
In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration

 ,
,  and
and
Abstract
:
1. Introduction
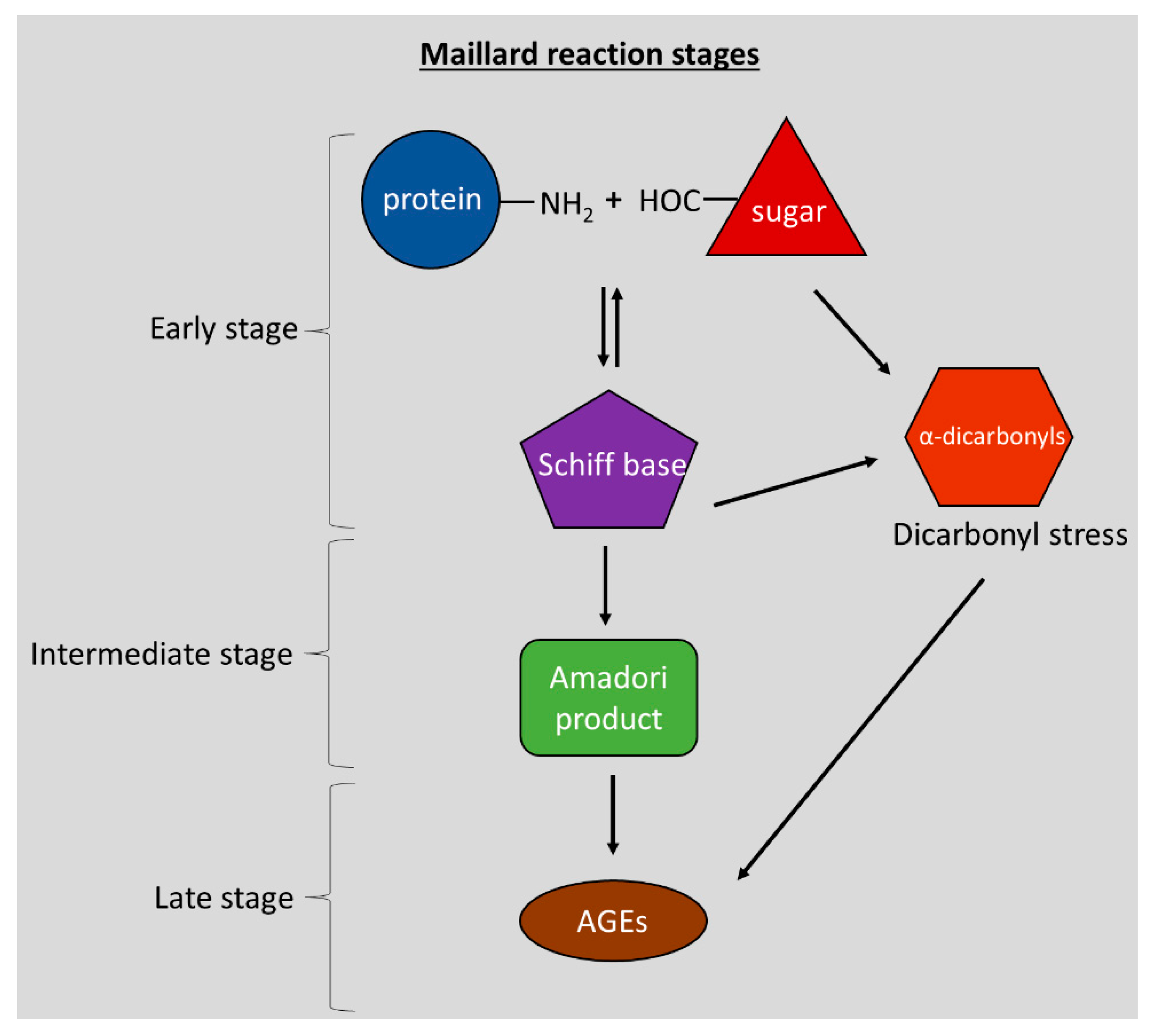
2. AGEs and Neurodegeneration
2.1. Evidence Associating AGEs with Neurodegeneration
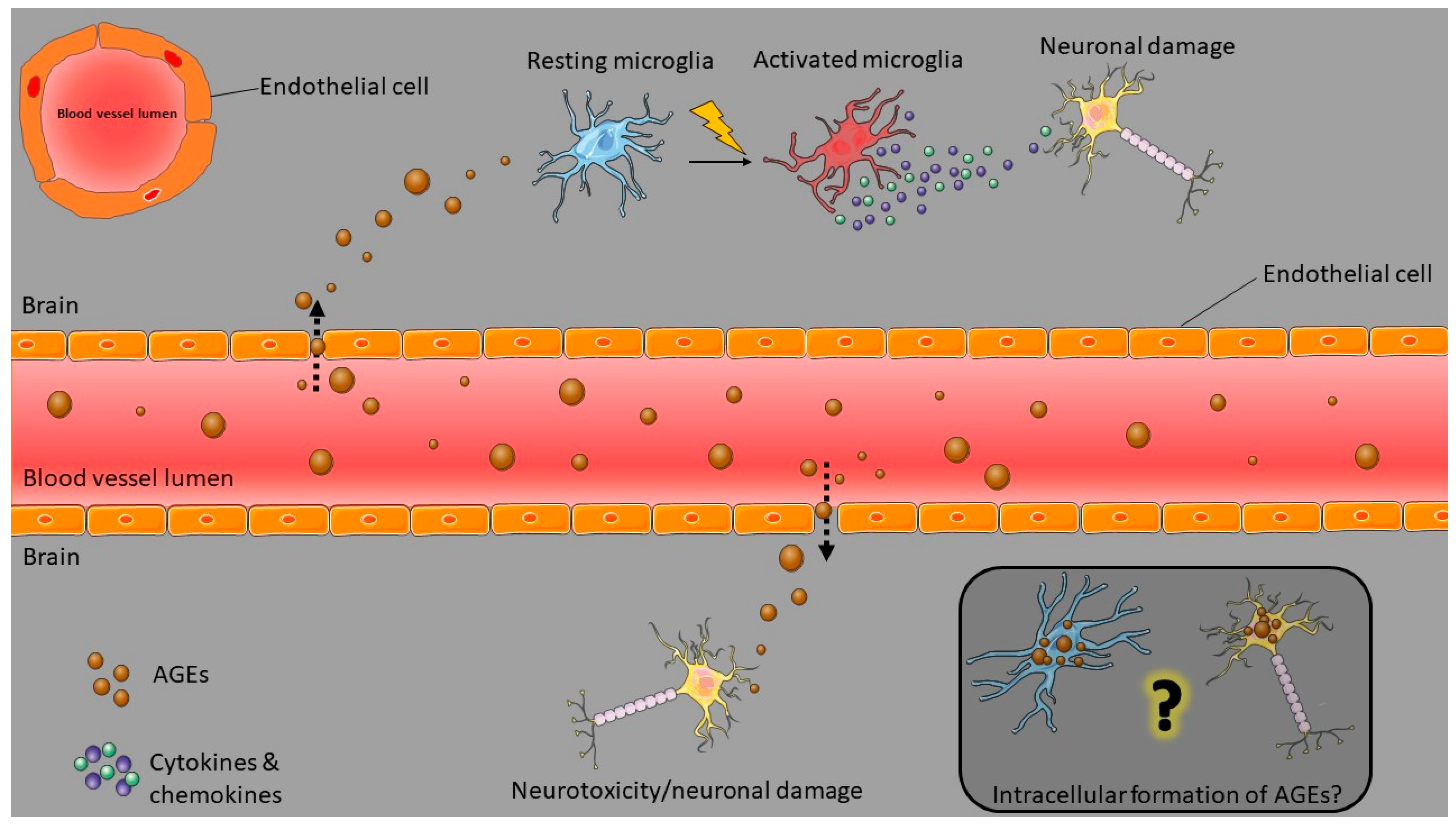
2.2. Concepts in AGE-Mediated Neurodegeneration
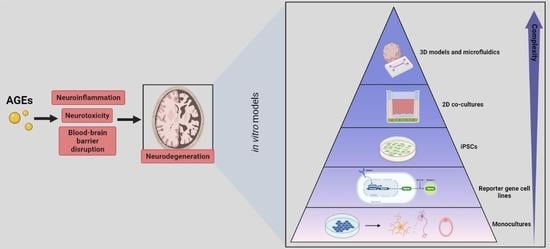
3. In Vitro Models to Study the Effect of AGEs on the Neuro-Immune Axis
3.1. Monocultures
3.1.1. Models of Brain Inflammation
Microglial Cell Models
- Immortalised microglial cell lines
- Animal-derived microglia cell lines
- Human derived microglial cell lines
- Human and animal-derived primary microglial cells
3.1.2. Other Inflammation Models
- Monocytes Cell Models
Immortalised Monocyte Cell Lines
- Human derived monocytic lines
- Human and animal-derived primary monocytes
3.1.3. Neuronal Cell Lines
- Animal neuronal cell lines
- Human neuronal cell lines
- Primary neuronal cells
3.1.4. Brain Endothelial Cells
- Animal-derived immortalised cell lines
- Human derived immortalised cell lines
- Primary endothelial cells
3.2. Reporter Cell Lines
3.3. iPSCs
3.4. Two-Dimensional Co-Cultures
3.5. Three-Dimensional Models and Microfluidics
4. Advisable In Vitro Models for AGE-Related Research

{kind=link}
{kind=link}
{kind=link}
| Advisable Model per Endpoint | Advantages | Disadvantages | References | |
|---|---|---|---|---|
| Neuroinflammation | HMC3 microglial line | Human line, RAGE expression, M1 and M2 phenotype, existing literature on AGEs effects | No aggregate formation | [128,129,132,133,310] |
| Co-culture of neurons with microglia in transwell | Well-established models, they give insights into how AGE indirectly affects neuron viability by activating microglia, easy to establish and interpret, comparative studies on the responses, i.e., SH-SY5Y/HMC3 | No aggregate formation | [285,286,287,288,290,309] | |
| iPSC-derived microglia | Patient-derived cells with diseased genotypic background, formation of aggregates | Non-high throughput, expensive, laborious procedure, low efficiency of differentiated cells | [263,264] | |
| NF-κΒ reporter cell lines | Very informative to understand AGE signalling | Most of the available lines are no brain-related lines | [154,159,244,246,247,254] | |
| Neurotoxicity | SH-SY5Y | Human line, extensively used in AGE-studies, RAGE expression, intracellular formation of AGEs, differentiated to dopaminergic neurons, Lewy body formation observed | Multiple differentiation protocols exist, possible unstable genome due to cancerous origin | [100,170,171,188,189,192,195,197,200,205] |
| iPSC derived neurons | Patient-derived cells with diseased genotypic background, formation of aggregates | Non-high throughput, expensive, laborious, low efficiency | [258,265] | |
| Nrf-2 reporter cell lines | Very informative to understand AGE signalling and the potential protective effects of AGE inhibitors | No brain-related lines are available | [249,252] | |
| BBB | hCMEC/D3 | Human line, expression of endothelial junctional markers and transporters, RAGE expression, applied in AGE and ND research, widely used and characterised BBB model | Exhibit lower TEER than primary endothelial cells | [311] |
