strength, muscle quality, and physical function. Kidney Int. 2003, 63, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Maltais, M.; Desroches, J.; Dionne, I.J. Changes in muscle mass and strength after menopause. J. Musculoskelet. Neuronal Interact. 2009, 9, 186–197. [Google Scholar] [PubMed]
- Rizzoli, R.; Stevenson, J.C.; Bauer, J.M.; van Loon, L.J.; Walrand, S.; Kanis, J.A.; Cooper, C.; Brandi, M.-L.; Diez-Perez, A.; Reginster, J.-Y. The role of dietary protein and vitamin D in maintaining musculoskeletal health in postmenopausal women: A consensus statement from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Maturitas 2014, 79, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, S.; Narici, M.; Kjaer, M.; Pöllänen, E.; Atkinson, R.A.; Hansen, M.; Kovanen, V. Sex hormones and skeletal muscle weakness. Biogerontology 2013, 14, 231–245. [Google Scholar] [CrossRef]
- Tiidus, P.M.; Lowe, D.A.; Brown, M. Estrogen replacement and skeletal muscle: Mechanisms and population health. J. Appl. Physiol. 2013, 115, 569–578. [Google Scholar] [CrossRef]
- Kitajima, Y.; Ono, Y. Estrogens maintain skeletal muscle and satellite cell functions. J. Endocrinol. 2016, 229, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Capllonch-Amer, G.; Sbert-Roig, M.; Galmes-Pascual, B.M.; Proenza, A.M.; Llado, I.; Gianotti, M.; Garcia-Palmer, F.J. Estradiol stimulates mitochondrial biogenesis and adiponectin expression in skeletal muscle. J. Endocrinol. 2014, 221, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Thoma, A.; Lightfoot, A.P. NF-kB and inflammatory cytokine signalling: Role in skeletal muscle atrophy. In Muscle Atrophy Advances in Experimental Medicine and Biology; Xiao, J., Ed.; Springer: Singapore, 2018; pp. 267–279. [Google Scholar]
- Li, Y.-P.; Reid, M.B. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am. J. Phy. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Kny, M.; Riediger, F.; Busch, K.; Schmidt, S.; Luft, F.C.; Slevogt, H.; Fielitz, J. Deletion of Nlrp3 protects from inflammation-induced skeletal muscle atrophy. Intensive Care Med. Exp. 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.-S.; Kim, H.J.; Hwang, J.T.; Ko, B.S. Dioscorea nipponica extracts enhance recovery from skeletal muscle atrophy by suppressing NF-κB expression. J. Funct. Foods 2020, 73, 104109. [Google Scholar] [CrossRef]
- Jackman, R.W.; Kandarian, S.C. The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2004, 287, C834–C843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.O.; Ko, W.K.; Kim, J.Y.; Ro, H.S. Paeoniflorin: An antihyperlipidemic agent from Paeonia lactiflora. Fitoterapia 2004, 75, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, Y.; Li, S.; Shi, J. A new paeoniflorin derivative isolated from the root bark ethanol extract of Paeonia suffruticosa. Zhongguo Zhong Yao Zhi 2005, 30, 759–761. [Google Scholar]
- Xin, Q.; Yuan, R.; Shi, W.; Zhu, Z.; Wang, Y.; Cong, W. A review for the anti-inflammatory effects of paeoniflorin in inflammatory disorders. Life Sci. 2019, 237, 116925. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, W.; Jiang, Y.; Wen, J.; Wei, S.; Zhao, Y. Paeoniflorin, a natural product with multiple targets in liver diseases—a mini review. Front. Pharmacol. 2020, 11, 531. [Google Scholar] [CrossRef]
- Wang, S.; Jia, D.; Lu, H.; Qu, X. Paeoniflorin improves myocardial injury via p38 MAPK/NF-KB p65 inhibition in lipopolysaccharide-induced mouse. Ann. Transl. Med. 2021, 9, 18. [Google Scholar] [CrossRef]
