B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders


1
Department of Neurology, University Medical Center, 37075 Göttingen, Germany
2
Department of Cardiology, University Medical Center, 97080 Würzburg, Germany
3
Institute of Neuropathology, University Medical Center, 37075 Göttingen, Germany
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(1), 37; https://doi.org/10.3390/ph14010037
Submission received: 10 December 2020
/
Revised: 2 January 2021
/
Accepted: 3 January 2021
/
Published: 6 January 2021
(This article belongs to the Special Issue Therapeutic Agents for Neurological Disorders)
Abstract
:The first description of neuromyelitis optica by Eugène Devic and Fernand Gault dates back to the 19th century, but only the discovery of aquaporin-4 autoantibodies in a major subset of affected patients in 2004 led to a fundamentally revised disease concept: Neuromyelits optica spectrum disorders (NMOSD) are now considered autoantibody-mediated autoimmune diseases, bringing the pivotal pathogenetic role of B cells and plasma cells into focus. Not long ago, there was no approved medication for this deleterious disease and off-label therapies were the only treatment options for affected patients. Within the last years, there has been a tremendous development of novel therapies with diverse treatment strategies: immunosuppression, B cell depletion, complement factor antagonism and interleukin-6 receptor blockage were shown to be effective and promising therapeutic interventions. This has led to the long-expected official approval of eculizumab in 2019 and inebilizumab in 2020. In this article, we review current pathogenetic concepts in NMOSD with a focus on the role of B cells and autoantibodies as major contributors to the propagation of these diseases. Lastly, by highlighting promising experimental and future treatment options, we aim to round up the current state of knowledge on the therapeutic arsenal in NMOSD.
1. Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are a heterogeneous group of monophasic or recurrent autoinflammatory diseases of the central nervous system (CNS). Characteristically, symptoms like severe loss of vision, weakness or paralysis of extremities, loss of sensation, bowel and bladder dysfunction, area postrema clinical syndrome or respiratory failure are the result of rapidly sequential or bilateral optic neuritis, longitudinally extensive myelitis and inflammatory brain lesions affecting the circumventricular organs [1]. The first description of a patient suffering from amaurosis associated with autoptic-proven myelitis by Antoine Portal dates back to the year 1804, while the systematic description of ‘myelitis optica’ by Devic and Gault appeared by the end of the 19th century [2]. For many years, NMOSD were considered prognostically unfavorable variants of multiple sclerosis. When specific pathogenic aquaporin-4 (AQP4) autoantibodies were detected in a major subset of patients at the beginning of the 21st century, NMOSD were finally considered to belong to a disease entity distinct from multiple sclerosis [3].
According to the current diagnostic guidelines of 2015, the presence of at least one core manifestation is sufficient for NMOSD diagnosis in anti-AQP4 seropositive patients, which are now considered classical neuromyelitis optica patients, while two manifestations including optic neuritis, myelitis or area postrema syndrome are required for seronegative patients [4]. Within the population of AQP4 negative patients, a subset has been identified expressing antibodies against myelin oligodendrocyte glycoprotein (MOG) [5]. Based on clinical, radiological and immunologic features, this condition, now called ‘MOG-immunoglobulin G (IgG)-associated encephalomyelitis’, is considered a separate disease entity, distinct from AQP4-positive neuromyelitis optica as well as from multiple sclerosis [6,7].
2. The Role of B Cells and Antibodies in NMOSD
The integral transmembrane protein AQP4 was discovered in 1984 and is capable of conducting water through the cell membrane. It is the most abundant type of water channel in the CNS, located specifically at the astrocytic foot processes, which form the blood-brain barrier (BBB), but also at ependyma and glia limitans [8]. Outside the CNS, it can be found in lower concentrations on the epithelial cells of many organs like kidney and stomach, but AQP4 tetramers aggregate different than within the CNS [9]. According to the current disease concept of seropositive NMOSD, autoantibodies directly targeting AQP4 penetrate the BBB or derive from peripheral plasmablasts within the CNS: Pathogenic binding of AQP4-IgG to AQP4 causes complement-dependent cytotoxicity and subsequent chemotaxis of eosinophils, neutrophils, macrophages and lymphocytes. This leads to a direct destruction of astrocytes with a secondary, irreversible demise of oligodendrocytes, axons and neurons [10]. In contrast to multiple sclerosis, cortical demyelination is only seen after years of ongoing disease activity [11]. Serum AQP4 autoantibody titers were shown to correlate with the size of spinal cord lesions, clinical disease activity and therapeutic response, as they decrease after immunotherapy and remain low during remissions [12].
Since antibody-producing cells like plasmablasts and plasma cells are derived from B lymphocytes, the role of this very lymphocyte compartment is crucial for the pathophysiology of NMOSD [13]. This goes along with the finding that in affected patients, the number of antibody-producing plasmablasts is strongly elevated in the peripheral blood, peaking at relapses [14]. It is worth noting that, in contrast to multiple sclerosis, plasmablasts can barely be detected in the cerebrospinal fluid (CSF) of NMOSD patients [15,16]. Accordingly, NMOSD is now considered a humoral autoimmune disorder, where anti-AQP4 antibodies are mainly produced in the periphery [10,17]. In line, oligoclonal bands are rarely seen in NMOSD patients, often disappearing during disease development [8]. An inflammation-induced opening of the BBB is supposed to be a prerequisite for the entry of peripherally secreted autoantibodies into the CNS. This is supported by the observation that a subset of NMOSD patients shows signs of viral infections just prior to clinical relapses [18]. Astonishingly, the permeability of the BBB has also been reported to be increased by IgG antibodies themselves [19]. Upon binding to their target on the astrocytic endfeet, AQP4 antibodies trigger the complement cascade, leading to the formation of a membrane attack complex with subsequent astrocytic edema, dysfunction, destruction and secondary neuronal injury [20]. It must be noted that T cells are also thought to be required for the disruption of the BBB and the orchestration of the immune attack in the CNS [21].
Besides antibody-mediated effects, B lymphocytes also inherit two potent, cellular functions contributing to disease activity in NMOSD: Firstly, activated B cells are capable of secreting pro-inflammatory cytokines like interleukin-(IL-)6 and tumor necrosis factor, further fostering inflammatory processes, especially by generating T helper 17 cells [22,23]. Interestingly, IL-6 in turn also supports B cell activation and growth [23]. NMOSD patients indeed display elevated IL-6 in the CSF along with an elevated number of circulating T helper 17 and IL-17-producing cytotoxic T cells [21,24]. Secondly, B lymphocytes act as antigen-presenting cells by specifically binding antigens on their B cell receptor, before internalizing, processing and presenting them to T helper cells via major histocompatibility factor II molecules on their surface [25]. It was shown that the antigen-presenting function of B cells is required for the induction of experimental CNS autoimmunity independent of their humoral involvement [26].
In conclusion, B cells uniquely contribute to disease progression in NMOSD by the provision of autoantibodies, the secretion of cytokines and the presentation of antigen. This placed in focus, we here review the interactions of rapidly evolving current and future NMOSD pharmaceuticals with B lymphocytes and autoantibodies.
3. Current and Evolving Therapeutic Strategies in NMOSD
Until very recently, off-label therapies were the only treatment options in NMOSD with unknown benefits and side effects. The choice of an appropriate therapy in order to prevent clinical relapses is essential to avoid irreversible disability progression, especially as recovery from clinical relapses is often incomplete in NMOSD [27]. Unfortunately, several effective drugs in the treatment of multiple sclerosis are not effective or even harmful when deliberately or unintendingly prescribed to NMOSD patients (alemtuzumab, dimethyl fumarate, fingolimod, glatiramer acetate, interferon-β and natalizumab) [28,29]. This emphasizes the importance of professional and correct diagnostic procedures before the initiation of therapeutic interventions in neuroinflammatory diseases.
The following section reviews current and potential future pharmaceutical treatment options in NMOSD with a focus on B cells, which, as shown above, are thought to be major contributors to NMOSD pathophysiology. While general immunosuppression used to be the standard therapy for many years, recently evolving treatment options include the targeted depletion of B cells, IL-6, complement antagonism and many more. Table 1 summarizes the currently available data on clinical trials in NMOSD, Figure 1 provides a graphical overview of the most important treatment strategies.
3.1. Immunosuppression
Although the involvement of specific AQP4 antibodies in NMOSD was uncovered only 16 years ago, an autoimmune pathogenesis of NMOSD was assumed before and several immunosuppressive agents have been tested within the last decades [30]. However, due to inconsistent diagnostic criteria, small case and open label studies, no formal approval for NMOSD treatment was granted to any of those substances.
3.1.1. Glucocorticoids
In NMOSD, acute relapses are usually treated for three to five days with a pulsed intravenous methylprednisolone therapy. In severe and steroid refractory cases, plasma exchange alone or in combination with glucocorticoids is often applied as escalation therapy [31,32]. Sometimes, a prolonged oral methylprednisolone therapy is used to ‘bridge’ patients until preventive immunomodulatory or immunosuppressive treatment with one of the below mentioned agents is initiated and effective.
Glucocorticoids cause an acute lymphopenia due to a redistribution of lymphocytes to lymph nodes, spleen and bone marrow [33,34]. While no immediate change in the number of circulating B cells was observed, they were found to be slightly reduced upon prolonged glucocorticoid administration [35,36]. Interestingly, a recent study revealed a decrease of naïve B cells and an increase of memory B cells after six months of glucocorticoid treatment in IgG4-related disease [37]. However, the number of circulating plasmablasts decreased upon treatment, possibly explaining beneficial effects in NMOSD [38]. Accumulating data demonstrate that glucocorticoids down-regulate interleukin-6 serum levels [39]. It has been reported that IgG and IgA levels decrease by up to 20 percent over the first few weeks of short-term moderate-to-high dose glucocorticoid treatment, then return to normal levels over weeks to months [40,41]. In immune thrombocytopenic purpura, glucocorticoid treatment significantly reduced B cell activating factor (BAFF) serum levels after long-term treatment [42].
3.1.2. Azathioprine
Although not officially approved, this widely-used purine synthesis inhibitor is traditionally considered a first-line preventive treatment of NMOSD due to substantially reduced relapse rates and disability after long-term treatment [43,44]. However, poor tolerability and a delayed mode of action cause discontinuation rates of up to 50% after 18 months of treatment [45].
Its active metabolites methylthioinosine monophosphate and thiodeoxyguanosine triphosphate inhibit purine synthesis in metabolic active cells, causing a dose-dependent decrease of total lymphocyte and B cell counts for several weeks to months [46,47,48]. While plasmablast counts seem to remain unchanged, transitional and memory B cell counts as well as IL-6 and immunoglobulin serum levels were observed to significantly drop upon azathioprine treatment [49,50,51,52,53]. The opposite is the case for serum BAFF levels, which were reported to increase in patients receiving azathioprine [54].
3.1.3. Cyclophosphamide
In a limited number of patients, this alkylate showed beneficial effects in case reports and a small retrospective study of four patients with a median improvement in the expanded disability status scale (EDSS) score from 8.0 to 5.75 [55,56]. In a case report about a patient with systemic lupus erythematosus-associated NMOSD, cyclophosphamide successfully halted relapses [57]. However, other studies report adverse effects in 80% of treated patients with no significant improvement of the EDSS score [58]. This explains why cyclophosphamide is only an alternative choice for very difficult cases [59].
Cyclophosphamide is immunosuppressive but not myeloablative as the active metabolite phosphoramide mustard forms deoxyribonucleic acid crosslinks in cells with low levels of the differentially expressed enzyme aldehyde dehydrogenase [60,61]. Along with strongly decreased lymphocyte and B cell counts, an inhibition of B cell activation, proliferation and differentiation was observed after intravenous cyclophosphamide administration. After longer treatment periods, cyclophosphamide was also found to reduce memory B cell and plasmablast counts [62,63,64]. Hypogammaglobulinemia in systemic lupus erythematosus (SLE) and increased BAFF serum levels in cancer chemoimmunotherapy are thought to be a consequence of cyclophosphamide treatment [65].
3.1.4. Mitoxantrone
According to the Neuromyelitis Optica Study Group, this type II topoisomerase inhibitor may be considered as second-line treatment [66]. Mitoxantrone improved EDSS scores in a small study with only five patients, but two patients experienced a relapse [67]. In a more recent, larger study with 20 patients and in line with another report, median annualized relapse rates declined from 2.8 to 0.7 and mean EDSS scores declined from 5.6 to 4.4 under mitoxantrone treatment [68,69].