| iPSC derived brain endothelial cells | Patient-derived cells with diseased genotypic background, formation of aggregates, high TEER values | Non-high-throughput, expensive, laborious, low efficiency, reproducibility not confirmed | [267,272] | |
| Co-cultures of brain endothelial cells with microglia in transwells | Well-established models, paracrine communication between cell types, more representative of BBB physiology, easy to establish and interpret | No aggregate formation, complicated | [57,226,273,292,293,294] | |
| BBB-on-a-chip | 3D, fluid shear stress, ECM, paracrine and juxtacrine signalling, combination of cell types | Complex | [304,305] |
5. Conclusions and Main Challenges in AGE In Vitro Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maillard, L.C. Action des acides amines sur les sucres; formation des melanoidines par voie methodique. Comptes R. Acad. Sci. Paris 1912, 154, 66–68. [Google Scholar]
- Hodge, J.E. Dehydrated Foods, Chemistry of Browning Reactions in Model Systems. J. Agric. Food Chem. 1953, 1, 928–943. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Fogliano, V. Dietary Advanced Glycosylation End-Products (dAGEs) and Melanoidins Formed through the Maillard Reaction: Physiological Consequences of their Intake. Annu. Rev. Food Sci. Technol. 2018, 9, 271–291. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Dicarbonyl Intermediates in the Maillard Reaction. Ann. N. Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J. The Maillard reaction in the human body. The main discoveries and factors that affect glycation. Pathol. Biol. 2010, 58, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, C. Toxicity of the AGEs generated from the Maillard reaction: On the relationship of food-AGEs and biological-AGEs. Mol. Nutr. Food Res. 2006, 50, 1140–1149. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diab. Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starowicz, M.; Zieliński, H. How Maillard Reaction Influences Sensorial Properties (Color, Flavor and Texture) of Food Products? Food Rev. Int. 2019, 35, 707–725. [Google Scholar] [CrossRef]
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843. [Google Scholar] [CrossRef]
- Kunkel, H.G.; Wallenius, G. New hemoglobin in normal adult blood. Science 1955, 122, 288. [Google Scholar] [CrossRef] [Green Version]
- Yatscoff, R.W.; Tevaarwerk, G.J.; MacDonald, J.C. Quantification of nonenzymically glycated albumin and total serum protein by affinity chromatography. Clin. Chem. 1984, 30, 446–449. [Google Scholar] [CrossRef]
- Roth, M. “Glycated hemoglobin,” not “glycosylated” or “glucosylated”. Clin. Chem. 1983, 29, 1991. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 963–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnier, V.M.; Cerami, A. Nonenzymatic browning in vivo: Possible process for aging of long-lived proteins. Science 1981, 211, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Degen, J.; Hellwig, M.; Henle, T. 1,2-Dicarbonyl Compounds in Commonly Consumed Foods. J. Agric. Food Chem. 2012, 60, 7071–7079. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.D.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Battah, S.; Karachalias, N.; Babaei-Jadidi, R.; Horányi, M.; Baróti, K.; Hollan, S.; Thornalley, P.J. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim. Biophys. Acta - Mol. Basis Dis. 2003, 1639, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, Y.; Fu, L. Dietary advanced glycation end-products: Perspectives linking food processing with health implications. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2559–2587. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.J.M.; Hanssen, N.M.J.; van Greevenbroek, M.M.; Van der Kallen, C.J.; Feskens, E.J.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. Dietary intake of advanced glycation endproducts is associated with higher levels of advanced glycation endproducts in plasma and urine: The CODAM study. Clin. Nutr. 2018, 37, 919–925. [Google Scholar] [CrossRef]
- Tessier, F.J.; Niquet-Léridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.-M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound 13C-labeled Nɛ-carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Roncero-Ramos, I.; Niquet-Léridon, C.; Strauch, C.; Monnier, V.M.; Tessier, F.J.; Navarro, M.P.; Delgado-Andrade, C. An advanced glycation end product (AGE)-rich diet promotes Nε-carboxymethyl-lysine accumulation in the cardiac tissue and tendons of rats. J. Agric. Food Chem. 2014, 62, 6001–6006. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary glycotoxins: Inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef]
- Aragonès, G.; Rowan, S.; Francisco, S.G.; Whitcomb, E.A.; Yang, W.; Perini-Villanueva, G.; Schalkwijk, C.G.; Taylor, A.; Bejarano, E. The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells 2021, 10, 1852. [Google Scholar] [CrossRef] [PubMed]
- Jack, M.; Wright, D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl. Res. 2012, 159, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trellu, S.; Courties, A.; Jaisson, S.; Gorisse, L.; Gillery, P.; Kerdine-Römer, S.; Vaamonde-Garcia, C.; Houard, X.; Ekhirch, F.-P.; Sautet, A.; et al. Impairment of glyoxalase-1, an advanced glycation end-product detoxifying enzyme, induced by inflammation in age-related osteoarthritis. Arthritis Res. Ther. 2019, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Lüth, H.-J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Münch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wollmer, M.A.; Hoerndli, F.; Münch, G.; Kuhla, B.; Rogaev, E.I.; Tsolaki, M.; Papassotiropoulos, A.; Götz, J. Role for glyoxalase I in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 7687–7692. [Google Scholar] [CrossRef] [Green Version]
- Lüth, H.-J.; Ogunlade, V.; Kuhla, B.; Kientsch-Engel, R.; Stahl, P.; Webster, J.; Arendt, T.; Münch, G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb. Cortex 2005, 15, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Münch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [Green Version]
