Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Demographics and Whole Genome Sequencing (WGS) In-Silico Analysis of Methicillin-Resistant Staphylococcus Aureus (MRSA)
2.2. Phenotypic and Genotypic Characteristics of the 11 MRSA Belonging to the ST612-CC8-t1257-SCCmec_IVd(2B) and Sister Lineages
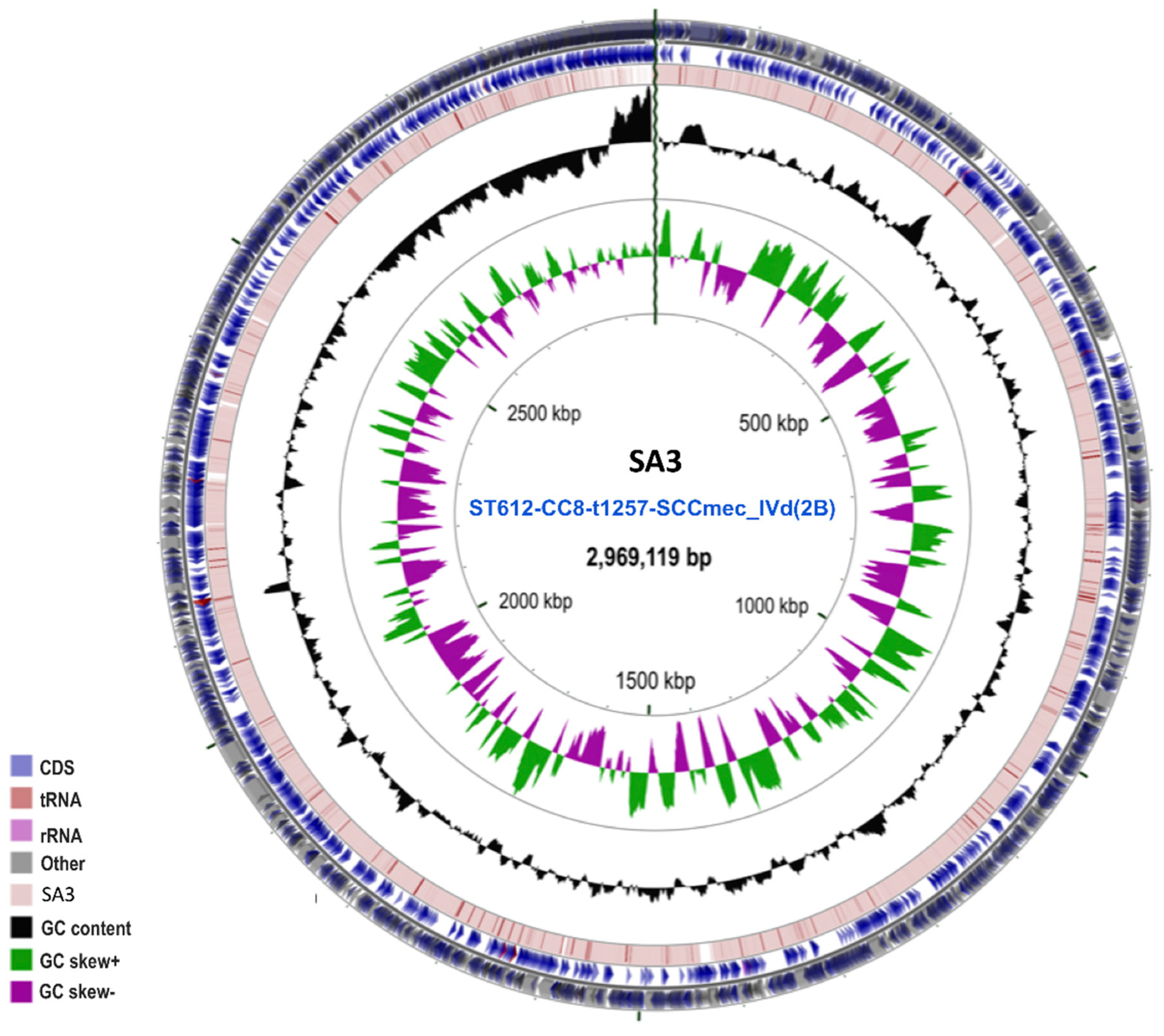
2.3. General Features of Endemic Clonal Subtype ST612-CC8-t1257-SCCmec_IVd(2B)
2.4. In-Silico Prediction of Clustered, Regularly Interspaced, Short Palindromic Repeats (CRISPRs), Restriction-Modification System (R-M system), Accessory Gene Regulator (Agr) Type, and Arginine Catabolic Mobile Element (ACME)
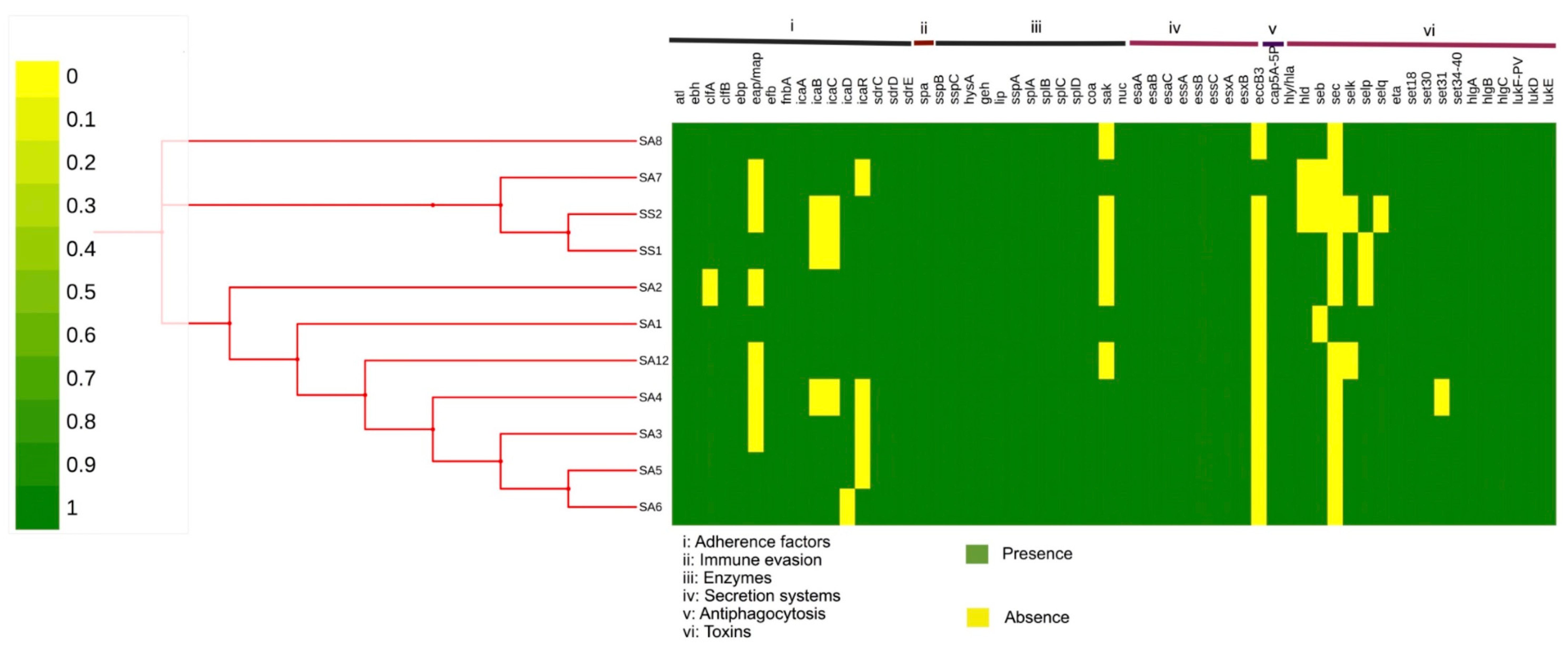
2.5. Pathogenicity and Virulome Insights of the Endemic Clone
2.6. Genomic Encoding Mechanisms and Pathways of Bacterial Persistence and Tolerance
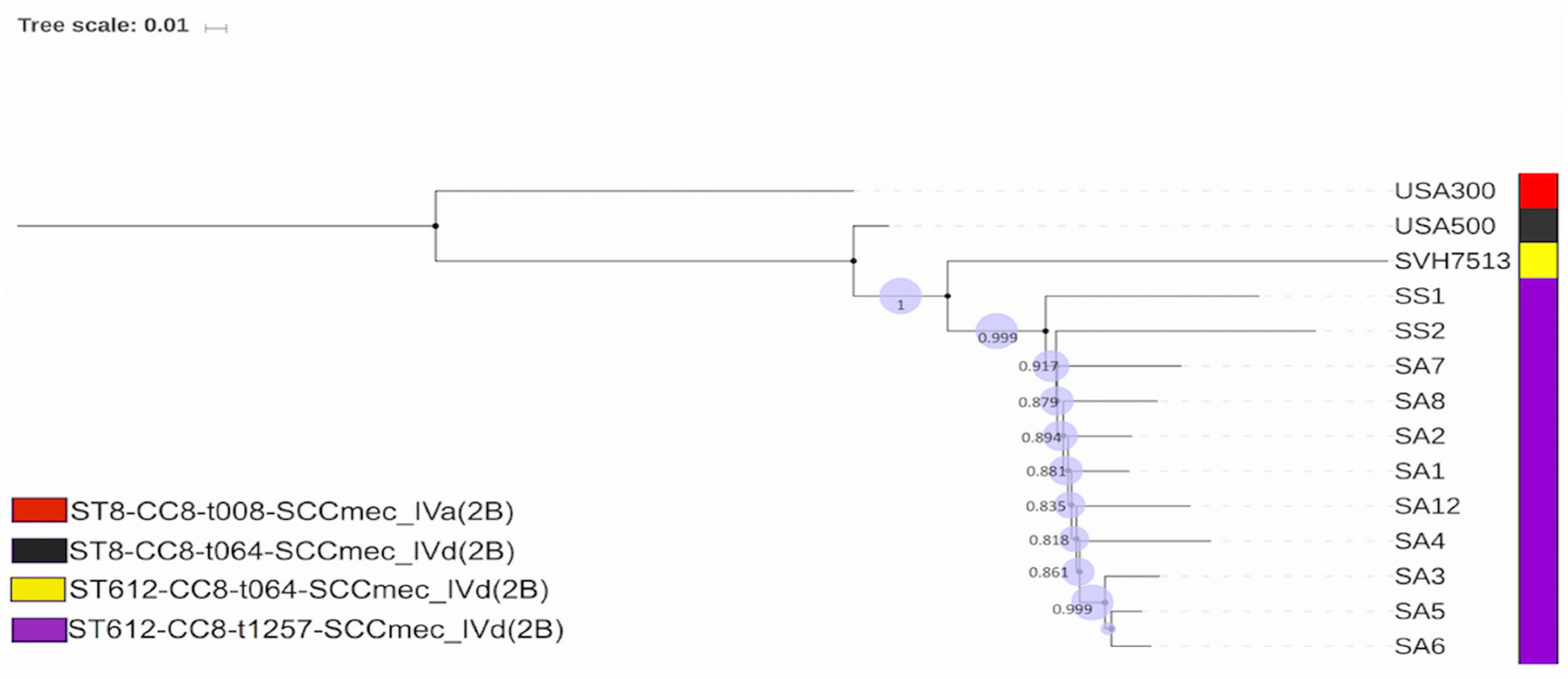
2.7. Comparative Phylogenomic Insights of the Endemic Clone
3. Discussion
4. Materials and Methods
4.1. Study Design and Identification of MRSA
4.1.1. Isolation, Identification and Molecular confirmation of S. aureus
4.1.2. Detection of MRSA and Antibiotic resistance testing
4.2. Whole Genome Sequencing (WGS) Analysis and Characterisation
4.2.1. Purification, Sequencing and Pre-Processing of Genomic Data
4.2.2. WGS-Based Molecular Typing of the Obtained MRSA
4.2.3. In-Silico Resistome Profiling
4.3. Genome Visualization and Gene Annotation
4.4. Detection of CRISPR Array, Restriction-Modification System (R-M system), Accessory Gene Regulator (agr) Type, Arginine Catabolic Mobile Element (ACME)
4.5. Genomic Insights of the Isolates in the Endemic Clone
4.5.1. Pathogenicity and Virulome Predictions
4.5.2. Genomic Prediction of Mechanisms of Bacterial Persistence and Tolerance
4.6. Comparative Phylogenomic Analysis and Metadata Insights
5. Conclusions
6. Ethical Considerations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACME | Arginine Catabolic Mobile Element |
| COGs | Clusters of Orthologous Groups |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat |
| PGAP | Prokaryotic Genome Annotation Pipeline |
| RAST | Rapid Annotation using Subsystem Technology |
| R-M System | Restriction-Modification System |
| MSCRAMMs | Microbial Surface Components Recognizing Adhesive Matrix Molecules |
| MDR | Multi Drug Resistance |
| MRSA | Methicillin Resistance Staphylococcus aureus |
| SCC | Staphylococcal Cassette Chromosome |
| MLST | Multilocus Sequence Typing |
| CC | Clonal Complexes |
References
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria To Guide Research, Discovery, And Development Of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Abdulgader, S.M.; Shittu, A.O.; Nicol, M.P.; Kaba, M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: A systematic review. Front. Microbiol. 2015, 6, 348. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.M.; Coombs, G.W.; Holden, M.T.G.; Lindsay, J.A.; Nimmo, G.R.; Tattevin, P.; Skov, R.L. Genomic insights into the emergence and spread of international clones of healthcare-, community- and livestock-associated methicillin-resistant Staphylococcus aureus: blurring of the traditional definitions. J. Glob. Antimicrob. Resist. 2016, 6, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lim, V.W.; Khan, A.; Pettigrew, K.; Lye, D.C.B.; Kanagasabai, K.; Phua, K.; Krishnan, P.; Ang, B.; Marimuthu, K.; et al. MRSA transmission dynamics among interconnected acute, intermediate-term, and long-term healthcare facilities in Singapore. Clin. Infect. Dis. 2017, 64, S76–S81. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Carrel, M.; Perencevich, E.N.; David, M.Z.; Goel, N. Community-Associated Methicillin-Resistant Staphylococcus aureus Infections in the Athlete. Emerg. Infect. Dis. 2009, 21, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Dierikx, C.M.; Hengeveld, P.D.; Veldman, K.T.; de Haan, A.; van der Voorde, S.; Dop, P.Y.; Bosch, T.; van Duijkeren, E. Ten years later: Still a high prevalence of MRSA in slaughter pigs despite a significant reduction in antimicrobial usage in pigs the Netherlands. J. Antimicrob. Chemother. 2016, 71, 2414–2418. [Google Scholar] [CrossRef] [PubMed]