- Guo, R.-B.; Wang, G.-F.; Zhao, A.-P.; Gu, J.; Sun, X.-L.; Hu, G. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-κB-mediated inflammatory responses. PLoS ONE 2012, 7, e49701. [Google Scholar] [CrossRef]
- Tohnai, G.; Adachi, H.; Katsuno, M.; Doi, H.; Matsumoto, S.; Kondo, N.; Miyazaki, Y.; Iida, M.; Nakatsuji, H.; Qiang, Q. Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2014, 23, 3552–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhao, J.; Jiang, L.; Xie, Y.; Xia, Y.; Lv, R.; Dong, L. Paeoniflorin improves menopause depression in ovariectomized rats under chronic unpredictable mild stress. Int. J. Clin. Exp. Med. 2015, 8, 5103. [Google Scholar]
- Takeuchi, T.; Nishii, O.; Okamura, T.; Yaginuma, T. Effect of paeoniflorin, glycyrrhizin and glycyrrhetic acid on ovarian androgen production. Am. J. Chin. Med. 1991, 19, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.; Cheng, J.; Jin, X.; Lao, W.; Johnson, M.; Tan, Y.; Qu, X. Paeoniflorin extract reverses dexamethasone-induced testosterone over-secretion through downregulation of cytochrome P450 17A1 expression in primary murine theca cells. J. Ethnopharmacol. 2019, 229, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef]
- Dirks, A.J.; Leeuwenburgh, C. The role of apoptosis in age-related skeletal muscle atrophy. Sports Med. 2005, 35, 473–483. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, T.C.; Germain, D. mtDNA, metastasis, and the mitochondrial unfolded protein response (UPRmt). Front. Cell. Dev. Biol. 2017, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esqueda, M.E.D.; Craig, T.; Hinojosa-Laborde, C. Effect of ovariectomy on renal estrogen receptor-α and estrogen receptor-β in young salt-sensitive and-resistant rats. Hypertension 2007, 50, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-Y.; Chen, J.-S.; Wu, X.-B.; Shyu, W.-C.; Chaunchaiyakul, R.; Zhao, X.-L.; Kuo, C.-H.; Cheng, Y.-J.; Yang, A.-L.; Lee, S.-D. Combined effects of 17β-estradiol and exercise training on cardiac apoptosis in ovariectomized rats. PLoS ONE 2018, 13, e0208633. [Google Scholar] [CrossRef]
- Wang, S.; Zuo, H.; Jin, J.; Lv, W.; Xu, Z.; Fan, Y.; Zhang, J.; Zuo, B. Long noncoding RNA Neat1 modulates myogenesis by recruiting Ezh2. Cell Death Dis. 2019, 10, 505. [Google Scholar] [CrossRef] [Green Version]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Leduc-Gaudet, J.-P.; Hussain, S.N.; Barreiro, E.; Gouspillou, G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef]
- Collins, B.C.; Laakkonen, E.K.; Lowe, D.A. Aging of the musculoskeletal system: How the loss of estrogen impacts muscle strength. Bone 2019, 123, 137–144. [Google Scholar] [CrossRef]
- Fragala, M.S.; Cadore, E.L.; Dorgo, S.; Izquierdo, M.; Kraemer, W.J.; Peterson, M.D.; Ryan, E.D. Resistance training for older adults: Position statement from the national strength and conditioning association. J. Strength Cond. Res. 2019, 33, 2019–2052. [Google Scholar] [CrossRef]
- Cohen-Solal, M.; Graulet, A.; Denne, M.; Gueris, J.; Baylink, D.; De Vernejoul, M. Peripheral monocyte culture supernatants of menopausal women can induce bone resorption: Involvement of cytokines. J. Clin. Endocrinol. Metab. 1993, 77, 1648–1653. [Google Scholar] [PubMed]
- Bertone-Johnson, E.R.; Manson, J.E.; Purdue-Smithe, A.C.; Hankinson, S.E.; Rosner, B.A.; Whitcomb, B.W. A prospective study of inflammatory biomarker levels and risk of early menopause. Menopause 2019, 26, 32. [Google Scholar] [CrossRef] [PubMed]