- Münch, G.; Lüth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.; Riederer, P. Crosslinking of alpha-synuclein by advanced glycation endproducts—An early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 20, 253–257. [Google Scholar] [CrossRef]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, V.V.; Laffont, I.; Serot, J.-M.; Fujii, J.; Taniguchi, N.; Siest, G. Increased protein glycation in cerebrospinal fluid of Alzheimer’s disease. Neurobiol. Aging 2001, 22, 397–402. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Meli, M.; Perier, C.; Ferron, C.; Parssegny, F.; Denis, C.; Gonthier, R.; Laurent, B.; Reynaud, E.; Frey, J.; Chamson, A. Serum pentosidine as an indicator of Alzheimer’s disease. J. Alzheimers Dis. 2002, 4, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, S.; Vanmierlo, T.; Scheijen, J.L.J.M.; van Horssen, J.; Amor, S.; Somers, V.; Schalkwijk, C.G.; Hendriks, J.J.A.; Wouters, K. Methylglyoxal-Derived Advanced Glycation Endproducts Accumulate in Multiple Sclerosis Lesions. Front. Immunol. 2019, 10, 855. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Smith, M.A.; Richey, G.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Shaikh, S.; Nicholson, L.F.B. Advanced glycation end products induce in vitro cross-linking of α-synuclein and accelerate the process of intracellular inclusion body formation. J. Neurosci. Res. 2008, 86, 2071–2082. [Google Scholar] [CrossRef]
- Münch, G.; Mayer, S.; Michaelis, J.; Hipkiss, A.R.; Riederer, P.; Müller, R.; Neumann, A.; Schinzel, R.; Cunningham, A.M. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of β-amyloid peptide. Biochim. Biophys. Acta -Mol. Basis Dis. 1997, 1360, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.-Y.; Lin, Y.-S.; Lin, Y.-S.; Chang, S.-S. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic. Biol. Med. 2010, 49, 474–480. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci. Rep. 2020, 10, 7274. [Google Scholar] [CrossRef] [PubMed]
- Shahriyary, L.; Riazi, G.; Lornejad, M.R.; Ghezlou, M.; Bigdeli, B.; Delavari, B.; Mamashli, F.; Abbasi, S.; Davoodi, J.; Saboury, A.A. Effect of glycated insulin on the blood-brain barrier permeability: An in vitro study. Arch. Biochem. Biophys. 2018, 647, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.A.; Kanda, T. Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-β by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Dobi, A.; Rosanaly, S.; Devin, A.; Baret, P.; Meilhac, O.; Harry, G.J.; D’Hellencourt, C.L.; Rondeau, P. Advanced glycation end-products disrupt brain microvascular endothelial cell barrier: The role of mitochondria and oxidative stress. Microvasc. Res. 2021, 133, 104098. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An overview of in vitro methods to study microglia. Front. Cell. Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.; Biber, K.; Vinet, J.; Wgm Boddeke, H. Microglia Phenotype Diversity. CNS Neurol. Disord. -Drug Targets 2011, 10, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerschensteiner, M.; Meinl, E.; Hohlfeld, R. Neuro-immune crosstalk in CNS diseases. Neuroscience 2009, 158, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Sabogal-Guáqueta, A.M.; Marmolejo-Garza, A.; de Pádua, V.P.; Eggen, B.; Boddeke, E.; Dolga, A.M. Microglia alterations in neurodegenerative diseases and their modeling with human induced pluripotent stem cell and other platforms. Prog. Neurobiol. 2020, 190, 101805. [Google Scholar] [CrossRef] [PubMed]
- Dheen, S.T.; Kaur, C.; Ling, E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef]