- Sallam, K.I.; Abd-Elghany, S.M.; Elhadidy, M.; Tamura, T. Molecular Characterization and Antimicrobial Resistance Profile of Methicillin-Resistant Staphylococcus aureus in Retail Chicken. J. Food Prot. 2015, 78, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Silva, J.; Teixeira, P. Staphylococcus aureus, a Food Pathogen: Virulence Factors and Antibiotic Resistance. In Foodborne Diseases; Elsevier: Amsterdam, The Netherlands, 2018; pp. 213–238. ISBN 9780128114964. [Google Scholar]
- Kadariya, J.; Smith, T.C.; Thapaliya, D. Staphylococcus aureus and staphylococcal food-borne disease: An ongoing challenge in public health. Biomed Res. Int. 2014, 2014, 827965. [Google Scholar] [CrossRef]
- Thapaliya, D.; Forshey, B.M.; Kadariya, J.; Quick, M.K.; Farina, S.; O’Brien, A.; Nair, R.; Nworie, A.; Hanson, B.; Kates, A.; et al. Prevalence and molecular characterization of Staphylococcus aureus in commercially available meat over a one-year period in Iowa, USA. Food Microbiol. 2017, 65, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Pantosti, A. Methicillin-resistant Staphylococcus aureus associated with animals and its relevance to human health. Front. Microbiol. 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Petinaki, E.; Spiliopoulou, I. Methicillin-resistant Staphylococcus aureus colonization and infection risks from companion animals: Current perspectives. Vet. Med. (Auckl. N. Z.) 2015, 6, 373–382. [Google Scholar] [CrossRef]
- Ambrosio, C.M.S.; de Alencar, S.M.; de Sousa, R.L.M.; Moreno, A.M.; Da Gloria, E.M. Antimicrobial activity of several essential oils on pathogenic and beneficial bacteria. Ind. Crop. Prod. 2017, 97, 128–136. [Google Scholar] [CrossRef]
- Zhou, Y.P.; Wilder-Smith, A.; Hsu, L.Y. The role of international travel in the spread of methicillin-resistant staphylococcus aureus. J. Travel Med. 2014, 21, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Jansen van Rensburg, M.J.; Eliya Madikane, V.; Whitelaw, A.; Chachage, M.; Haffejee, S.; Gay Elisha, B. The dominant methicillin-resistant Staphylococcus aureus clone from hospitals in Cape Town has an unusual genotype: ST612. Clin. Microbiol. Infect. 2011, 17, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Antiabong, J.F.; Kock, M.M.; Maphanga, T.G.; Salawu, A.M.; Mbelle, N.M.; Ehlers, M.M. Trends in the Genetic Background of Methicillin-Resistant Staphylococcus Aureus Clinical Isolates in a South African Hospital: An Institutional-Based Observational Study. Open Microbiol. J. 2017, 11, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Harbarth, S. Control of endemic methicillin-resistant Staphylococcus aureus—Recent advances and future challenges. Clin. Microbiol. Infect. 2006, 12, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.R.; David, M.Z.; Salata, R.A. Current concepts on the virulence mechanisms of meticillin-resistant Staphylococcus aureus. J. Med. Microbiol. 2012, 61, 1179–1193. [Google Scholar] [CrossRef] [Green Version]
- DeLeo, F.R.; Diep, B.A.; Otto, M. Host defense and pathogenesis in Staphylococcus aureus infections. Infect. Dis. Clin. North Am. 2009, 23, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. MRSA virulence and spread. Cell. Microbiol. 2012, 14, 1513–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, K.Y.L.; Stinear, T.P.; Howden, B.P. Functional genomics of staphylococcus aureus. Brief. Funct. Genom. 2013, 12, 305–315. [Google Scholar] [CrossRef]
- Axon, J.E.; Carrick, J.B.; Barton, M.D.; Collins, N.M.; Russell, C.M.; Kiehne, J.; Coombs, G. Methicillin-resistant Staphylococcus aureus in a population of horses in Australia. Aust. Vet. J. 2011, 89, 221–225. [Google Scholar] [CrossRef]