By disrupting deoxyribonucleic acid synthesis and repair, this intravenous agent causes a strong decrease of lymphocyte, B cell and memory B cell counts [70,71,72,73]. While tumor necrosis factor alpha production of B cells was inhibited, the proportion of IL-10-producing regulatory B cell subsets was increased in mitoxantrone-treated patients [74]. Antibody-production was shown to be reduced and BAFF levels were increased in two other reports [74,75].
3.1.5. Mycophenolate Mofetil
Although this agent requires an extended period of time to affect the immune system and is discontinued by a significant proportion of treated patients due to side effects, mycophenolate mofetil effectively reduced relapse rates and EDSS after 24 months in a prospective study with 67 patients [76]. Similar observations were made in a retrospective study with 59 included patients [77]
By inhibiting the enzyme inosine-5′-monophosphate dehydrogenase, this agent suppresses the proliferation of B and T lymphocytes, causing a reduction of lymphocyte and B cell counts [78]. In vitro, antibody formation by polyclonally activated human B lymphocytes was almost completely inhibited by its active metabolite [79]. In another study, its active metabolite inhibited T cell-induced B cell activation, B cell expansion and plasma cell differentiation in vitro. Moreover, the authors found that inhibition of B cell activation and differentiation was not due to an induction of cell death but rather due to a cell cycle arrest. However, terminally differentiated plasma cells were not susceptible to inhibition as they express reduced levels of inosine-5′-monophosphate dehydrogenase. B cells for these experiments were isolated from rheumatoid arthritis patients [80]. Similar findings in patients with systemic lupus erythematosus showed that mycophenolic acid counteracts B cell proliferation and plasmablast formation [81]. In an experimental murine model of colitis, mycophenolate mofetil promoted down-regulation of expanded B cells and production of TNF-α [82]. In SLE patients treated with mycophenolate mofetil, serum BAFF levels were increased [54].
3.1.6. Methotrexate
In a small observational study including 51 patients, methotrexate dramatically decreased the frequency of relapses [69]. According to two other retrospective observational case series, the median annualized relapse rate was significantly reduced following treatment, suggesting that methotrexate is a good alternative to azathioprine [83,84]. In a retrospective assessment of treatment-recalcitrant, fulminant inflammatory CNS syndromes including NMOSD, treatment with high doses of methotrexate was observed to be safe and highly effective, producing a rapid and nearly complete cessation of disease activity [85].
Apart from the killing of proliferating cells through folate antagonism, potentially relevant modes of action of methotrexate in the treatment of autoinflammatory diseases include the inhibition of enzymes involved in purine metabolism, the inhibition of T cell activation, the selective downregulation of B cells and an increased cluster of differentiation (CD) 95 sensitivity of activated T cells [86]. Lately, circulating regulatory B cells were proposed as biomarkers for the therapeutic response to methotrexate in early rheumatoid arthritis [87]. Methotrexate has been associated with the inhibition of pro-inflammatory cytokines such as IL-1, IL-6 and tumor necrosis factor (TNF)-α [88]. In systemic lupus erythematosus, methotrexate treatment increased BAFF serum levels [54].
3.1.7. Intravenous Immunoglobulins
These preparations, comprised of pooled IgG antibodies from the serum of thousands of donors, have been proven to be effective in some antibody-mediated autoimmune diseases [89]. In a study with eight patients, it was observed that the mean relapse rate and the EDSS score declined significantly upon intravenous IgG treatment in NMOSD patients [90]. Another study with only six patients found a significant reduction of relapses, while EDSS scores did not change upon therapy with intravenous IgG [91]. For acute relapses, a sole intravenous IgG infusion was not recommended as first-line therapy option, while adding high-dosed steroids to immunoglobulin therapy was superior to high-dose steroids alone in patients with high onset EDSS scores [92].
After infusion, a reduction of lymphocyte counts has been reported [93]. In patients with Guillain–Barré syndrome, B cell counts were not significantly altered by intravenous immunoglobulins [94]. Another report observed a weak decrease in blood B cell numbers accompanied with a reduced B cell proliferation [95,96]. It was shown that the frequency of plasmablasts was temporarily elevated, peaking one week after infusion, while the frequency of interleukin-6 producing B cells and the expression of CD25, CD40, CD80 and major histocompatibility complex class II on B cells was decreased [94,97,98,99]. In line, the antigen-presenting function of B cells was found to be inhibited by intravenous IgG treatment [100]. While the frequency of memory B cells remained unaltered, IL-10 producing regulatory B cells were elevated and interleukin-6 serum levels decreased [94,101,102]. Inhibition of B cell immunoglobulin production by intravenous IgG was first described in vitro [103]. In addition, decreased IgE serum levels were observed in children with asthma after intravenous IgG treatment [104]. Interestingly, intravenous immunoglobulins were shown to contain certain IgG antibodies that were able to recognize BAFF. After specific antibody binding to BAFF, steric hindrance prevented the antiapoptotic effects of BAFF on B lymphocytes [105].
3.2. Anti-CD20 Antibodies
3.2.1. Rituximab
This widely-used monoclonal antibody binds to the CD20 surface protein on B cells causing antibody-dependent cell-mediated cytotoxicity as well as complement-dependent cytotoxicity [122]. Studies have shown profound beneficial effects on the annualized relapse rate and disability progression in NMOSD [123,124]. In a recent meta-analysis with 577 patients, the mean annualized relapse rate ratio was diminished by 1.56 and the mean EDSS score was reduced by -1.16 after rituximab therapy [125,126]. At the same time, rituximab therapy showed an overall good safety profile and an acceptable tolerability. However, one must also mention serious safety issues, including hypogammaglobinemia and prolonged neutropenia after long-term use [125].
Usually, a fast and complete depletion of circulating B cells in the blood compartment is observed upon treatment with rituximab, but the time course of B cell repopulation can vary considerably between patients. In most patients, absolute blood B cell counts are still low six to nine months after infusion [127,128]. Interestingly, it was recently shown that in the reappearing B cell pool, the frequency of immature B cells and the expression of activation markers was found to be increased [129,130]. Mature plasmablasts and plasma cells are largely preserved as they lack CD20 expression [127]. In SLE and rheumatoid arthritis patients, BAFF levels were found to be elevated during B-cell depletion and declined upon B-cell repopulation [131,132]. B cells returning after depletion secreted reduced levels of IL-6 in humans and IL-6 serum levels were shown to be unaffected six months after B cell depletion [22,133]. Further, the depletion of regulatory B cells was associated with an enhanced secretion of pro-inflammatory cytokines by CD11b+ antigen-presenting cells [134].
3.2.2. Ocrelizumab
This humanized anti-CD20 antibody has been recently approved for the treatment of relapsing-remitting MS and binds to an epitope that overlaps the one rituximab binds [122,128]. To date, there is no published evidence regarding the treatment of NMOSD with ocrelizumab, but the same mode of action suggests a similar effectivity. As in rituximab, decreases of immunoglobulin levels were seen in ocrelizumab-treated patients [135].
B-cell-depleting therapy after ocrelizumab infusion with an interval of at least 5 months led to transiently reduced absolute lymphocyte counts, which recovered after the second cycle [126]. Effects of ocrelizumab on BAFF or IL-6 levels have not been published so far.
3.2.3. Ofatumumab
This entirely human anti-CD20 antibody targets an epitope completely distinct from that of rituximab. Preceding its approval in the treatment of chronic lymphocytic leukemia, ofatumumab has been broadly investigated upon in the field of hematology and there are several ongoing studies regarding the treatment of B cell malignancies [136]. Besides an intravenous application, a subcutaneous route of administration has been tested in multiple sclerosis and rheumatoid arthritis [137,138]. In the MIRROR study, a dose-dependent B cell depletion after treatment with subcutaneous ofatumumab was observed and the efficacy of incomplete B cell depletion in multiple sclerosis was postulated [137]. Regarding NMOSD, there is a promising report about a marked reduction in relapse rate in a single patient [139].
3.2.4. Ublituximab
This novel monoclonal antibody is directed against a unique epitope of the CD20 antigen. It was glycoengineered for enhanced B cell targeting through antibody-dependent cellular cytotoxicity, which allows lower doses and shorter infusion times in comparison with other anti-CD20 monoclonal antibodies [140]. In a first Phase 1 open-label, standard-of-care, single treatment arm, unblinded, single center interventional trial including NMOSD patients, experimental subjects received one infusion of 450 mg of intravenous ublituximab at the onset of an NMOSD exacerbation in addition to standard of care treatment with daily intravenous glucocorticoids at a dose of 1000 mg for five days. B cell depletion for two months was achieved in four of five patients along with improved disability status scores [108].
3.2.5. BAT4406F
BAT4406F is a novel, fully humanized anti-CD20 monoclonal antibody. In an ongoing, Phase 1, open-label, dose-escalation study in NMOSD patients, the overall objective is to assess the safety, tolerability, and pharmacokinetics of BAT4406F injection in NMOSD patients (NCT04146285).
3.3. Targeting CD19
3.3.1. Inebilizumab
Targeting CD19, which is expressed for longer time on differentiated B cells than CD20, this antibody leads to a broad depletion of late B cell stages including plasmablasts and early plasma cells, which are thought to produce autoantibodies in NMOSD [141]. However, it must be noted, that CD19 is not expressed on terminally differentiated plasma cells. In a recent phase 2/3 trial, inebilizumab remarkably reduced the relapse rate by 73% in both seropositive and seronegative NMOSD patients and reduced disability worsening by 15.5% along with a favorable safety and tolerability profile [115]. In June 2020, inebilizumab received its first global approval in the USA for the treatment of NMOSD in adult seropositive patients [142].
3.3.2. Tandem Chimeric Antigen Receptors (CAR) T Cells Targeting CD19 and CD20
Combining both antigen-binding and T-cell activating functions into a single receptor, CAR are receptor proteins that have been engineered to give T cells the new ability to target a specific protein. Sustained B cell depletion by CD19/20-targeted CAR T cells is a highly effective treatment for murine lupus erythematosus and human acute B cell lymphoblastic leukemia [143,144]. Following this rationale, a promising phase 1 study tested safety and efficacy of anti-CD19/20 CAR T cells in the treatment of patients with AQP4-IgG seropositive NMOSD but was withdrawn due to insufficient patient recruitment (NCT03605238).
3.4. Brutons Tyrosine Kinase Inhibition
3.4.1. SHR1459
Bruton’s tyrosine kinase plays a crucial role in B cell development by transmitting intracellular signals from the pre-B cell receptor [145]. There are approvals for ibrutinib in chronic lymphocytic lymphoma and acalabrutinib as well as zanubrutinib in mantle cell lymphoma [146,147,148]. The inhibition of Bruton’s tyrosine kinase has lately shown benefits in other autoimmune diseases like multiple sclerosis and systemic lupus erythematosus [149]. Following this rationale, a very recently initiated study (NCT04670770) will investigate the effect of the Bruton’s tyrosine inhibitor SHR1459 in NMOSD patients for the first time.
3.5. IL-6 Receptor Antagonism
3.5.1. Tocilizumab
This monoclonal antibody specifically targets the IL-6 receptor, which is expressed on most immune cell subsets. IL-6 is produced by a large number of cells, including B and T cells, monocytes and fibroblasts and is considered a pro-inflammatory cytokine [150]. It has been found to be elevated in the CSF and serum of NMOSD patients and is thought to induce T cell activation, generation of autoreactive T helper 17 cells and AQP4-antibody secretion by plasmablasts [151,152]. Tocilizumab was shown to decrease the annualized relapse rate from 2.9 to 0.4 and significantly reduced EDSS, neuropathic pain, and general fatigue [153]. In a similar report, tocilizumab significantly decreased the annualized relapse rate from 4.0 to 0.4 and the median EDSS from 7.3 to 5.5 [154]. A larger, more recent trial with 118 included patients found that 92% of NMOSD patients treated with tocilizumab were relapse-free after 48 weeks compared to 69% in the azathioprine group with similar findings regarding disability progression [155]. A recent randomized phase 2 study found that tocilizumab significantly reduced the risk of a subsequent NMOSD relapse and was superior compared to with a treatment with azathioprine [117].
Treatment with tocilizumab had neither an impact on total lymphocyte count and absolute B cell, transitional B cell and plasmablast numbers, nor did it alter BAFF levels of treated patients [156,157]. However, tocilizumab was shown to reduce memory B cell subsets as well as IgA and IgG serum levels in rheumatoid arthritis patients [158]. Interestingly, it was found that tocilizumab treatment does not change interleukin-6 serum levels in rheumatoid arthritis patients [159].