- Calvani, R.; Joseph, A.-M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 2013, 394, 393–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [Green Version]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]
- Langen, R.C.; Van Der Velden, J.L.; Schols, A.M.; Kelders, M.C.; Wouters, E.F.; Janssen-Heininger, Y.M. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 2004, 18, 227–237. [Google Scholar] [CrossRef]
- Langen, R.C.; Schols, A.M.; Kelders, M.C.; Wouters, E.f.; Janssen-heininger, Y.M. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-κB. FASEB J. 2001, 15, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, L.; Morlá, M.; Iglesias, A.; Busquets, X.; Lladó, J.; Olmos, G. IFN-γ prevents TNF-α-induced apoptosis in C2C12 myotubes through down-regulation of TNF-R2 and increased NF-κB activity. Cell. Signal. 2005, 17, 1333–1342. [Google Scholar] [CrossRef]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.-S.; Mitra, A.; Rahat, B.; Kim, K.; Pfeifer, K. Loss of imprinting mutations define both distinct and overlapping roles for misexpression of IGF2 and of H19 lncRNA. Nucleic Acids Res. 2017, 45, 12766–12779. [Google Scholar] [CrossRef]
- Bajaj, P.; Reddy Jr, B.; Millet, L.; Wei, C.; Zorlutuna, P.; Bao, G.; Bashir, R. Patterning the differentiation of C2C12 skeletal myoblasts. Integr. Biol. 2011, 3, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Naz, F.; Juan, A.H.; Dell’Orso, S.; Sartorelli, V. Identification of skeletal muscle satellite cells by immunofluorescence with pax7 and Laminin Antibodies. J. Vis. Exp. 2018, 134, e57212. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; De Bari, C. Immunostaining of skeletal tissues. In Bone Research Protocols; Springer: New York, NY, USA, 2019; pp. 437–450. [Google Scholar]
- Liu, W.; Han, F.; Qu, S.; Yao, Y.; Zhao, J.; Akhtar, M.L.; Ci, Y.; Zhang, H.; Li, H.; Zhao, Y. MARVELD1 depletion leads to dysfunction of motor and cognition via regulating glia-dependent neuronal migration during brain development. Cell Death Dis. 2018, 9, 999. [Google Scholar] [CrossRef] [PubMed]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Measuring the strength of mice. J. Vis. Exp. 2013, e2610. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, K.; Park, K.-S.; Rahat, B.; Yu, Z.-X.; Noeker, J.; Mitra, A.; Knutsen, R.; Springer, D.; Kozel, B. Cardiac pathologies in mouse loss of imprinting models are due to misexpression of H19 long noncoding RNA. eLife 2021, 10, e67250. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-S.; Kim, H.; Kim, H.J.; Lee, K.-I.; Lee, S.-Y.; Kim, J. Paeoniflorin Alleviates Skeletal Muscle Atrophy in Ovariectomized Mice through the ERα/NRF1 Mitochondrial Biogenesis Pathway. Pharmaceuticals 2022, 15, 390. https://doi.org/10.3390/ph15040390
Park K-S, Kim H, Kim HJ, Lee K-I, Lee S-Y, Kim J. Paeoniflorin Alleviates Skeletal Muscle Atrophy in Ovariectomized Mice through the ERα/NRF1 Mitochondrial Biogenesis Pathway. Pharmaceuticals. 2022; 15(4):390. https://doi.org/10.3390/ph15040390
Chicago/Turabian StylePark, Ki-Sun, Hyungjun Kim, Hye Jin Kim, Kang-In Lee, Seo-Young Lee, and Jieun Kim. 2022. "Paeoniflorin Alleviates Skeletal Muscle Atrophy in Ovariectomized Mice through the ERα/NRF1 Mitochondrial Biogenesis Pathway" Pharmaceuticals 15, no. 4: 390. https://doi.org/10.3390/ph15040390