- Ji, Y. Methicillin-Resistant Staphylococcus Aureus (MRSA) protocols. In Methods in Molecular Biology; Ji, Y., Ed.; Humana Press: Totowa, NJ, USA, 2014; Volume 1085, ISBN 978-1-62703-663-4. [Google Scholar]
- Groves, M.D.; Crouch, B.; Coombs, G.W.; Jordan, D.; Pang, S.; Barton, M.D.; Giffard, P.; Abraham, S.; Trott, D.J. Molecular epidemiology of methicillin-resistant Staphylococcus aureus isolated from Australian veterinarians. PLoS ONE 2016, 11, e0146034. [Google Scholar] [CrossRef] [PubMed]
- Saputra, S.; Jordan, D.; Worthing, K.A.; Norris, J.M.; Wong, H.S.; Abraham, R.; Trott, D.J.; Abraham, S. Antimicrobial resistance in coagulase-positive staphylococci isolated from companion animals in Australia: A one year study. PLoS ONE 2017, 12, e0176379. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Otto, M. Improved understanding of factors driving methicillin-resistant Staphylococcus aureus epidemic waves. Clin. Epidemiol. 2013, 5, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen van Rensburg, M.J.; Whitelaw, A.C.; Elisha, B.G. Genetic basis of rifampicin resistance in methicillin-resistant Staphylococcus aureus suggests clonal expansion in hospitals in Cape Town, South Africa. BMC Microbiol. 2012, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Perovic, O.; Singh-Moodley, A.; Govender, N.P.; Kularatne, R.; Whitelaw, A.; Chibabhai, V.; Naicker, P.; Mbelle, N.; Lekalakala, R.; Quan, V.; et al. A small proportion of community-associated methicillin-resistant Staphylococcus aureus bacteraemia, compared to healthcare-associated cases, in two South African provinces. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Perovic, O.; Iyaloo, S.; Kularatne, R.; Lowman, W.; Bosman, N.; Wadula, J.; Seetharam, S.; Duse, A.; Mbelle, N.; Bamford, C.; et al. Prevalence and trends of staphylococcus aureus bacteraemia in hospitalized patients in South Africa, 2010 to 2012: Laboratory-based surveillance mapping of antimicrobial resistance and molecular epidemiology. PLoS ONE 2015, 10, e0145429. [Google Scholar] [CrossRef] [PubMed]
- Moodley, A.; Oosthuysen, W.F.; Dusé, A.G.; Marais, E.; South African MRSA Surveillance Group. Molecular characterization of clinical methicillin-resistant Staphylococcus aureus isolates in South Africa. J. Clin. Microbiol. 2010, 48, 4608–4611. [Google Scholar] [CrossRef] [PubMed]
- Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.; Essack, S.Y. Genomic analysis of methicillin-resistant Staphylococcus aureus isolated from poultry and occupational farm workers in Umgungundlovu District, South Africa. Sci. Total Environ. 2019, 670, 704–716. [Google Scholar] [CrossRef]
- Amoako, D.G.; Bester, L.A.; Somboro, A.M.; Baijnath, S.; Govind, C.N.; Essack, S.Y. Plasmid-mediated resistance and virulence mechanisms in the private health sector in KwaZulu-Natal, South Africa: An investigation of methicillin resistant Staphylococcus aureus (MRSA) clinical isolates collected during a three month period. Int. J. Infect. Dis. 2016, 46, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Stobbe, M.D.; Jansen, G.A.; Moerland, P.D.; van Kampen, A.H.C. Knowledge representation in metabolic pathway databases. Brief. Bioinform. 2014, 15, 455–470. [Google Scholar] [CrossRef]
- Poptsova, M.S.; Gogarten, J.P. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology 2010, 156, 1909–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.R.; Nookaew, I.; Hauser, L.; Gorin, A. Assessment of genome annotation using gene function similarity within the gene neighborhood. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaves, D.K.; Ginsburg, E.; Bang, J.J.; Fleming, J.M. Persistent organic pollutants and obesity: Are they potential mechanisms for breast cancer promotion? Endocr. Relat. Cancer 2015, 22, R69–R86. [Google Scholar] [CrossRef] [PubMed]