3.5.2. Satralizumab
Using antibody-recycling technology, this IL-6 receptor-antagonizing antibody was designed to have improved pharmacokinetics (longer half lifetime in vivo) compared to tocilizumab [160]. In two large phase 3 studies, satralizumab significantly reduced the risk of protocol-defined relapses but did not differ from placebo in its effect on pain or fatigue. Also, satralizumab was significantly less effective in seronegative patients, which requires further analysis [118,119]. In June 2020, based on these positive results, subcutaneous satralizumab received its first global approval in Canada for the treatment of NMOSD in adults and children aged ≥ 12 years who are AQP4-IgG seropositive. Satralizumab was subsequently approved in Japan and Switzerland. Satralizumab is under regulatory review in the European Union and United States of America, and is undergoing clinical development in several countries worldwide [161]. Its effects on B cells may be similar to those of tocilizumab, but still need to be examined.
3.6. Complement Factor Antagonism
3.6.1. Eculizumab
Preventing the cleavage of complement factor 5 (C5) into its subunits, this monoclonal antibody is thought to block the AQP4-antibody-triggered complement cascade in NMOSD. Thus, inflammation, the formation of the membrane attack complex along with subsequent astrocyte destruction and neuronal malfunction are suppressed by eculizumab [20,162]. Eculizumab has been approved for the treatment of myasthenia gravis, paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome for longer time [163]. Assuming beneficial effects in NMOSD, a phase 3 trial including 143 AQP4-antibody positive NMOSD patients, finally found that eculizumab significantly suppressed the adjudicated annualized relapse rate, while there were no benefits on disability progression during the short period of the trial [111]. These favorable outcomes of made eculizumab the first agent to be specifically approved for the treatment of seropositive NMOSD patients in the EU, USA, Canada and Japan in 2019 [164]. Despite these positive results, the drug requires a 35-min intravenous infusion every 2 weeks and C5 level activity begins to rise fairly rapidly two weeks after the last dose of eculizumab has been received [165]. Eculizumab decreased serum IL-6 levels in patients with paroxysmal nocturnal hemoglobinuria and increased B cell counts [166,167]. Effects on the B cell compartment in NMOSD patients are not yet assessed in detail and may strongly differ from those in paroxysmal nocturnal hemoglobinuria due to a completely different pathophysiology.
3.6.2. Ravulizumab
As a successor of eculizumab, this monoclonal antibody is designed for longer therapy intervals of up to eight weeks as its structure differs in four amino acids, leading to a four-fold longer half-life compared to eculizumab [168]. It was already approved for the treatment of paroxysmal nocturnal haemoglobinuria in 2018 and is now also considered a therapeutic option for NMOSD [169]. There is an ongoing phase 3 study to evaluate the efficacy and safety of ravulizumab for the treatment of adult participants with NMOSD (NCT04201262). To date, the effect of C5-inhibitors on peripheral B cells remains unclear.
3.7. Targeting Pathological AQP4 Antibodies
3.7.1. Aquaporumab
This antibody is designed to compete with AQP4-antibodies for AQP4 binding. Because of a mutated Fc portion, it does not activate complement- and cell-dependent cytotoxicity [170]. In a preclinical study, aquaporumab was already proven to efficiently compete with pathological AQP4-antibodies [171]. Very recently, high affinity aquaporumab, which was generated by affinity maturation using saturation mutagenesis, was shown to block cellular injury caused by NMOSD patient sera in AQP4-expressing cell cultures. Further development of aquaporumab for NMOSD therapy will require characterization and optimization of pharmacokinetics and other pharmacological properties, as well as evaluation of potential toxicity following long-term administration [172].
3.7.2. HBM9161
This human monoclonal antibody targets the neonatal fragment crystallizable (Fc) receptor and blocks the IgG-Fc binding site. Thereby, it is thought to accelerate the degradation of IgG and significantly reduce the total serum IgG levels (including pathological anti-AQP4 IgG in NMOSD). There is a recruiting Phase 1 study investigating safety, tolerability, pharmacodynamics, and efficacy of HBM 9161 in patients with an attack of NMOSD (NCT04227470).
3.8. Other Therapeutic Strategies
3.8.1. Bevacizumab
This well-established anti-vascular endothelial growth factor-antibody was tested in NMOSD patients with the intention to maintain the integrity of the BBB and thereby to block pathogenetic autoantibodies from entering the CNS. In a first small proof-of-concept trial with 10 patients, no patients required escalation to plasma exchange after high-dose corticosteroids and intravenous bevacizumab while an excellent safety profile was observed suggesting that bevacizumab might be considered for future studies as an add-on to currently available therapies [107]. It must be noted, that another investigation reported the emergence of a tumefactive demyelination during treatment with intravitreal bevacizumab [173].
3.8.2. Bortezomib
By binding the catalytic site of the 26S subunit, this agent selectively inhibits proteasome activity and thereby leads to an enhanced apoptosis rate of rapidly proliferating cells, including plasma cells, possibly by preventing degradation of pro-apoptotic factors. Therefore, Bortezomib is approved for the treatment of multiple myeloma and mantle cell lymphoma [114]. Stimulation of murine astroglia with AQP4-IgG resulted in the release of pro-granulocytic chemokines and could be inhibited by bortezomib [174]. In one sole human study, four of the five patients were relapse-free during the 1-year follow-up after bortezomib treatment initiation. Remission was closely associated with the reduction of autoimmune activity reflected by a decrease in serum AQP4 titers, peripheral plasma cell counts and precursor B cells with proteasome inhibition. For patients with highly recurrent NMOSD who are unresponsive to conventional immunosuppressive treatments, bortezomib may serve as a salvage therapy. However, additional studies are required to validate its benefits [175].
3.8.3. Cetirizine
The beneficial effect of this commonly used selective inhibitor of the histamine receptor 1 in a murine NMOSD model is thought to be related to a reduction of eosinophil infiltration into the CNS and lesion formation [176]. The fact that eosinophil infiltration is a prominent feature of NMOSD lesions and eosinophils were shown to be elevated in the CSF of NMOSD patients corroborates this hypothesis [177]. Astonishingly, cetirizine administration given as add-on therapy to standard treatment of NMOSD lead to a fourfold reduction of the annualized relapse rate after one year [112]. However, it must be noted that this study was performed with only 18 patients. Larger trials will be needed to confirm these results.
3.8.4. Sivelestat
3.8.5. Telitacicept
B-lymphocyte stimulator/B cell activating factor (BLyS, BAFF) and A proliferation-inducing ligand (APRIL) are cytokines of the TNF-superfamily, which are capable of inducing proliferation and differentiation of B cells by activating classical and noncanonical NF-κB signaling pathways. Both cytokines can bind BAFF-receptor (BAFFR), as well as B cell maturation antigen (BCMA), as well as transmembrane activator and calcium-modulator and cyclophilin-ligand activator (TACI) [181]. These receptors are mainly expressed on maturated B cells. This explains why Telitaticept, a novel recombinant TACI-Fc fusion protein, can target and neutralize both BAFF and APRIL, which were found to be elevated in the sera of NMOSD patients when compared to MS patients [182]. Based on the promising results of a pivotal phase 2b clinical trial (NCT02885610), which included 249 adults with moderate-to-severe SLE, the FDA has granted fast-track approval to telitacicept for the treatment of patients with SLE. Safety assessments also showed a good tolerability profile for telitacicept in this population [183]. In a still ongoing phase III, randomized, placebo-controlled study, telitacicept is tested in seropositive NMOSD patients without recent immunosuppressive treatment (NCT03330418).
3.8.6. C1-Esterase Inhibitor/Cinryze
As discussed above, there is compelling evidence for a central role of complement factors in NMOSD pathogenesis. Patients do not lack endogenous complement factor 1 (C1)-esterase inhibitor, but increased complement activation in acute lesions justifies its testing in NMOSD [184]. In a small study with 10 patients, the administration of a C1-esterase inhibitor, which is a nano filtered natural C1-esterase inhibitor concentrate, as add-on therapy to treatment with systemic corticosteroids was well tolerated and safe in acute NMOSD relapses. A promising benefit of C1-esterase inhibitor in NMOSD patients was suggested, as a reduction of neurologic damage and an improvement of clinical outcomes were observed, but due to the lack of a control group, the significance of these findings was limited [185]. However, in vitro data with human serum and in vivo data in rats suggest that the complement inhibition activity of C1-inhibitor in serum is too low to confer clinical benefit in NMOSD [106]. Also, chronically elevated endogenous plasma levels of C1-esterase inhibitor were found in NMOSD patients when compared to MS patients or controls [186].
3.8.7. Tolerogenic Dendritic Cells Loaded with Myelin Peptides
Dendritic cells are located in peripheral and lymphoid tissues and are essential for homeostasis of T cell-dependent immune responses by promoting both pro- and anti-inflammatory activities in response to different stimuli. Within these cells, the so-called tolerogenic dendritic cells represent a heterogenous pool of cells with immunosuppressive effects regarding the expression of costimulatory molecules, the production of anti-inflammatory cytokines such as IL-10 and a reduced capacity to stimulate a T-cell response [109]. For the generation of human tolerogenic dendritic cells, isolated CD14+ peripheral monocytes are stimulated with granulocyte-macrophage colony-stimulating factor. After harvesting untouched, suspended cells, additional treatment with lipopolysaccharides, tumor necrosis factor-alpha or interferon-gamma generates dendritic cells. Thereafter, stimulation with cytokines (e.g. interleukin-10), organic molecules (e.g. vitamin D3), chemicals, drugs (e.g. dexamethasone) and other factors can turn them into tolerogenic dendritic cells without the existence of a standardized protocol [187]. The use of tolerogenic dendritic cells in several phase I clinical trials (type I diabetes, rheumatoid arthritis and Crohn’s disease) was safe with promising clinical and immunomodulatory results [188]. A first in human study to assess the tolerability and safety profile of a treatment with dendritic cell in patients with MS or NMOSD including 20 patients has just been completed (NCT02283671).
3.8.8. Hematopoietic Stem Cell Transplantation
Autologous hematopoietic stem cell transplantation is a treatment option occasionally used in severe cases of NMOSD. Currently available data suggest that inflammatory activity is reduced in the short term, but a clear majority of the patients will relapse within 5 years [189]. A recent study with 11 NMOSD patients found prolonged drug-free remission with AQP4-IgG seroconversion to negative following nonmyeloablative autologous hematopoietic stem cell transplantation, warranting further investigation [110]. A more recent meta-analysis investigating the effect of autologous hematopoietic stem cell transplantation on NMOSD and including a total of 31 patients computed progression-free survival in 76% and transplant-related mortality in 0% of treated subjects [190]. Low- and intermediate-intensity regimens and RRMS patients showed optimal benefit from this therapy. Still, comparatively severe side effects and periprocedural complications further emphasize the need for less dangerous treatment options.
Allogenic hematopoietic stem cell transplantation with emerging therapeutic effects such as an induction of tolerance to auto-antigens and graft versus autoimmunity effects may point toward future treatment perspectives in this field [191].
3.8.9. Mesenchymal Stromal Cells
Derived from the human umbilical cord, mesenchymal stromal cells are able to produce many different immunomodulatory molecules. In doing so, they were shown to suppress B cell proliferation through an arrest in the G0/G1 phase of the cell cycle suggesting beneficial effects in B cell-mediated autoimmune disorders [192]. In a first small study with 5 NMOSD patients, clinical disease parameters improved and relapse frequencies were reduced after initial intravenous infusion of 4 × 107 human umbilical cord-derived mesenchymal stromal cells followed by 2 × 107 cells both intravenously and intrathecally on days 7, 14 and 21 [193]. Here, B cells were inhibited while T cells increased after treatment, indicating an immune-related mechanism. In a case report, an NMOSD patient consented for local application of autologous mesenchymal stromal cells on pressure ulcers surprisingly showed an improvement of disability for 6 years and was free of relapses [194].
3.8.10. Dalfampridine
By magnifying the axonal conductance, this potassium channel inhibitor is used in MS patients to improve cognition, fatigue, and dexterity [195]. In a randomized, double-blind, placebo-controlled crossover study of dalfampridine in transverse myelitis patients, intervention improved walking speed and other neurological functions [113]. This suggests that NMOSD patients with gait affecting transverse myelitis might also benefit from dalfampridine treatment.