- Takeda, A.; Yasuda, T.; Miyata, T.; Goto, Y.; Wakai, M.; Watanabe, M.; Yasuda, Y.; Horie, K.; Inagaki, T.; Doyu, M. Advanced glycation end products co-localized with astrocytes and microglial cells in Alzheimer’s disease brain. Acta Neuropathol. 1998, 95, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Sinicropi, S.; Yen, S.-H.; Ko, L.-W.; Mattiace, L.A.; Bucala, R.; Vlassara, H. Glycation and microglial reaction in lesions of Alzheimer’s disease. Neurobiol. Aging 1996, 17, 733–743. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A. Activated microglia in dementia with Lewy bodies. Neurology 2000, 55, 132–134. [Google Scholar] [CrossRef]
- Hall, E.D.; Oostveen, J.A.; Gurney, M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 1998, 23, 249–256. [Google Scholar] [CrossRef]
- McGeer, P.L.; Kawamata, T.; Walker, D.G.; Akiyama, H.; Tooyama, I.; McGeer, E.G. Microglia in degenerative neurological disease. Glia 1993, 7, 84–92. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the: Substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef]
- Schumacher, U.; Kretzschmar, H.; Pfüller, U. Staining of cerebral amyloid plaque glycoproteins in patients with Alzheimer’s disease with the microglia-specific lectin from mistletoe. Acta Neuropathol. 1994, 87, 422–424. [Google Scholar] [CrossRef]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Sato, K. Activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front. Cell. Neurosci. 2018, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Xu, L.; Tang, X.N.; Qiao, Y.; Giffard, R.G. Microglia potentiate damage to blood-brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke 2006, 37, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, R.; Fujiwara, Y.; Saito, M.; Arakawa, S.; Shirakawa, J.-I.; Yamanaka, M.; Komohara, Y.; Marumo, K.; Nagai, R. Intracellular Accumulation of Advanced Glycation End Products Induces Osteoblast Apoptosis Via Endoplasmic Reticulum Stress. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2020, 35, 1992–2003. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Lee, J.H.; Gaire, B.P.; Kim, S.Y. Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-κB, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells. Antioxidants 2020, 9, 792. [Google Scholar] [CrossRef]
- He, F.Q.; Qiu, B.Y.; Li, T.K.; Xie, Q.; Cui, D.J.; Huang, X.L.; Gan, H.T. Tetrandrine suppresses amyloid-β-induced inflammatory cytokines by inhibiting NF-κB pathway in murine BV2 microglial cells. Int. Immunopharmacol. 2011, 11, 1220–1225. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-B Pathway. Biomed Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wang, Z.; Zhang, J.H.; Tang, J.; Liu, X.; Tan, L.; Huang, Q.Y.; Feng, H. Receptor for Advanced Glycation End-Product Antagonist Reduces Blood-Brain Barrier Damage after Intracerebral Hemorrhage. Stroke 2015, 46, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.C.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Kang, W.C.; Oh, S.; Bayarsaikhan, D.; Ahn, H.; Lee, J.; Park, H.; Lee, S.; Choi, J.; Lee, H.S. Advanced glycation end-product (AGE)-albumin from activated macrophage is critical in human mesenchymal stem cells survival and post-ischemic reperfusion injury. Sci. Rep. 2017, 7, 11593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.M.; Byun, K.; Cho, K.; Kim, J.Y.; Yoo, J.S.; Kim, D.; Paek, S.H.; Kim, S.U.; Simpson, R.J.; Lee, B.; et al. Human microglial cells synthesize albumin in brain. PLoS ONE 2008, 3, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Kim, C.Y.; Mook-Jung, I.; Paek, S.H.; Kim, S.U.; Yamamoto, T.; Won, M.H.; Song, B.J.; et al. Induction of neuronal death by microglial AGE-albumin: Implications for Alzheimer’s disease. PLoS ONE 2012, 7, e37917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Son, M.; Hong, J.; Jeong, G.