- Klompe, S.E.; Sternberg, S.H. Harnessing “A Billion Years of Experimentation”: The Ongoing Exploration and Exploitation of CRISPR–Cas Immune Systems. Cris. J. 2018, 1, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Lasa, A.; Gibas, C.J.; Romalde, J.L. Comparative Genomic Analysis of Two Vibrio toranzoniae Strains with Different Virulence Capacity Reveals Clues on Its Pathogenicity for Fish. Front. Microbiol. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakr Shabbir, M.A.; Hao, H.; Shabbir, M.Z.; Hussain, H.I.; Iqbal, Z.; Ahmed, S.; Sattar, A.; Iqbal, M.; Li, J.; Yuan, Z. Survival and evolution of CRISPR-Cas system in prokaryotes and its applications. Front. Immunol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pleška, M.; Qian, L.; Okura, R.; Bergmiller, T.; Wakamoto, Y.; Kussell, E.; Guet, C.C. Bacterial Autoimmunity Due to a Restriction-Modification System. Curr. Biol. 2016, 26, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Li, S.R.; Jiang, B.; Hu, X.M.; Li, S. Therapeutic Targeting of the Staphylococcus aureus Accessory Gene Regulator (agr) System. Front. Microbiol. 2018, 9, 55. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, 1–103. [Google Scholar] [CrossRef]
- Oosthuysen, W.F.; Orth, H.; Lombard, C.J.; Sinha, B.; Wasserman, E. Population structure analyses of Staphylococcus aureus at Tygerberg Hospital, South Africa, reveals a diverse population, a high prevalence of Panton-Valentine leukocidin genes, and unique local methicillin-resistant S. aureus clones. Clin. Microbiol. Infect. 2014, 20, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.J.; LaRussa, S.J.; Dana, A.; Smith, H.; Xu, A.; Ryan, C.; Uhlemann, A.-C.; Boundy, S.; Goldberg, J.; Narechania, A.; et al. Emergence of the Epidemic Methicillin-Resistant Staphylococcus aureus Strain USA300 Coincides with Horizontal Transfer of the Arginine Catabolic Mobile Element and speG-mediated Adaptations for Survival on Skin. MBio 2013, 4, e00889-13. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.U.; Bizzarro, M.J.; Baltimore, R.S.; Dembry, L.M.; Gallagher, P.G. Clinical and molecular epidemiology of methicillin-resistant staphylococcus aureus in a neonatal intensive care unit in the decade following implementation of an active detection and isolation program. J. Clin. Microbiol. 2015, 53, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Salam, A.M.; Quave, C.L. Targeting Virulence in Staphylococcus aureus by Chemical Inhibition of the Accessory Gene Regulator System In Vivo. mSphere 2018, 3, e00500-17. [Google Scholar] [CrossRef] [PubMed]
- Shore, A.C.; Rossney, A.S.; Brennan, O.M.; Kinnevey, P.M.; Humphreys, H.; Sullivan, D.J.; Goering, R.V.; Ehricht, R.; Monecke, S.; Coleman, D.C. Characterization of a novel arginine catabolic mobile element (ACME) and staphylococcal chromosomal cassette mec composite island with significant homology to Staphylococcus epidermidis ACME type II in methicillin-resistant Staphylococcus aureus genotype. Antimicrob. Agents Chemother. 2011, 55, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Schaumburg, F.; Alabi, A.S.; Peters, G.; Becker, K. New epidemiology of Staphylococcus aureus infection in Africa. Eur. Soc. Clin. Infect. Dis. 2014, 20, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Martinez, J.L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 2015, 6, 1–6. [Google Scholar] [CrossRef]
- Deneke, C.; Rentzsch, R.; Renard, B.Y. PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef]
- Speziale, P.; Pietrocola, G.; Rindi, S.; Provenzano, M.; Provenza, G.; Di Poto, A.; Visai, L.; Arciola, C.R. Structural and functional role of Staphylococcus aureus surface components recognizing adhesive matrix molecules of the host. Future Microbiol. 2009, 4, 1337–1352. [Google Scholar] [CrossRef]