3.8.11. Alpha-1 Antitrypsin
A phase 1 study evaluated whether the use of alpha1-antitrypsin, an approved medication for patients with congenital deficiency of alpha-1 antitrypsin associated with emphysema, is beneficial in acute attacks of NMO, improving patient disability and quality of life (NCT02087813). However, it was withdrawn without any results due to recruitment issues.
4. Outlook
Beyond the listed agents, which are already being investigated upon in clinical trials, several promising novel treatment strategies should be monitored in the future:
4.1. Calcineurin Inhibition
In 2014, a patient with neuromyelitis optica spectrum disorder combined with Sjögren’s syndrome was found to be relapse-free following tacrolimus treatment [196]. Following this case, a first study with 25 included patients revealed that tacrolimus could reduce the relapse rate by 86.2% and improve the EDSS score (4.5 vs. 2.3; p < 0.001) significantly [197]. In a more recent report, the combined use of the calcineurin inhibitor tacrolimus with prednisolone clearly suppressed relapses in both anti-AQP4 antibody-positive and -negative NMOSD [198].
4.2. Targeting Eosinophils
4.2.1. Anti-IL5 Antibodies
As mentioned above, eosinophil counts were found to be elevated in the CSF of NMOSD patients and eosinophil infiltration is a main feature of intracerebral lesions [177]. In this line, the second-generation antihistamine ketotifen greatly reduced AQP4-IgG/eosinophil-dependent cytotoxicity and NMOSD pathology in mice [176]. In experimental models of NMOSD, the inhibition of eosinophil granulocytes by anti-IL-5 antibodies or gene depletion significantly reduced lesion severity [176]. Therefore, IL-5 antibodies like mepolizumab and reslizumab may present promising future treatment options.
4.2.2. Anti-IgE Antibodies
Case reports about highly elevated IgE-levels in patients with NMOSD support the concept of a crucial role of this immunoglobulin in NMOSD pathology [177]. The anti-IgE antibody omalizumab, already being approved for allergic asthma, might therefore also represent a considerable treatment option in patients with NMOSD [199].
4.3. Blocking of Pathogenic Autoantibodies
In addition to aquaporumab, a Fc-mutated anti-AQP4 antibody preventing endogenous pathogenic antibodies to bind to their target [172], small drug-like molecules like arbidol, tamarixetin and berbamine alkaloids were shown to compete with AQP4 binding and therefore may exhibit beneficial effects in NMOSD in future clinical trials [9]. Moreover, there is an interesting report about the enzyme endoglycosidase S, which specifically removes Fc glycans, rendering AQP4-antibodies non-pathogenic [200].
4.4. Targeting the BAFF/APRIL System
The crucial role of B cells and their effector functions in NMOSD also make them favorable targets for future therapies. Beyond the above-described telitacicept, several other monoclonal antibodies target the BAFF/APRIL complex: atacicept, an anti-BAFF antibody, surprisingly exacerbated MS disease activity in a phase 2 study [201], but may be beneficial in NMOSD because of a different pathophysiology. Other agents like tabalumab, belimumab and anti-BAFF-receptor ianalumab are already tested in clinical trials including systemic lupus erythematosus, pemphigus vulgaris, rheumatoid arthritis and multiple myeloma patients [202,203,204]. Due to the fact that they are also autoantibody-dependent diseases, NMOSD patients might also benefit from these novel agents.
4.5. Microbiota
The trigger of AQP4-antibody formation remains unknown up to the present day. Interestingly, AQP4-specific T cells also recognized the ABC transporter of Clostridium perfringens, which comprises both pathogenic and commensal strains in the human gut [205]. This is in line with the idea that recruitment and activation of autoantibody-producing B cells from the endogenous immune repertoire depends on the availability of the target autoantigen on commensal microbiota [206]. These observations identify a sequence of events triggering organ-specific autoimmune disease and these processes may offer novel therapeutic targets.
5. Conclusions
In summary, B cells and pathological autoantibodies are major targets when it comes to effective treatment strategies of NMOSD. Compared to MS, where B cells intrathecally secrete IgG, AQP4-tagreting IgG in NMOSD are thought to be mainly produced in the periphery, placing in focus plasmablast- and plasma cell-targeting therapies. Compared to CD20 depleting agents, inebilizumab can bind these antibody-producing cells, which might contribute to its astonishing success and approval within the last year. While general immunosuppressants inherit serious side effects, the era of monoclonal antibodies has paved the way towards a specific modification of NMOSD pathophysiology. Targeting CD19, IL-6 receptors and C5 complement has reformed the therapeutic arsenal in the rare condition of NMOSD, where clinical studies are hard to perform due to high costs, complicated diagnostic procedures and recruitment issues. The large bouquet of promising agents with differing modes of action in development may further improve the prognosis of patients suffering from this devastating disease in the future.
Author Contributions
J.T. wrote the manuscript and drafted the illustrations. L.H. wrote and reviewed the manuscript. M.S.W. conceptualized and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| APRIL | A proliferation-inducing ligand |
| AQP4 | Aquaporin 4 |
| BAFF | B cell activating factor |
| BAFFR | BAFF receptor |
| BBB | Blood-brain barrier |
| BCMA | B cell maturation antigen |
| BLyS | B lymphocyte stimulator |
| C1 | Complement factor 1 |
| C5 | Complement factor 5 |
| CAR | Chimeric antigen receptor |
| CD | Cluster of differentiation |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EDSS | Expanded disability status scale |
| Fc | Fragment crystallizable |
| Ig | Immunoglobulin |
| IL | Interleukin |
| MOG | Myelin oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| NMOSD | Neuromyelitis optica spectrum disorders |
| SLE | Systemic lupus erythematosus |
| TACI | Transmembrane activator and calcium-modulator and cyclophilin-ligand activator |
| TNF | Tumor necrosis factor |
References
- Prasad, S.; Chen, J. What you need to know about aqp4, mog, and nmosd. Semin. Neurol. 2019, 39, 718–731. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B. The history of neuromyelitis optica. J. Neuroinflamm. 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Weber, M.S.; Derfuss, T.; Metz, I.; Bruck, W. Defining distinct features of anti-mog antibody associated central nervous system demyelination. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418762083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis optica spectrum disorder and anti-mog syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.S.; Derfuss, T.; Bruck, W. Anti-myelin oligodendrocyte glycoprotein antibody-associated central nervous system demyelination-a novel disease entity? JAMA Neurol. 2018, 75, 909–910. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Waters, P.; Zipp, F.; Hohlfeld, R.; Vincent, A.; Wildemann, B. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat. Clin. Pract. Neurol. 2008, 4, 202–214. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Kinzel, S.; Weber, M.S. The role of peripheral cns-directed antibodies in promoting inflammatory cns demyelination. Brain Sci. 2017, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Wildemann, B.; Jarius, S.; Orellano, B.; Sasidharan, S.; Weber, M.S.; Stuve, O. Immunopathogenesis of neuromyelitis optica. Adv. Immunol. 2014, 121, 213–242. [Google Scholar] [PubMed]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of nmo: A study on antibody titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausser-Kinzel, S.; Weber, M.S. The role of b cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front. Immunol. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Horn, K.; Kinzel, S.; Weber, M.S. Deciphering the role of b cells in multiple sclerosis-towards specific targeting of pathogenic function. Int. J. Mol. Sci. 2017, 18, 2048. [Google Scholar] [CrossRef] [Green Version]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Hausler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate t cell-mediated cns autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, A.; Bergamaschi, R.; Martinelli, V.; Trojano, M.; Tola, M.R.; Merelli, E.; Mancardi, L.; Gallo, P.; Filippi, M.; Zaffaroni, M.; et al. Clinical characteristics, course and prognosis of relapsing devic’s neuromyelitis optica. J. Neurol. 2004, 251, 47–52. [Google Scholar] [CrossRef]
- Vincent, T.; Saikali, P.; Cayrol, R.; Roth, A.D.; Bar-Or, A.; Prat, A.; Antel, J.P. Functional consequences of neuromyelitis optica-igg astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J. Immunol. 2008, 181, 5730–5737. [Google Scholar] [CrossRef]
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Investig. 2019, 129, 2000–2013. [Google Scholar] [CrossRef] [Green Version]
- Mitsdoerffer, M.; Kuchroo, V.; Korn, T. Immunology of neuromyelitis optica: A t cell-b cell collaboration. Ann. N. Y. Acad. Sci. 2013, 1283, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of il-6-producing b cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. Il-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pitarokoili, K.; Gold, R. Dimethyl fumarate for patients with neuromyelitis optica spectrum disorder. Mult. Scler. 2018, 24, 364–365. [Google Scholar] [CrossRef]
- Weber, M.S.; Hemmer, B. Cooperation of b cells and t cells in the pathogenesis of multiple sclerosis. Results Probl. Cell Differ. 2010, 51, 115–126. [Google Scholar]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. Mhc class ii-dependent b cell apc function is required for induction of cns autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef]
- Sahraian, M.A.; Moghadasi, A.N.; Azimi, A.R.; Asgari, N.; Akhoundi, F.H.; Abolfazli, R.; Alaie, S.; Ashtari, F.; Ayromlou, H.; Baghbanian, S.M.; et al. Diagnosis and management of neuromyelitis optica spectrum disorder (nmosd) in iran: A consensus guideline and recommendations. Mult. Scler. Relat. Disord. 2017, 18, 144–151. [Google Scholar] [CrossRef]
- Traub, J.; Hausser-Kinzel, S.; Weber, M.S. Differential effects of ms therapeutics on b cells-implications for their use and failure in aqp4-positive nmosd patients. Int. J. Mol. Sci. 2020, 21, 5021. [Google Scholar] [CrossRef]
- Kinzel, S.; Weber, M.S. B cell-directed therapeutics in multiple sclerosis: Rationale and clinical evidence. CNS Drugs 2016, 30, 1137–1148. [Google Scholar] [CrossRef]
- Selmaj, K.; Selmaj, I. Novel emerging treatments for nmosd. Neurol. Neurochir. Pol. 2019, 53, 317–326. [Google Scholar] [CrossRef]
- Abboud, H.; Petrak, A.; Mealy, M.; Sasidharan, S.; Siddique, L.; Levy, M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult. Scler. 2016, 22, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, C.L.; Wegner, C.; Sprenger, T.; Weber, M.S.; Bruck, W.; Hemmer, B.; Sellner, J. Favourable response to plasma exchange in tumefactive cns demyelination with delayed b-cell response. Mult. Scler. 2012, 18, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.T.; Yu, D.T.; Clements, P.J.; Fowlston, S.; Eisman, J.; Bluestone, R. Effect of corticosteroids on the human immune response: Comparison of one and three daily 1 gm intravenous pulses of methylprednisolone. J. Lab. Clin. Med. 1978, 91, 625–634. [Google Scholar] [PubMed]
- Fauci, A.S.; Dale, D.C.; Balow, J.E. Glucocorticosteroid therapy: Mechanisms of action and clinical considerations. Ann. Intern. Med. 1976, 84, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Olnes, M.J.; Kotliarov, Y.; Biancotto, A.; Cheung, F.; Chen, J.; Shi, R.; Zhou, H.; Wang, E.; Tsang, J.S.; Nussenblatt, R.; et al. Effects of systemically administered hydrocortisone on the human immunome. Sci. Rep. 2016, 6, 23002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, J.D.; Hepburn, B. Prednisone-induced alterations of circulating human lymphocyte subsets. J. Lab. Clin. Med. 1983, 101, 479–487. [Google Scholar]