-B.; Paek, S.H.; Won, M.-H.; Lee, B. Activated microglial cells synthesize and secrete AGE-albumin. Anat. Cell Biol. 2012, 45, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, G.; Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Kuhla, B.; Heinrich, K.; Riederer, P.; Huttunen, H.J.; Founds, H.; Sajithlal, G. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp. Brain Res. 2003, 150, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [Green Version]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- MacLean, M.; Juranek, J.; Cuddapah, S.; López-Díez, R.; Ruiz, H.H.; Hu, J.; Frye, L.; Li, H.; Gugger, P.F.; Schmidt, A.M. Microglia RAGE exacerbates the progression of neurodegeneration within the SOD1G93A murine model of amyotrophic lateral sclerosis in a sex-dependent manner. J. Neuroinflamm. 2021, 18, 139. [Google Scholar] [CrossRef]
- Kopec, K.K.; Carroll, R.T. Alzheimer’s beta-amyloid peptide 1-42 induces a phagocytic response in murine microglia. J. Neurochem. 1998, 71, 2123–2131. [Google Scholar] [CrossRef]
- Bignami, A.; Eng, L.F.; Dahl, D.; Uyeda, C.T. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972, 43, 429–435. [Google Scholar] [CrossRef]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; DeLeo, J.A. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.; Lin, C.; Liu, Q.; He, Y.; Ruganzu, J.B.; Jin, H.; Peng, X.; Ji, S.; Ma, Y.; Yang, W. Tanshinone IIA attenuates neuroinflammation via inhibiting RAGE/NF-κB signaling pathway in vivo and in vitro. J. Neuroinflamm. 2020, 17, 302. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, J.S.; Zhang, D.D.; Yang, Y.Q.; Huang, L.T.; Yu, Z.; Chen, R.D.; Yang, H.K.; Hang, C.H. Inhibition of the Receptor for Advanced Glycation End-Products (RAGE) Attenuates Neuroinflammation While Sensitizing Cortical Neurons Towards Death in Experimental Subarachnoid Hemorrhage. Mol. Neurobiol. 2017, 54, 755–767. [Google Scholar] [CrossRef]
- Deng, X.-L.; Feng, L.; Wang, Z.-X.; Zhao, Y.-E.; Zhan, Q.; Wu, X.-M.; Xiao, B.; Shu, Y. The Runx1/Notch1 Signaling Pathway Participates in M1/M2 Microglia Polarization in a Mouse Model of Temporal Lobe Epilepsy and in BV-2 Cells. Neurochem. Res. 2020, 45, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, K.; Fiddler, J.; Soh, T.; Clarke, S. BV-2 Microglial Cells Used in a Model of Neuroinflammation. FASEB J. 2015, 29, 608.2. [Google Scholar] [CrossRef]
- Righi, M.; Mori, L.; De Libero, G.; Sironi, M.; Biondi, A.; Mantovani, A.; Donini, S.D.; Ricciardi-Castagnoli, P. Monokine production by microglial cell clones. Eur. J. Immunol. 1989, 19, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Alizadeh, D.; Zhao, D.; Farrukh, O.; Lin, J.; Badie, S.A.; Badie, B. S100B attenuates microglia activation in gliomas: Possible role of STAT3 pathway. Glia 2011, 59, 486–498. [Google Scholar] [CrossRef] [Green Version]
- Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Deuther-Conrad, W.; Münch, G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J. Neurochem. 2003, 87, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.A.; Moreira, R.; Brites, D. Phenotypic effects of wild-type and mutant SOD1 expression in n9 murine microglia at steady state, inflammatory and immunomodulatory conditions. Front. Cell. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corradin, S.B.; Mauël, J.; Donini, S.D.; Quattrocchi, E.; Ricciardi-Castagnoli, P. Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia 1993, 7, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, D.; Shanmugam, K.; Low, M.; Bennett, L.; Govindaraghavan, S.; Head, R.; Ooi, L.; Münch, G. Determination of anti-inflammatory activities of standardised preparations of plant- and mushroom-based foods. Eur. J. Nutr. 2014, 53, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.; Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Schinzel, R.; Wiesinger, H.; Riederer, P.; Münch, G.; Biology, N.C.; Leipzig, I.; Riederer, P.; et al. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur. J. Neurosci. 2001, 14, 1961–1967. [Google Scholar] [CrossRef]
- Khazaei, M.R.; Habibi-Rezaei, M.; Karimzadeh, F.; Moosavi-Movahedi, A.A.; Sarrafnejhad, A.A.; Sabouni, F.; Bakhti, M. Microglial Cell Death Induced by Glycated Bovine Serum Albumin: Nitric Oxide Involvement. J. Biochem. 2008, 144, 197–206. [Google Scholar] [CrossRef]
- Berbaum, K.; Shanmugam, K.; Stuchbury, G.; Wiede, F.; Körner, H.; Münch, G. Induction of novel cytokines and chemokines by advanced glycation endproducts determined with a cytometric bead array. Cytokine 2008, 41, 198–203. [Google Scholar] [CrossRef]
- Nagai, A.; Nakagawa, E.; Hatori, K.; Choi, H.B.; McLarnon, J.G.; Lee, M.A.; Kim, S.U. Generation and characterization of immortalized human microglial cell lines: Expression of cytokines and chemokines. Neurobiol. Dis. 2001, 8, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaikhan, E.; Bayarsaikhan, D.; Lee, J.; Son, M.; Oh, S.; Moon, J.; Park, H.-J.; Roshini, A.; Kim, S.U.; Song, B.-J.; et al. Microglial AGE-albumin is critical for neuronal death in Parkinson’s disease: A possible implication for theranostics. Int. J. Nanomed. 2016, 10, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Janabi, N.; Peudenier, S.; Héron, B.; Ng, K.H.; Tardieu, M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Lett. 1995, 195, 105–108. [Google Scholar] [CrossRef]
- Martin, S.; Vincent, J.P.; Mazella, J. Involvement of the neurotensin receptor-3 in the neurotensin-induced migration of human microglia. J. Neurosci. 2003, 23, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhart, E.; Kollroser, M.; Rechberger, G.; Reicher, H.; Heinemann, A.; Schratl, P.; Hallström, S.; Wintersperger, A.; Nusshold, C.; DeVaney, T.; et al. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010, 10, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Janabi, N.; Di Stefano, M.; Wallon, C.; Hery, C.; Chiodi, F.; Tardieu, M. Induction of human immunodeficiency virus type 1 replication in human glial cells after proinflammatory cytokines stimulation: Effect of IFNγ, IL1β, and TNFα on differentiation and chemokine production in glial cells. Glia 1998, 23, 304–315. [Google Scholar] [CrossRef]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive il-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef]