- Oyama, T.; Miyazaki, M.; Yoshimura, M.; Takata, T.; Ohjimi, H.; Jimi, S. Biofilm-Forming Methicillin-Resistant Staphylococcus aureus Survive in Kupffer Cells and Exhibit High Virulence in Mice. Toxins 2016, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Cihalova, K.; Chudobova, D.; Michalek, P.; Moulick, A.; Guran, R.; Kopel, P.; Adam, V.; Kizek, R. Staphylococcus aureus and MRSA growth and biofilm formation after treatment with antibiotics and SeNPs. Int. J. Mol. Sci. 2015, 16, 24656–24672. [Google Scholar] [CrossRef] [PubMed]
- Tasse, J.; Trouillet-Assant, S.; Josse, J.; Martins-Simões, P.; Valour, F.; Langlois-Jacques, C.; Badel-Berchoux, S.; Provot, C.; Bernardi, T.; Ferry, T.; et al. Association between biofilm formation phenotype and clonal lineage in Staphylococcus aureus strains from bone and joint infections. PLoS ONE 2018, 13, e0200064. [Google Scholar] [CrossRef] [PubMed]
- Thiran, E.; Di Ciccio, P.A.; Graber, H.U.; Zanardi, E.; Ianieri, A.; Hummerjohann, J. Biofilm formation of Staphylococcus aureus dairy isolates representing different genotypes. J. Dairy Sci. 2017, 101, 1000–1012. [Google Scholar] [CrossRef]
- Vanhommerig, E.; Moons, P.; Pirici, D.; Lammens, C.; Hernalsteens, J.P.; De Greve, H.; Kumar-Singh, S.; Goossens, H.; Malhotra-Kumar, S. Comparison of biofilm formation between major clonal lineages of methicillin resistant Staphylococcus aureus. PLoS ONE 2014, 9, e104561. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Li, Q.; Zhang, Y.; Song, J.; Shi, X.; Shi, C. Biofilm formation and antibiotic resistance pattern of dominant Staphylococcus aureus clonal lineages in China. J. Food Saf. 2017, 37, 1–7. [Google Scholar] [CrossRef]
- Naicker, P.R.; Karayem, K.; Hoek, K.G.P.; Harvey, J.; Wasserman, E. Biofilm formation in invasive Staphylococcus aureus isolates is associated with the clonal lineage. Microb. Pathog. 2016, 90, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, A.; Stapels, D.A.C.; Weerwind, L.T.; Ko, Y.P.; Ruyken, M.; Lee, J.C.; van Kessel, K.P.M.; Rooijakkers, S.H.M. The Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology 2016, 162, 1185–1194. [Google Scholar] [CrossRef]
- Burts, M.L.; Williams, W.A.; DeBord, K.; Missiakas, D.M. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. USA 2005, 102, 1169–1174. [Google Scholar] [CrossRef]
- Ates, L.S.; Houben, E.N.G.; Bitter, W. Type VII Secretion: A Highly Versatile Secretion System. Microbiol. Spectr. 2016, 357–384. [Google Scholar] [CrossRef]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Wardenburg, J.B. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumann, D.; Nübel, U.; Bröker, B.M. Staphylococcus aureus toxins—Their functions and genetics. Infect. Genet. Evol. 2014, 21, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Argudín, M.Á.; Mendoza, M.C.; Rodicio, M.R. Food Poisoning and Staphylococcus aureus Enterotoxins. Toxins 2010, 2, 1751–1773. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; van Strijp, J.A.G.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447. [Google Scholar] [CrossRef]
- Cupane, L.; Pugacova, N.; Berzina, D.; Cauce, V.; Gardovska, D.; Miklaševics, E. Patients with Panton-Valentine leukocidin positive Staphylococcus aureus infections run an increased risk of longer hospitalisation. Int. J. Mol. Epidemiol. Genet. 2012, 3, 48–55. [Google Scholar]
- Vandenesch, F.; Lina, G.; Henry, T. Staphylococcus aureus Hemolysins, bi-component Leukocidins, and Cytolytic Peptides: A Redundant Arsenal of Membrane-Damaging Virulence Factors? Front. Cell. Infect. Microbiol. 2012, 2, 1–15. [Google Scholar] [CrossRef]
- Karayem, K.J. A Phenotypic and Genotypic Characterisation of Strain Types, Virulence Factors and Agr Groups of Colonising Staphylococcus Aureus Associated with Bloodstream Infection. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, March 2015. [Google Scholar]