- Lanzillotta, M.; Della-Torre, E.; Milani, R.; Bozzolo, E.; Bozzalla-Cassione, E.; Rovati, L.; Arcidiacono, P.G.; Partelli, S.; Falconi, M.; Ciceri, F.; et al. Effects of glucocorticoids on b-cell subpopulations in patients with igg4-related disease. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), 159–166. [Google Scholar]
- Lin, W.; Zhang, P.; Chen, H.; Chen, Y.; Yang, H.; Zheng, W.; Zhang, X.; Zhang, F.; Zhang, W.; Lipsky, P.E. Circulating plasmablasts/plasma cells: A potential biomarker for igg4-related disease. Arthritis Res. Ther. 2017, 19, 25. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; LaForge, K.S.; Sehgal, P.B. On the mechanism for efficient repression of the interleukin-6 promoter by glucocorticoids: Enhancer, tata box, and rna start site (inr motif) occlusion. Mol. Cell. Biol. 1990, 10, 5736–5746. [Google Scholar] [CrossRef] [Green Version]
- Settipane, G.A.; Pudupakkam, R.K.; McGowan, J.H. Corticosteroid effect on immunoglobulins. J. Allergy Clin. Immunol. 1978, 62, 162–166. [Google Scholar] [CrossRef]
- Lack, G.; Ochs, H.D.; Gelfand, E.W. Humoral immunity in steroid-dependent children with asthma and hypogammaglobulinemia. J. Pediatr. 1996, 129, 898–903. [Google Scholar] [CrossRef]
- Kamhieh-Milz, J.; Ghosoun, N.; Sterzer, V.; Salama, A. Effect of glucocorticoid treatment on baff and april expression in patients with immune thrombocytopenia (itp). Clin. Immunol. 2018, 188, 74–80. [Google Scholar] [CrossRef]
- Sellner, J.; Boggild, M.; Clanet, M.; Hintzen, R.Q.; Illes, Z.; Montalban, X.; Du Pasquier, R.A.; Polman, C.H.; Sorensen, P.S.; Hemmer, B. Efns guidelines on diagnosis and management of neuromyelitis optica. Eur. J. Neurol. 2010, 17, 1019–1032. [Google Scholar] [CrossRef]
- Kimbrough, D.J.; Fujihara, K.; Jacob, A.; Lana-Peixoto, M.A.; Leite, M.I.; Levy, M.; Marignier, R.; Nakashima, I.; Palace, J.; de Seze, J.; et al. Treatment of neuromyelitis optica: Review and recommendations. Mult. Scler. Relat. Disord. 2012, 1, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Elsone, L.; Kitley, J.; Luppe, S.; Lythgoe, D.; Mutch, K.; Jacob, S.; Brown, R.; Moss, K.; McNeillis, B.; Goh, Y.Y.; et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: A multicentre retrospective observational study from the UK. Mult. Scler. 2014, 20, 1533–1540. [Google Scholar] [CrossRef]
- Evans, W.E. Pharmacogenetics of thiopurine s-methyltransferase and thiopurine therapy. Ther. Drug Monit. 2004, 26, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Maltzman, J.S.; Koretzky, G.A. Azathioprine: Old drug, new actions. J. Clin. Investig. 2003, 111, 1122–1124. [Google Scholar] [CrossRef]
- Leandro, M.J.; Cambridge, G.; Edwards, J.C.; Ehrenstein, M.R.; Isenberg, D.A. B-cell depletion in the treatment of patients with systemic lupus erythematosus: A longitudinal analysis of 24 patients. Rheumatology 2005, 44, 1542–1545. [Google Scholar] [CrossRef] [Green Version]
- Hayry, P.; von Willebrand, E.; Ahonen, J.; Eklund, B.; Salmela, K.; Hockerstedt, K.; Pettersson, E.; Koskimies, S. Effects of cyclosporine, azathioprine, and steroids on the renal transplant, on the cytologic patterns of intragraft inflammation, and on concomitant rejection-associated changes in recipient blood. Transplant. Proc. 1988, 20, 153–162. [Google Scholar]
- Bottomley, M.J.; Chen, M.; Fuggle, S.; Harden, P.N.; Wood, K.J. Application of operational tolerance signatures are limited by variability and type of immunosuppression in renal transplant recipients: A cross-sectional study. Transplant. Direct 2017, 3, e125. [Google Scholar] [CrossRef]
- Thiel, J.; Salzer, U.; Hassler, F.; Effelsberg, N.M.; Hentze, C.; Sic, H.; Bartsch, M.; Miehle, N.; Peter, H.H.; Warnatz, K.; et al. B cell homeostasis is disturbed by immunosuppressive therapies in patients with anca-associated vasculitides. Autoimmunity 2013, 46, 429–438. [Google Scholar] [CrossRef]
- Roekevisch, E.; Szegedi, K.; Hack, D.P.; Schram, M.E.; Res, P.; Bos, J.D.; Leeflang, M.M.G.; Luiten, R.M.; Kezic, S.; Spuls, P.I.; et al. Effect of immunosuppressive treatment on biomarkers in adult atopic dermatitis patients. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Sokollik, C.; McLin, V.A.; Vergani, D.; Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G. Juvenile autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2018, 95, 69–76. [Google Scholar] [CrossRef]
- Hernandez-Breijo, B.; Gomez, A.; Soukka, S.; Johansson, P.; Parodis, I. Antimalarial agents diminish while methotrexate, azathioprine and mycophenolic acid increase baff levels in systemic lupus erythematosus. Autoimmun. Rev. 2019, 18, 102372. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Stuve, O. Cyclophosphamide in multiple sclerosis: Scientific rationale, history and novel treatment paradigms. Ther. Adv. Neurol. Disord. 2009, 2, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, H.; Sakushima, K.; Takahashi, I.; Nishimura, H.; Yashima-Yamada, M.; Nakamura, M.; Tsuzaka, K.; Maruo, Y.; Takahashi, T.; Yabe, I.; et al. Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Intern. Med. 2013, 52, 969–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C.; To, C.H.; Mak, A.; Poon, W.L. Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J. Rheumatol. 2008, 35, 172–174. [Google Scholar]
- Torres, J.; Pruitt, A.; Balcer, L.; Galetta, S.; Markowitz, C.; Dahodwala, N. Analysis of the treatment of neuromyelitis optica. J. Neurol. Sci. 2015, 351, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Saida, T. [treatment of nmo]. Rinsho Shinkeigaku 2009, 49, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.G.; Tilby, M.J. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992, 6, 163–173. [Google Scholar] [CrossRef]
- Brodsky, R.A. High-dose cyclophosphamide for autoimmunity and alloimmunity. Immunol. Res. 2010, 47, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Cupps, T.R.; Whalen, G.; Fauci, A.S. Selective effects of cyclophosphamide therapy on activation, proliferation, and differentiation of human b cells. J. Clin. Investig. 1987, 79, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schluter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorner, T.; Jacobi, A.M.; Lipsky, P.E. B cells in autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Moschella, F.; Torelli, G.F.; Valentini, M.; Urbani, F.; Buccione, C.; Petrucci, M.T.; Natalino, F.; Belardelli, F.; Foa, R.; Proietti, E. Cyclophosphamide induces a type i interferon-associated sterile inflammatory response signature in cancer patients’ blood cells: Implications for cancer chemoimmunotherapy. Clin. Cancer Res. 2013, 19, 4249–4261. [Google Scholar] [CrossRef] [Green Version]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T.; et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the neuromyelitis optica study group (nemos). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Weinstock-Guttman, B.; Ramanathan, M.; Lincoff, N.; Napoli, S.Q.; Sharma, J.; Feichter, J.; Bakshi, R. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (devic disease). Arch. Neurol. 2006, 63, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, W.; Park, M.S.; Sohn, E.H.; Li, X.F.; Kim, H.J. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch. Neurol. 2011, 68, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Cabre, P.; Olindo, S.; Marignier, R.; Jeannin, S.; Merle, H.; Smadja, D.; Aegis of French National Observatory of Multiple, S. Efficacy of mitoxantrone in neuromyelitis optica spectrum: Clinical and neuroradiological study. J. Neurol. Neurosurg. Psychiatry 2013, 84, 511–516. [Google Scholar] [CrossRef]
- Mazerski, J.; Martelli, S.; Borowski, E. The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: Molecular dynamics simulations. Acta Biochim. Pol. 1998, 45, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, O.; Wiendl, H.; Kieseier, B.C.; Archelos, J.J.; Hemmer, B.; Stuve, O.; Hartung, H.P. Multiple sclerosis: Mitoxantrone promotes differential effects on immunocompetent cells in vitro. J. Neuroimmunol. 2005, 168, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Chanvillard, C.; Millward, J.M.; Lozano, M.; Hamann, I.; Paul, F.; Zipp, F.; Dorr, J.; Infante-Duarte, C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS ONE 2012, 7, e39625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, J.M.; DeJoy, S.Q.; Gibbons, J.J., Jr. Selective immunomodulation by the antineoplastic agent mitoxantrone. I. Suppression of b lymphocyte function. J. Immunol. 1986, 137, 727–732. [Google Scholar]
- Fox, E.J. Mechanism of action of mitoxantrone. Neurology 2004, 63, S15–S18. [Google Scholar] [CrossRef] [PubMed]
- Kannel, K.; Alnek, K.; Vahter, L.; Gross-Paju, K.; Uibo, R.; Kisand, K.V. Changes in blood b cell-activating factor (baff) levels in multiple sclerosis: A sign of treatment outcome. PLoS ONE 2015, 10, e0143393. [Google Scholar] [CrossRef]
- Montcuquet, A.; Collongues, N.; Papeix, C.; Zephir, H.; Audoin, B.; Laplaud, D.; Bourre, B.; Brochet, B.; Camdessanche, J.P.; Labauge, P.; et al. Effectiveness of mycophenolate mofetil as first-line therapy in aqp4-igg, mog-igg, and seronegative neuromyelitis optica spectrum disorders. Mult. Scler. 2017, 23, 1377–1384. [Google Scholar] [CrossRef]
- Huh, S.Y.; Kim, S.H.; Hyun, J.W.; Joung, A.R.; Park, M.S.; Kim, B.J.; Kim, H.J. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol. 2014, 71, 1372–1378. [Google Scholar] [CrossRef]
- Walkiewicz-Pielaszek, K.; Swacha, M.; Bullo-Piontecka, B.; Rutkowski, B.; Olesinska, M. Mycophenolate mofetil--20 years of experience in treatment of rheumatic diseases. Postepy Hig. Med. Dosw. Online 2015, 69, 176–187. [Google Scholar]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Karnell, J.L.; Karnell, F.G., 3rd; Stephens, G.L.; Rajan, B.; Morehouse, C.; Li, Y.; Swerdlow, B.; Wilson, M.; Goldbach-Mansky, R.; Groves, C.; et al. Mycophenolic acid differentially impacts b cell function depending on the stage of differentiation. J. Immunol. 2011, 187, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Eickenberg, S.; Mickholz, E.; Jung, E.; Nofer, J.R.; Pavenstadt, H.J.; Jacobi, A.M. Mycophenolic acid counteracts b cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, M.S.; Kim, E.Y.; Park, H.J.; Chang, C.Y.; Park, K.S.; Jung, D.Y.; Kwon, C.H.; Joh, J.W.; Kim, S.J. Mycophenolate mofetil promotes down-regulation of expanded b cells and production of tnf-alpha in an experimental murine model of colitis. Cytokine 2008, 44, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kitley, J.; Elsone, L.; George, J.; Waters, P.; Woodhall, M.; Vincent, A.; Jacob, A.; Leite, M.I.; Palace, J. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.S.; Malhotra, K.; Scott, T. Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol. 2014, 14, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beh, S.C.; Kildebeck, E.; Narayan, R.; Desena, A.; Schell, D.; Rowe, E.S.; Rowe, V.; Burns, D.; Whitworth, L.; Frohman, T.C.; et al. High-dose methotrexate with leucovorin rescue: For monumentally severe cns inflammatory syndromes. J. Neurol. Sci. 2017, 372, 187–195. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Fortea-Gordo, P.; Villalba, A.; Nuno, L.; Santos-Bornez, M.J.; Peiteado, D.; Monjo, I.; Puig-Kroger, A.; Sanchez-Mateos, P.; Martin-Mola, E.; Balsa, A.; et al. Circulating cd19+cd24hicd38hi regulatory b cells as biomarkers of response to methotrexate in early rheumatoid arthritis. Rheumatology 2020, 59, 3081–3091. [Google Scholar] [CrossRef]
- Aggarwal, A.; Misra, R. Methotrexate inhibits interleukin-6 production in patients with juvenile rheumatoid arthritis. Rheumatol. Int. 2003, 23, 134–137. [Google Scholar] [CrossRef]
- Hughes, R.A.; Donofrio, P.; Bril, V.; Dalakas, M.C.; Deng, C.; Hanna, K.; Hartung, H.P.; Latov, N.; Merkies, I.S.; van Doorn, P.A.; et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ice study): A randomised placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Magraner, M.J.; Coret, F.; Casanova, B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 2013, 28, 65–72. [Google Scholar] [CrossRef]