- Kong, C.; Neoh, H.M.; Nathan, S. Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy. Toxins 2016, 8, 72. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 2. [Google Scholar] [CrossRef]
- Fang, F.C.; Frawley, E.R.; Tapscott, T.; Vázquez-Torres, A. Bacterial Stress Responses during Host Infection. Cell Host Microbe 2016, 20, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marles-Wright, J.; Lewis, R.J. Stress responses of bacteria. Curr. Opin. Struct. Biol. 2007, 17, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Grayson, P.; Schulten, K. Glycerol Conductance and Physical Asymmetry of the Escherichia coli Glycerol Facilitator GlpF. Biophys. J. 2003, 85, 2977–2987. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Soto, C.G.; Valenzuela-Soto, E.M. Glycine betaine rather than acting only as an osmolyte also plays a role as regulator in cellular metabolism. Biochimie 2018, 147, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Schwan, W.R.; Wetzel, K.J. Osmolyte transport in Staphylococcus aureus and the role in pathogenesis. World J. Clin. Infect. Dis. 2016, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.-F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Glickman, M.S. Function of site-2 proteases in bacteria and bacterial pathogens. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2808–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroos, L.; Akiyama, Y. Biochemical and structural insights into intramembrane metalloprotease mechanisms. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2873–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I. Cells in stress: Transcriptional activation of heat shock genes. Science 1993, 259, 1409–1410. [Google Scholar] [CrossRef]
- Weibezahn, J.; Schlieker, C.; Tessarz, P.; Mogk, A.; Bukau, B. Novel insights into the mechanism of chaperone-assisted protein disaggregation. Biol. Chem. 2005, 386, 739–744. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Schürch, A.C.; Arredondo-Alonso, S.; Willems, R.J.L.; Goering, R.V. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin. Microbiol. Infect. 2018, 24, 350–354. [Google Scholar] [CrossRef]
- Salipante, S.J.; Sengupta, D.J.; Cummings, L.A.; Land, T.A.; Hoogestraat, D.R.; Cookson, T. Application of Whole-Genome Sequencing for Bacterial Strain Typing in Molecular Epidemiology. J. Clin. Microbiol. 2015, 53, 1072–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Integrated Surveillance of Antimicrobial Resistance in Foodborne Bacteria: Application of a One Health Approach: Guidance from the WHO Advisory Group on Integrated Surveillanec of Antimicrobial Resistance (AGISAR); WHO: Geneva, Switzerland, 2017; ISBN 978-92-4-151241-1. [Google Scholar]
- Kateete, D.P.; Kimani, C.N.; Katabazi, F.A.; Okeng, A.; Okee, M.S.; Nanteza, A.; Joloba, M.L.; Najjuka, F.C. Identification of Staphylococcus aureus: DNase and Mannitol salt agar improve the efficiency of the tube coagulase test. Ann. Clin. Microbiol. Antimicrob. 2010, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Chenoll, E.; Aznar, R. Identification and typing of food-borne Staphylococcus aureus by PCR-based techniques. Syst. Appl. Microbiol. 2005, 28, 340–352. [Google Scholar] [CrossRef]
- Datta, P.; Gulati, N.; Singla, N.; Vasdeva, H.R.; Bala, K.; Chander, J.; Gupta, V. Evaluation of various methods for the detection of meticillin-resistant Staphylococcus aureus strains and susceptibility patterns. J. Med. Microbiol. 2011, 60, 1613–1616. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: 27th Edition Informational Supplement M100-S27; CLSI: Wayne, PA, USA, 2017. [Google Scholar]
- Kuntová, L.; Pantuček, R.; Rájová, J.; Ružičková, V.; Petráš, P.; Mašlaňová, I.; Doškař, J. Characteristics and distribution of plasmids in a clonally diverse set of methicillin-resistant staphylococcus aureus strains. Arch. Microbiol. 2012, 194, 607–614. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing (EUCAST). Breakpoint Tables for Interpretation of MICs and Zone Diameters, Version 8.0, 2017. Available online: http://www.eucast.org/clinical_breakpoints/ (accessed on 16 September 2019).
- Osei Sekyere, J.; Amoako, D.G. Genomic and Phenotypic Characterisation of Fluoroquinolone Resistance Mechanisms in Enterobacteriaceae in Durban, South Africa. PLoS ONE 2017, 12, e0178888. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Kaya, H.; Hasman, H.; Larsen, J.; Stegger, M.; Johannesen, T.B.; Allesøe, R.L.; Lemvigh, C.K.; Aarestrup, F.M.; Lund, O.; Larsen, A.R. SCCmecFinder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. mSphere 2018, 3, e00612-17. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing whole-genome sequencing with sanger sequencing for spa typing of methicillin-resistant staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring Patterns of Evolutionary Descent among Clusters of Related Bacterial Genotypes from Multilocus Sequence Typing Data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder:a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- CLC Genomics Workbench, 11.0.0; Qiagen: Aarhus, Denmark, 2017; Available online: https://www.qiagenbioinformatics.com/ (accessed on 16 September 2019).
- Varghese, N.J.; Mukherjee, S.; Ivanova, N.; Konstantinidis, K.T.; Mavrommatis, K.; Kyrpides, N.C.; Pati, A. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015, 43, 6761–6771. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2017, 34, 292–293. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clone (ST612-CC8-t1257-SCCmec_IVd(2B) a | Genome Characteristics | Classification | e Pathogenicity Score(Number of Pathogenic Families) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number | Strain ID | Point | Host | Source | Tandem Repeats | CRISPRs (Cas Cluster) | RM-System b | Agr type c | ACME d | |
| 1 | SA1 | Farm | Animal | Faecal | 135 | 8 (1) | Type I and IV | Type I | II | 0.926 (939) f |
| 2 | SA2 | Farm | Animal | Faecal | 127 | 8 (0) | Type I and IV | Type I | II | 0.930 (895) f |
| 3 | SA3 | Farm | Animal | Faecal | 128 | 8 (1) | Type I and IV | Type I | II | 0.925 (910) f |
| 4 | SA4 | Farm | Animal | Faecal | 133 | 9 (0) | Type I and IV | Type I | II | 0.926 (878) f |
| 5 | SA5 | Farm | Human | Nasal | 136 | 6 (1) | Type I and IV | Type I | II | 0.925 (931) f |
| 6 | SA6 | Farm | Human | Nasal | 129 | 6 (0) | Type I and IV | Type I | II | 0.926 (907) f |
| 7 | SA7 | Farm | Human | Nasal | 145 | 7 (1) | Type I and IV | Type I | II | 0.925 (912) f |
| 8 | SA8 | Abattoir | Animal | Rinsate | 143 | 8 (0) | Type I and IV | Type I | II | 0.928 (870) g |
| 9 | SA12 | Abattoir | Animal | Rinsate | 129 | 7 (1) | Type I and IV | Type I | II | 0.926 (903) f |
| 10 | SS1 | Retail point | Animal | Carcass | 133 | 8 (2) | Type I and IV | Type I | II | 0.931 (841) f |
| 11 | SS2 | Retail point | Animal | Carcass | 258 | 7 (0) | Type I and IV | Type I | II | 0.929 (828) f |
| ST8-CC8-t008-SCCmec_IVa (2B) | ||||||||||
| 12 | USA300 | - h | Human | - | 134 | 8 (0) | Type I and IV | Type I | I | 0.924 (1094) f |
| ST8-CC8-t064-SCCmec_IVd (2B) | ||||||||||
| 13 | USA500 | - | Human | - | 133 | 7 (0) | Type I and IV | Type I | III | 0.921 (1021) f |
| ST612-CC8-t064-SCCmec_IVd (2B) | ||||||||||
| 14 | SHV713 | - | Animal | - | 122 | 7 (0) | Type I and IV | Type I | II | 0.924 (956) f |
| Isolate | Resistance Mechanisms |
|---|---|
| SA1 | mecA, blaZ, aac(6’)-aph(2’’), erm(C), tet(M), gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA2 | mecA, blaZ, aac(6’)-aph(2’’), erm(C), mrs(A) tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA3 | mecA, blaZ, aac(6’)-aph(2’’), msr(A), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA4 | mecA, blaZ, aac(6’)-aph(2’’), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA5 | mecA, blaZ, msr(A), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB(H481N) |