- Viswanathan, S.; Wong, A.H.; Quek, A.M.; Yuki, N. Intravenous immunoglobulin may reduce relapse frequency in neuromyelitis optica. J. Neuroimmunol. 2015, 282, 92–96. [Google Scholar] [CrossRef]
- Li, X.; Tian, D.C.; Fan, M.; Xiu, Y.; Wang, X.; Li, T.; Jia, D.; Xu, W.; Song, T.; Shi, F.D.; et al. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (nmosd). Mult. Scler. Relat. Disord. 2020, 44, 102325. [Google Scholar] [CrossRef]
- Koffman, B.M.; Dalakas, M.C. Effect of high-dose intravenous immunoglobulin on serum chemistry, hematology, and lymphocyte subpopulations: Assessments based on controlled treatment trials in patients with neurological diseases. Muscle Nerve 1997, 20, 1102–1107. [Google Scholar] [CrossRef]
- Brem, M.D.; Jacobs, B.C.; van Rijs, W.; Fokkink, W.J.R.; Tio-Gillen, A.P.; Walgaard, C.; van Doorn, P.A.; Ijspeert, H.; van der Burg, M.; Huizinga, R. Ivig-induced plasmablasts in patients with guillain-barre syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Rigal, D.; Vermot-Desroches, C.; Heitz, S.; Bernaud, J.; Alfonsi, F.; Monier, J.C. Effects of intravenous immunoglobulins (ivig) on peripheral blood b, nk, and t cell subpopulations in women with recurrent spontaneous abortions: Specific effects on lfa-1 and cd56 molecules. Clin. Immunol. Immunopathol. 1994, 71, 309–314. [Google Scholar] [CrossRef]
- de Grandmont, M.J.; Racine, C.; Roy, A.; Lemieux, R.; Neron, S. Intravenous immunoglobulins induce the in vitro differentiation of human b lymphocytes and the secretion of igg. Blood 2003, 101, 3065–3073. [Google Scholar] [CrossRef] [Green Version]
- Kessel, A.; Peri, R.; Haj, T.; Snir, A.; Slobodin, G.; Sabo, E.; Rosner, I.; Shoenfeld, Y.; Toubi, E. Ivig attenuates tlr-9 activation in b cells from sle patients. J. Clin. Immunol. 2011, 31, 30–38. [Google Scholar] [CrossRef]
- Seite, J.F.; Guerrier, T.; Cornec, D.; Jamin, C.; Youinou, P.; Hillion, S. Tlr9 responses of b cells are repressed by intravenous immunoglobulin through the recruitment of phosphatase. J. Autoimmun. 2011, 37, 190–197. [Google Scholar] [CrossRef]
- Toyoda, M.; Zhang, X.M.; Petrosian, A.; Wachs, K.; Moudgil, A.; Jordan, S.C. Inhibition of allospecific responses in the mixed lymphocyte reaction by pooled human gamma-globulin. Transpl. Immunol. 1994, 2, 337–341. [Google Scholar] [CrossRef]
- Paquin Proulx, D.; Aubin, E.; Lemieux, R.; Bazin, R. Inhibition of b cell-mediated antigen presentation by intravenous immunoglobulins (ivig). Clin. Immunol. 2010, 135, 422–429. [Google Scholar] [CrossRef]
- Kabuto, M.; Fujimoto, N.; Tanaka, T. Increase of interleukin-10-producing b cells associated with long-term remission after i.V. Immunoglobulin treatment for pemphigus. J. Dermatol. 2016, 43, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Mori, M.; Kobayashi, Y.; Miyamae, T.; Imagawa, T.; Okuyama, T.; Kurozumi, H.; Yokota, S. Intravenous gamma-globulin therapy improves hypercytokinemia in the acute phase of kawasaki disease. Mod. Rheumatol. 2004, 14, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Ozawa, T.; Mushiake, K.; Motoyoshi, F.; Kameyama, T.; Kasahara, K.; Kaneko, H.; Yamashina, M.; Kato, Y.; Orii, T. Suppression of immunoglobulin production of lymphocytes by intravenous immunoglobulin. J. Clin. Immunol. 1991, 11, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Mazer, B.D.; Gelfand, E.W. An open-label study of high-dose intravenous immunoglobulin in severe childhood asthma. J. Allergy Clin. Immunol. 1991, 87, 976–983. [Google Scholar] [CrossRef]
- Le Pottier, L.; Bendaoud, B.; Dueymes, M.; Daridon, C.; Youinou, P.; Shoenfeld, Y.; Pers, J.O. Baff, a new target for intravenous immunoglobulin in autoimmunity and cancer. J. Clin. Immunol. 2007, 27, 257–265. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Asavapanumas, N.; Phuan, P.W.; Verkman, A.S. Potential therapeutic benefit of c1-esterase inhibitor in neuromyelitis optica evaluated in vitro and in an experimental rat model. PLoS ONE 2014, 9, e106824. [Google Scholar] [CrossRef]
- Mealy, M.A.; Shin, K.; John, G.; Levy, M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin. Exp. Neuroimmunol. 2015, 6, 413–418. [Google Scholar] [CrossRef]
- Mealy, M.A.; Levy, M. A pilot safety study of ublituximab, a monoclonal antibody against cd20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine 2019, 98, e15944. [Google Scholar] [CrossRef]
- Florez-Grau, G.; Zubizarreta, I.; Cabezon, R.; Villoslada, P.; Benitez-Ribas, D. Tolerogenic dendritic cells as a promising antigen-specific therapy in the treatment of multiple sclerosis and neuromyelitis optica from preclinical to clinical trials. Front. Immunol. 2018, 9, 1169. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.K.; Balabanov, R.; Han, X.; Burns, C.; Gastala, J.; Jovanovic, B.; Helenowski, I.; Jitprapaikulsan, J.; Fryer, J.P.; Pittock, S.J. Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 2019, 93, e1732–e1741. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I.; Fabian, M.T.; Telford, R.; Kraus, T.A.; Chehade, M.; Masilamani, M.; Moran, T.; Farrell, C.; Ebel, S.; Cook, L.J.; et al. Open-label, add-on trial of cetirizine for neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, K.; Wymbs, N.F.; Huang, H.; Mealy, M.A.; Pardo, C.A.; Zackowski, K.; Levy, M. Randomized, placebo-controlled crossover study of dalfampridine extended-release in transverse myelitis. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317740145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, D.C.; Yang, C.S.; Han, B.; Wang, J.; Yang, L.; Shi, F.D. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 2017, 74, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (n-momentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Nikoo, Z.; Badihian, S.; Shaygannejad, V.; Asgari, N.; Ashtari, F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: A randomized clinical trial. J. Neurol. 2017, 264, 2003–2009. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Qiu, W.; Ma, H.; Zhang, X.; Zhu, Z.; Yang, C.S.; Jia, D.; Zhang, T.X.; Yuan, M.; et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (tango): An open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020, 19, 391–401. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Absoud, M.; Brex, P.; Ciccarelli, O.; Diribe, O.; Giovannoni, G.; Hellier, J.; Howe, R.; Holland, R.; Kelly, J.; McCrone, P.; et al. A multicentre randomised controlled trial of intravenous immunoglobulin compared with standard therapy for the treatment of transverse myelitis in adults and children (strive). Health Technol. Assess. 2017, 21, 1–50. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Wang, J.; Zhou, Y.; Yang, H.; Wang, Z.; Yan, Z.; Long, Y.; Yin, J.; Feng, H.; Li, C.; et al. Low-dose mycophenolate mofetil for treatment of neuromyelitis optica spectrum disorders: A prospective multicenter study in south china. Front. Immunol. 2018, 9, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinley, M.P.; Moss, B.P.; Cohen, J.A. Safety of monoclonal antibodies for the treatment of multiple sclerosis. Expert Opin. Drug Saf. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Cabre, P.; Mejdoubi, M.; Jeannin, S.; Merle, H.; Plumelle, Y.; Cavillon, G.; Smadja, D.; Marignier, R.; Francophone Society of Multiple, S.; investigators, O. Treatment of neuromyelitis optica with rituximab: A 2-year prospective multicenter study. J. Neurol. 2018, 265, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, B.; Jordan, A.; Imitola, J.; Racke, M.K. Safety and efficacy of rituximab: Experience of a single multiple sclerosis center. Clin. Neuropharmacol. 2018, 41, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chai, B.; Gu, C.; Wu, R.; Dong, T.; Yao, Y.; Zhang, Y. Effectiveness of rituximab in neuromyelitis optica: A meta-analysis. BMC Neurol. 2019, 19, 36. [Google Scholar] [CrossRef] [Green Version]
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral cd19(+) b-cell counts and infusion intervals as a surrogate for long-term b-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Milo, R. Therapies for multiple sclerosis targeting b cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Jakimovski, D.; Weinstock-Guttman, B.; Ramanathan, M.; Kolb, C.; Hojnacki, D.; Minagar, A.; Zivadinov, R. Ocrelizumab: A b-cell depleting therapy for multiple sclerosis. Expert Opin. Biol. Ther. 2017, 17, 1163–1172. [Google Scholar] [CrossRef]
- Nissimov, N.; Hajiyeva, Z.; Torke, S.; Grondey, K.; Bruck, W.; Hausser-Kinzel, S.; Weber, M.S. B cells reappear less mature and more activated after their anti-cd20-mediated depletion in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 25690–25699. [Google Scholar] [CrossRef]
- Hausler, D.; Hausser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A.; Zamvil, S.S.; Bruck, W.; Lehmann-Horn, K.; Weber, M.S. Functional characterization of reappearing b cells after anti-cd20 treatment of cns autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Vallerskog, T.; Heimburger, M.; Gunnarsson, I.; Zhou, W.; Wahren-Herlenius, M.; Trollmo, C.; Malmstrom, V. Differential effects on baff and april levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenstein, M.R.; Wing, C. The baffling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ning, Q.; Jin, K.; Xie, J.; Ye, J. Does rituximab improve clinical outcomes of patients with thyroid-associated ophthalmopathy? A systematic review and meta-analysis. BMC Ophthalmol. 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Horn, K.; Schleich, E.; Hertzenberg, D.; Hapfelmeier, A.; Kumpfel, T.; von Bubnoff, N.; Hohlfeld, R.; Berthele, A.; Hemmer, B.; Weber, M.S. Anti-cd20 b-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J. Neuroinflamm. 2011, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Dorner, T.; Burmester, G.R. New approaches of b-cell-directed therapy: Beyond rituximab. Curr. Opin. Rheumatol. 2008, 20, 263–268. [Google Scholar] [CrossRef]
- Soe, Z.N.; Allsup, D. The use of ofatumumab in the treatment of b-cell malignancies. Future Oncol. 2017, 13, 2611–2628. [Google Scholar] [CrossRef]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The mirror study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef]
- Kurrasch, R.; Brown, J.C.; Chu, M.; Craigen, J.; Overend, P.; Patel, B.; Wolfe, S.; Chang, D.J. Subcutaneously administered ofatumumab in rheumatoid arthritis: A phase i/ii study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J. Rheumatol. 2013, 40, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Lovett-Racke, A.E.; Gormley, M.; Liu, Y.; Petracca, M.; Cocozza, S.; Shubin, R.; Wray, S.; Weiss, M.S.; Bosco, J.A.; et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-cd20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult. Scler. 2020. [Google Scholar] [CrossRef]
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a b cell-depleting anti-cd19 antibody for the treatment of autoimmune neurological diseases: Insights from preclinical studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Inebilizumab: First approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Bargou, R.C.; Nagorsen, D.; Friberg, G.R.; Baeuerle, P.A.; Forman, S.J. Redirecting t cells to eradicate b-cell acute lymphoblastic leukemia: Bispecific t-cell engagers and chimeric antigen receptors. Leukemia 2017, 31, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic efficacy of anti-cd19 car-t cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 2020. [CrossRef]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. Btk signaling in b cell differentiation and autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [PubMed]
- Deeks, E.D. Ibrutinib: A review in chronic lymphocytic leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Dhillon, S. Acalabrutinib: First global approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-controlled trial of an oral btk inhibitor in multiple sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Kleiter, I.; Schroder, A.; Hellwig, K.; Chan, A.; Yamamura, T.; Gold, R. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-cd20 therapy. JAMA Neurol. 2013, 70, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Mori, M.; Uzawa, A.; Masuda, H.; Muto, M.; Ohtani, R.; Kuwabara, S. Increased cerebrospinal fluid metalloproteinase-2 and interleukin-6 are associated with albumin quotient in neuromyelitis optica: Their possible role on blood-brain barrier disruption. Mult. Scler. 2017, 23, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Kishimoto, T. Il-6-dependent and -independent pathways in the development of interleukin 17-producing t helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Matsuoka, T.; Miyamoto, K.; Kusunoki, S.; Okamoto, T.; Murata, M.; Miyake, S.; Aranami, T.; Yamamura, T. Efficacy of the anti-il-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology 2014, 82, 1302–1306. [Google Scholar] [CrossRef] [Green Version]
- Ringelstein, M.; Ayzenberg, I.; Harmel, J.; Lauenstein, A.S.; Lensch, E.; Stogbauer, F.; Hellwig, K.; Ellrichmann, G.; Stettner, M.; Chan, A.; et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015, 72, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Ectrims 2019 committees. Mult. Scler. 2019, 25, 1–2. [CrossRef]
- Moura, R.A.; Quaresma, C.; Vieira, A.R.; Goncalves, M.J.; Polido-Pereira, J.; Romao, V.C.; Martins, N.; Canhao, H.; Fonseca, J.E. B-cell phenotype and igd-cd27- memory b cells are affected by tnf-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS ONE 2017, 12, e0182927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiao, D.M.; Qin, W.; Xie, B.H.; Wang, T.H.; Huang, H.; Zhao, B.J.; Han, X.; Sun, Q.Q.; Wu, X.D.; et al. The clinical value of hematological markers in rheumatoid arthritis patients treated with tocilizumab. J. Clin. Lab. Anal. 2019, 33, e22862. [Google Scholar] [CrossRef]
- Roll, P.; Muhammad, K.; Schumann, M.; Kleinert, S.; Einsele, H.; Dorner, T.; Tony, H.P. In vivo effects of the anti-interleukin-6 receptor inhibitor tocilizumab on the b cell compartment. Arthritis Rheum. 2011, 63, 1255–1264. [Google Scholar] [CrossRef]
- Choi, I.A.; Lee, S.J.; Park, W.; Park, S.H.; Shim, S.C.; Baek, H.J.; Yoo, D.H.; Kim, H.A.; Lee, S.K.; Lee, Y.J.; et al. Effects of tocilizumab therapy on serum interleukin-33 and interleukin-6 levels in patients with rheumatoid arthritis. Arch. Rheumatol. 2018, 33, 389–394. [Google Scholar] [CrossRef]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered ph-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Heo, Y.A. Satralizumab: First approval. Drugs 2020, 80, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Brachet, G.; Bourquard, T.; Gallay, N.; Reiter, E.; Gouilleux-Gruart, V.; Poupon, A.; Watier, H. Eculizumab epitope on complement c5: Progress towards a better understanding of the mechanism of action. Mol. Immunol. 2016, 77, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Eculizumab: A review in generalized myasthenia gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Eculizumab: A review in neuromyelitis optica spectrum disorder. Drugs 2020, 80, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, F.; Murphy, O.; Pardo, S.; Levy, M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin. Investig. Drugs 2018, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Weitz, I.C.; Razavi, P.; Rochanda, L.; Zwicker, J.; Furie, B.; Manly, D.; Mackman, N.; Green, R.; Liebman, H.A. Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with pnh independent of its effects on hemolysis and microparticle formation. Thromb. Res. 2012, 130, 361–368. [Google Scholar] [CrossRef]
- Alfinito, F.; Ruggiero, G.; Sica, M.; Udhayachandran, A.; Rubino, V.; Della Pepa, R.; Palatucci, A.T.; Annunziatella, M.; Notaro, R.; Risitano, A.M.; et al. Eculizumab treatment modifies the immune profile of pnh patients. Immunobiology 2012, 217, 698–703. [Google Scholar] [CrossRef]
- Sheridan, D.; Yu, Z.X.; Zhang, Y.; Patel, R.; Sun, F.; Lasaro, M.A.; Bouchard, K.; Andrien, B.; Marozsan, A.; Wang, Y.; et al. Design and preclinical characterization of alxn1210: A novel anti-c5 antibody with extended duration of action. PLoS ONE 2018, 13, e0195909. [Google Scholar] [CrossRef] [Green Version]
- Ravulizumab. In Drugs and Lactation Database (Lactmed); National Library of Medicine (US): Bethesda, MD, USA, 2006.
- Verkman, A.S.; Phuan, P.W.; Asavapanumas, N.; Tradtrantip, L. Biology of aqp4 and anti-aqp4 antibody: Therapeutic implications for nmo. Brain Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Smith, A.J.; Phuan, P.W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef]
- Duan, T.; Tradtrantip, L.; Phuan, P.W.; Bennett, J.L.; Verkman, A.S. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 2020, 162, 107827. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Rossiter, D.; Fehmi, J.; Stevens, J.C.; Renowden, S.A.; Cohen, N.; Bailey, C.; Scolding, N.J. Tumefactive demyelination presenting during bevacizumab treatment. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker-Caulfield, M.E.; Guo, Y.; Johnson, R.K.; McCarthy, C.B.; Fitz-Gibbon, P.D.; Lucchinetti, C.F.; Howe, C.L. Nfkappab signaling drives pro-granulocytic astroglial responses to neuromyelitis optica patient igg. J. Neuroinflamm. 2015, 12, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H. Bortezomib for neuromyelitis optica spectrum disorder: A new therapeutic option for the more severe forms? JAMA Neurol. 2018, 75, 128–129. [Google Scholar] [CrossRef]
- Zhang, H.; Verkman, A.S. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J. Clin. Investig. 2013, 123, 2306–2316. [Google Scholar] [CrossRef]
- Muroishi, T.; Sakai, K.; Yanase, D.; Ikeda, Y.; Machiya, T.; Kato-Motozaki, Y.; Samuraki, M.; Yamada, M. Serum anti-aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder presenting as acute eosinophilic encephalomyelitis. J. Clin. Neurosci. 2018, 48, 93–94. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin g-induced damage in mouse brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef]
- Herges, K.; de Jong, B.A.; Kolkowitz, I.; Dunn, C.; Mandelbaum, G.; Ko, R.M.; Maini, A.; Han, M.H.; Killestein, J.; Polman, C.; et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult. Scler. 2012, 18, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The baff/april system in sle pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Okada, K.; Matsushita, T.; Kira, J.; Tsuji, S. B-cell activating factor of the tnf family is upregulated in neuromyelitis optica. Neurology 2010, 74, 177–178. [Google Scholar] [CrossRef] [PubMed]
- 2019 acr/arp annual meeting abstract supplement. Arthritis Rheumatol. 2019, 71 (Suppl. S10), 1–5362. [CrossRef] [PubMed]
- Chamberlain, J.L.; Huda, S.; Whittam, D.H.; Matiello, M.; Morgan, B.P.; Jacob, A. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: A brief review. J. Neurol. 2019. [Google Scholar] [CrossRef]
- Levy, M.; Mealy, M.A. Purified human c1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Hakobyan, S.; Luppe, S.; Evans, D.R.; Harding, K.; Loveless, S.; Robertson, N.P.; Morgan, B.P. Plasma complement biomarkers distinguish multiple sclerosis and neuromyelitis optica spectrum disorder. Mult. Scler. 2017, 23, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Ha, S.J. Generation of tolerogenic dendritic cells and their therapeutic applications. Immune Netw. 2016, 16, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase i (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.; Tolf, A.; Hagglund, H.; Askmark, H. Autologous haematopoietic stem cell transplantation for neurological diseases. J. Neurol. Neurosurg. Psychiatry 2018, 89, 147–155. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, B. Effect of autologous hematopoietic stem cell transplantation on multiple sclerosis and neuromyelitis optica spectrum disorder: A prisma-compliant meta-analysis. Bone Marrow Transplant. 2020, 55, 1928–1934. [Google Scholar] [CrossRef]
- Ceglie, G.; Papetti, L.; Valeriani, M.; Merli, P. Hematopoietic stem cell transplantation in neuromyelitis optica-spectrum disorders (nmo-sd): State-of-the-art and future perspectives. Int. J. Mol. Sci. 2020, 21, 5304. [Google Scholar] [CrossRef]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate b-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ye, D.; Qian, L.; Zhu, L.; Wang, C.; Guan, D.; Zhang, X.; Xu, Y. Human umbilical cord mesenchymal stem cell therapy on neuromyelitis optica. Curr. Neurovasc. Res. 2012, 9, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Dulamea, A.O.; Sirbu-Boeti, M.P.; Bleotu, C.; Dragu, D.; Moldovan, L.; Lupescu, I.; Comi, G. Autologous mesenchymal stem cells applied on the pressure ulcers had produced a surprising outcome in a severe case of neuromyelitis optica. Neural. Regen. Res. 2015, 10, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Korsen, M.; Kunz, R.; Schminke, U.; Runge, U.; Kohlmann, T.; Dressel, A. Dalfampridine effects on cognition, fatigue, and dexterity. Brain Behav. 2017, 7, e00559. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, X.; Liu, X.; Mu, W.; Yang, W.; Liu, Y.; Ge, P.; Li, H. Patient with neuromyelitis optica spectrum disorder combined with sjogren’s syndrome relapse free following tacrolimus treatment. Intern. Med. 2014, 53, 2377–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wu, Q.; Ke, G.; Bu, B. Efficacy and safety of tacrolimus treatment for neuromyelitis optica spectrum disorder. Sci. Rep. 2017, 7, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, M.; Oji, S.; Tanaka, S.; Izaki, S.; Hashimoto, B.; Fukaura, H.; Nomura, K. Tacrolimus is effective for neuromyelitis optica spectrum disorders with or without anti-aqp4 antibody. Mult. Scler. Relat. Disord. 2019, 39, 101907. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Frati, F.; Ridolo, E.; Greco, A.; de Vincentiis, M.; Masieri, S.; Makri, E.; Incorvaia, C. The spectrum of therapeutic activity of mepolizumab. Expert Rev. Clin. Immunol. 2019, 15, 959–967. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Ratelade, J.; Zhang, H.; Verkman, A.S. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin g into therapeutic antibody. Ann. Neurol. 2013, 73, 77–85. [Google Scholar] [CrossRef]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radu, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (atams): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Hoffman, R.W.; Merrill, J.T.; Alarcon-Riquelme, M.M.; Petri, M.; Dow, E.R.; Nantz, E.; Nisenbaum, L.K.; Schroeder, K.M.; Komocsar, W.J.; Perumal, N.B.; et al. Gene expression and pharmacodynamic changes in 1,760 systemic lupus erythematosus patients from two phase iii trials of baff blockade with tabalumab. Arthritis Rheumatol. 2017, 69, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Duggan, S.T. Belimumab: A review in systemic lupus erythematosus. Drugs 2018, 78, 355–366. [Google Scholar] [CrossRef]
- Dorner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.A.; Simonett, C.; Mooney, L.; et al. Treatment of primary sjogren’s syndrome with ianalumab (vay736) targeting b cells by baff receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.; Zamvil, S.S. Aquaporin 4-specific t cells in neuromyelitis optica exhibit a th17 bias and recognize clostridium abc transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Approved (*) and experimental B cell- and antibody-targeting treatment options in neuromyelitis optica spectrum disorders. B cell development begins in the bone marrow, where hematopoietic stem cells proliferate into pro- and pre-B cells. Immature B cells, the earliest B cell subset in the peripheral blood undergo maturation into transitional, mature and memory B cells upon stimuli like interlukin-6 (IL-6), B cell activating factor (BAFF) and A proliferation-inducing ligand (APRIL). Plasmablasts and plasma cells are capable of producing immunoglobulins, including pathological anti-aquaporin-4 (AQP4) immunoglobulin G (IgG). After binding AQP4 on astrocytic foot processes at the blood-brain barrier, complement activation (including complement factor 5(C5)) causes cytotoxicity by forming membrane attack complexes with subsequent neuronal injury. BAFFR = BAFF receptor; BCMA = B cell maturation antigen; BTK = Bruton’s tyrosine kinase; CAR = chimeric antigen receptors; CD = cluster of differentiation; IL-6R = interleukin-6 receptor; TACI = transmembrane activator and calcium-modulator and cyclophilin-ligand activator.
Figure 1.