| SA6 | mecA, blaZ, msr(A), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA7 | mecA, blaZ, aac(6’)-aph(2’’), tet(M), dfrC, rpoB (H481N) |
| SA8 | mecA, blaZ, aac(6’)-aph(2’’), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SA12 | mecA, blaZ, aac(6’)-aph(2’’), erm(C), tet(M), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SS1 | mecA, blaZ, aac(6’)-aph(2’’), erm(C), mph(C), msr(A), tet(M), tet(K), dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| SS2 | mecA, blaZ, aac(6’)-aph(2’’), tetM, dfrC, gyrA(S84L), parC(S80Y), parE(D434N), rpoB (H481N) |
| USA300 | mecA |
| USA500 | mecA, blaZ, tetM, gyrA(S84L), parC(S80F) |
| SHV713 | mecA, blaZ, tetM, rpoB (H481N) |
| Isolate | Sequence Type (Clonal Complexe) | Pathogenicity Score (Number of Pathogenic Families) | Accession Number |
|---|---|---|---|
| ISU935 | ST5 (CC5) | 0.933 (899) | CP017090.1 |
| JKD6008 | ST239 (CC8) | 0.927 (1036) | CP002120.1 |
| M013 | ST59 (CC59) | 0.930 (337) | CP003166.2 |
| Type | Associated Proteins/Enzymes/Genes |
|---|---|
| Stress response | |
| Protection from Reactive Oxygen Species | Superoxide dismutase (Fe) (EC 1.15.1.1) |
| Oxidative stress | Superoxide dismutase (Mn) (EC 1.15.1.1) |
| Ferric uptake regulation protein FUR | |
| Peroxide stress regulator (PerR), FUR family | |
| Organic hydroperoxide resistance protein | |
| CoA disulfide thiol-disulfide redox system | CoA-disulfide reductase (EC 1.8.1.14) |
| Glutathione: Redox cycle | Glutathione peroxidase (EC 1.11.1.9) |
| Osmotic stress | |
| Osmoregulation | Glycerol uptake facilitator protein (GlpF) |
| Choline and Betaine Uptake and Betaine Biosynthesis | Choline ABC transport system, permease protein (OpuBB) |
| Choline ABC transport system, permease protein (OpuAC) | |
| Choline ABC transport system, permease protein (OpuAA) | |
| Betaine aldehyde dehydrogenase (EC 1.2.1.8) | |
| Choline ABC transport system, permease protein (OpuBC) | |
| Glycine betaine transporter (OpuD) | |
| Choline ABC transport system, permease protein (OpuBD) | |
| Glycine betaine ABC transport system, permease protein (OpuAB) | |
| Choline ABC transport system, permease protein (OpuBA) | |
| Choline dehydrogenase (EC 1.1.99.1) | |
| Periplasmic stress | |
| Periplasmic Stress Response | Intramembrane protease (RasP/YluC) |
| Others | |
| SigmaB stress response regulation | Serine-protein kinase (RsbW) (EC 2.7.11.1) |
| Serine phosphatase (RsbU), regulator of sigma subunit | |
| RNA polymerase sigma factor (SigB) | |
| Anti-sigma B factor antagonist (RsbV) | |
| Bacterial hemoglobins | Hemoglobin-like protein (HbO) |
| High frequency lysogen (Hfl) operon | RNA-binding protein (Hfq) |
| Heat shock | GroES, GroEL, S4 paralog and GrpE |
| Bacteriocins | Two-component response regulator (BceR) Bacitracin export ATP-binding protein (BceA) Bacitracin export permease protein (BceB) Two-component sensor histidine kinase (BceS) |
| Detoxification | Nudix proteins (nucleoside triphosphate hydrolases): 8-oxo-dGTPase Bsu (YtkD) and ADP-ribose pyrophosphatase (EC 3.6.1.13) |
| Housecleaning nucleoside triphosphate pyrophosphatases: Nucleotidase (YfbR), HD superfamily and Dimeric dUTPase (EC 3.6.1.23) | |
| Nucleoside triphosphate pyrophospho-hydrolase (MazG) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.A.; Essack, S.Y. Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa. Pathogens 2019, 8, 166. https://doi.org/10.3390/pathogens8040166
Amoako DG, Somboro AM, Abia ALK, Allam M, Ismail A, Bester LA, Essack SY. Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa. Pathogens. 2019; 8(4):166. https://doi.org/10.3390/pathogens8040166
Chicago/Turabian StyleAmoako, Daniel Gyamfi, Anou M. Somboro, Akebe Luther King Abia, Mushal Allam, Arshad Ismail, Linda A. Bester, and Sabiha Y. Essack. 2019. "Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa" Pathogens 8, no. 4: 166. https://doi.org/10.3390/pathogens8040166