Approved (*) and experimental B cell- and antibody-targeting treatment options in neuromyelitis optica spectrum disorders. B cell development begins in the bone marrow, where hematopoietic stem cells proliferate into pro- and pre-B cells. Immature B cells, the earliest B cell subset in the peripheral blood undergo maturation into transitional, mature and memory B cells upon stimuli like interlukin-6 (IL-6), B cell activating factor (BAFF) and A proliferation-inducing ligand (APRIL). Plasmablasts and plasma cells are capable of producing immunoglobulins, including pathological anti-aquaporin-4 (AQP4) immunoglobulin G (IgG). After binding AQP4 on astrocytic foot processes at the blood-brain barrier, complement activation (including complement factor 5(C5)) causes cytotoxicity by forming membrane attack complexes with subsequent neuronal injury. BAFFR = BAFF receptor; BCMA = B cell maturation antigen; BTK = Bruton’s tyrosine kinase; CAR = chimeric antigen receptors; CD = cluster of differentiation; IL-6R = interleukin-6 receptor; TACI = transmembrane activator and calcium-modulator and cyclophilin-ligand activator.


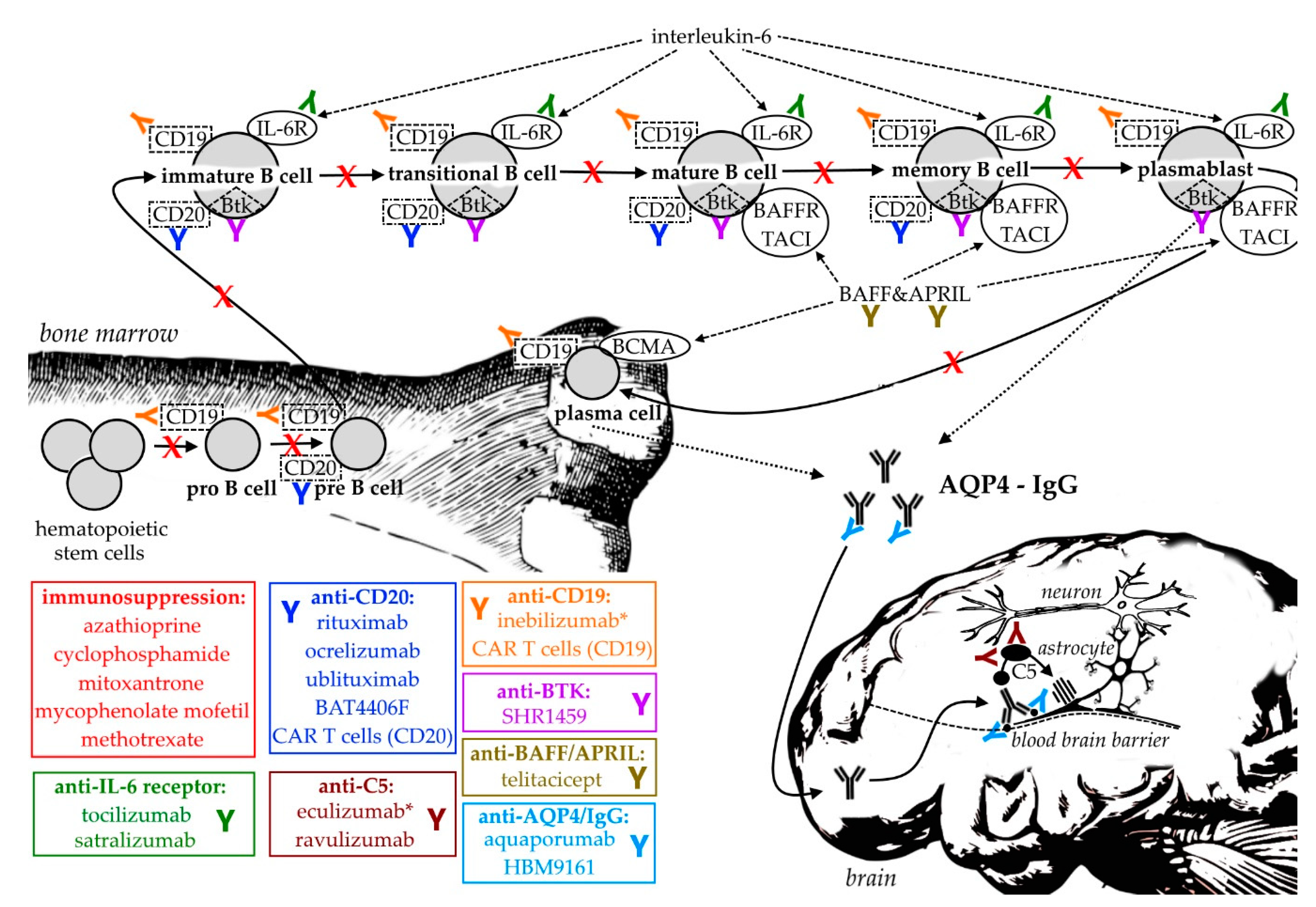{kind=link}
Table 1.
Interventional clinical studies including neuromyelitis optica spectrum disorders (NMOSD) patients as listed on clinicaltrials.gov (accessed December 2020). APRIL = A proliferation-inducing ligand; BAFF = B cell activating factor; BTK = Bruton’s tyrosine kinase; C1 = complement factor 1; C5 = complement factor 5; CAR = chimeric antigen receptors; CD = cluster of differentiation; H1 = histamine receptor 1; IL-6 = interleukin-6; K+ = potassium; VEGF = vascular epithelial growth factor.
Table 1.
Interventional clinical studies including neuromyelitis optica spectrum disorders (NMOSD) patients as listed on clinicaltrials.gov (accessed December 2020). APRIL = A proliferation-inducing ligand; BAFF = B cell activating factor; BTK = Bruton’s tyrosine kinase; C1 = complement factor 1; C5 = complement factor 5; CAR = chimeric antigen receptors; CD = cluster of differentiation; H1 = histamine receptor 1; IL-6 = interleukin-6; K+ = potassium; VEGF = vascular epithelial growth factor.
| Clinical Trials.gov Identifier | Phase | Year Study Started | Year Approved | Intervention | Target | Participants | Status (12/2020) | Results | Section |
|---|---|---|---|---|---|---|---|---|---|
| Phase 1 | |||||||||
| NCT00501748 | 1 | 2004 | n/a | Rituximab | CD20 | 20 | completed | n/a | Section 3.2.1 |
| NCT01759602 | 1 | 2013 | n/a | C1 esterase inhibitor | C1 esterase | 10 | completed | [106] | Section 3.8.6 |
| NCT01777412 | 1 | 2013 | n/a | Bevacizumab | VEGF | 10 | completed | [107] | Section 3.8.1 |
| NCT02087813 | 1 | 2014 | n/a | Alpha1-antitrypsin | n/a | 0 | withdrawn | n/a | Section 3.8.11 |
| NCT02276963 | 1 | 2016 | n/a | Ublituximab | CD20 | 6 | completed | [108] | Section 3.2.4 |
| NCT02283671 | 1 | 2015 | n/a | Tolerogenic dendritic cells | n/a | 10 | completed | [109] | Section 3.8.7 |
| NCT03605238 | 1 | 2018 | n/a | Tandem CAR T cells | CD19/CD20 | 0 | withdrawn | n/a | Section 3.3.2 |
| NCT04146285 | 1 | 2019 | n/a | BAT4406F | CD20 | 48 | not yet recruiting | n/a | Section 3.2.5 |
| NCT04227470 | 1 | 2020 | n/a | HBM9161 | CD20 | 12 | recruiting | n/a | Section 3.7.2 |
| Phase 1/2 | |||||||||
| NCT00787722 | 1/2 | 2009 | n/a | Autologous stem cells | n/a | 13 | completed | [110] | Section 3.8.8 |
| NCT00904826 | 1/2 | 2009 | 2019 | Eculizumab | C5 | 14 | completed | [111] | Section 3.6.1 |
| NCT01339455 | 1/2 | 2011 | n/a | Autologous stem cells | n/a | 3 | terminated | n/a | Section 3.8.8 |
| NCT01364246 | 1/2 | 2010 | n/a | Mesenchymal stromal cells | n/a | 20 | unknown | n/a | Section 3.8.9 |
| NCT02865018 | 1/2 | 2014 | n/a | Cetirizine | H1 | 16 | completed | [112] | Section 3.8.3 |
| NCT03062579 | 1/2 | 2017 | n/a | Tocilizumab | IL-6 receptor | 10 | completed | n/a | Section 3.5.1 |
| Phase 2 | |||||||||
| NCT00716066 | 2 | 2008 | n/a | Autologous stem cells | n/a | 40 | recruiting | n/a | Section 3.8.8 |
| NCT01845584 | 2 | 2013 | n/a | Intravenous IgG | n/a | 7 | completed | n/a | Section 3.1.6 |
| NCT02166346 | 2 | 2014 | n/a | Dalfampridine | K+ channel | 24 | completed | [113] | Section 3.8.10 |
| NCT02249676 | 2 | 2013 | n/a | Mesenchymal stromal cells | n/a | 15 | completed | n/a | Section 3.8.9 |
| NCT02893111 | 2 | 2015 | n/a | Bortezomib | proteasome | 5 | completed | [114] | Section 3.8.2 |
| NCT04064944 | 2 | 2019 | n/a | Immunoabsorption | n/a | 144 | not yet recruiting | n/a | Section 3.1.1 |
| NCT04670770 | 2 | 2020 | n/a | SHR1459 | BTK | 10 | not yet recruiting | n/a | Section 3.4.1 |
| Phase 2/3 | |||||||||
| NCT02200770 | 2/3 | 2015 | 2020 | Inebilizumab | CD19 | 231 | active, not recruiting | [115] | Section 3.3.1 |
| NCT03002038 | 2/3 | 2015 | n/a | Rituximab | CD20 | 76 | completed | [116] | Section 3.2.1 |
| NCT03350633 | 2/3 | 2017 | n/a | Tocilizumab | IL-6 receptor | 118 | completed | [117] | Section 3.5.1 |
| NCT03829566 | 2/3 | 2019 | n/a | Autologous stem cells | n/a | 0 | withdrawn | n/a | Section 3.8.8 |
| NCT04155424 | 2/3 | 2020 | 2019 | Eculizumab | C5 | 15 | recruiting | n/a | Section 3.6.1 |
| Phase 3 | |||||||||
| NCT00004645 | 3 | 1995 | n/a | Plasma exchange | n/a | 22 | unknown | n/a | Section 3.1.1 |
| NCT01892345 | 3 | 2014 | 2019 | Eculizumab | C5 | 143 | terminated | [111] | Section 3.6.1 |
| NCT02003144 | 3 | 2015 | 2019 | Eculizumab | C5 | 119 | active, not recruiting | n/a | Section 3.6.1 |
| NCT02028884 | 3 | 2014 | n/a | Satralizumab | IL-6 receptor | 83 | active, not recruiting | [118] | Section 3.5.2 |
| NCT02073279 | 3 | 2014 | n/a | Satralizumab | IL-6 receptor | 95 | active, not recruiting | [119] | Section 3.5.2 |
| NCT02398994 | 3 | 2015 | n/a | Intravenous IgG | n/a | 2 | terminated | [120] | Section 3.1.6 |
| NCT03330418 | 3 | 2017 | n/a | Telitacicept | APRIL/BAFF | 118 | recruiting | n/a | Section 3.8.5 |
| NCT04201262 | 3 | 2019 | n/a | Ravulizumab | C5 | 55 | recruiting | n/a | Section 3.6.2 |
| NCT04660539 | 3 | 2021 | n/a | Satralizumab | IL-6 receptor | 127 | not yet recruituing | n/a | Section 3.5.2 |
| Phase 4 | |||||||||
| NCT00304291 | 4 | 2001 | n/a | Mitoxantrone | n/a | 5 | completed | [67] | Section 3.1.4 |
| NCT02021825 | 4 | 2009 | n/a | Mitoxantrone | n/a | 50 | unknown | n/a | Section 3.1.4 |
| NCT02809079 | 4 | 2016 | n/a | Mycophenolate mofetil | n/a | 100 | unknown | [121] | Section 3.1.5 |
| NCT04256252 | 4 | 2014 | n/a | Rituximab | CD20 | 100 | terminated | n/a | Section 3.2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Traub, J.; Husseini, L.; Weber, M.S. B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders. Pharmaceuticals 2021, 14, 37. https://doi.org/10.3390/ph14010037
AMA Style
Traub J, Husseini L, Weber MS. B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders. Pharmaceuticals. 2021; 14(1):37. https://doi.org/10.3390/ph14010037
Chicago/Turabian StyleTraub, Jan, Leila Husseini, and Martin S. Weber. 2021. "B Cells and Antibodies as Targets of Therapeutic Intervention in Neuromyelitis Optica Spectrum Disorders" Pharmaceuticals 14, no. 1: 37. https://doi.org/10.3390/ph14010037
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.