- Liu, J.; Hjorth, E.; Zhu, M.; Calzarossa, C.; Samuelsson, E.-B.; Schultzberg, M.; Åkesson, E. Interplay between human microglia and neural stem/progenitor cells in an allogeneic co-culture model. J. Cell. Mol. Med. 2013, 17, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjorth, E.; Zhu, M.; Toro, V.C.; Vedin, I.; Palmblad, J.; Cederholm, T.; Freund-Levi, Y.; Faxen-Irving, G.; Wahlund, L.-O.; Basun, H.; et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J. Alzheimers Dis. 2013, 35, 697–713. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, C.; Crisby, M.; Winblad, B.; Schultzberg, M. Effects of statins on microglia. J. Neurosci. Res. 2005, 82, 10–19. [Google Scholar] [CrossRef]
- Hjorth, E.; Frenkel, D.; Weiner, H.; Schultzberg, M. Effects of immunomodulatory substances on phagocytosis of abeta(1-42) by human microglia. Int. J. Alzheimers. Dis. 2010, 2010, 798424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, S.B.; Uy, B.; Perera, A.; Nicholson, L.F.B. AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 2012, 60, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Bigl, K.; Gaunitz, F.; Schmitt, A.; Rothemund, S.; Schliebs, R.; Munch, G.; Arendt, T.; Münch, G.; Arendt, T. Cytotoxicity of advanced glycation endproducts in human micro- and astroglial cell lines depends on the degree of protein glycation. J. Neural Transm. 2008, 115, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Baker, T.J.J. Characterization of ameboid microglia isolated from developing mammalian brain. J. Neurosci. 1986, 6, 2163–2178. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Yao, K.; Zu, H. bing Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K. Diversity and plasticity of microglial cells in psychiatric and neurological disorders. Pharmacol. Ther. 2015, 154, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Boje, K.M.M.; Arora, P.K.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Takata, K.; Kitamura, Y.; Saeki, M.; Terada, M.; Kagitani, S.; Kitamura, R.; Fujikawa, Y.; Maelicke, A.; Tomimoto, H.; Taniguchi, T.; et al. Galantamine-induced amyloid-β clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 40180–40191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.-W. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Yuan, K.; Ji, B.; Han, Y.; Li, J. Protective effects of curcumin against neuroinflammation induced by Aβ25-35 in primary rat microglia: Modulation of high-mobility group box 1, toll-like receptor 4 and receptor for advanced glycation end products expression. Ann. Transl. Med. 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J. The human leukemia cell line, THP-1: A multifacetted model for the study of monocyte-macrophage differentiation. Experientia 1991, 47, 22–31. [Google Scholar] [CrossRef]
- Qin, Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 2012, 221, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Pertyńska-Marczewska, M.; Kiriakidis, S.; Wait, R.; Beech, J.; Feldmann, M.; Paleolog, E.M. Advanced glycation end products upregulate angiogenic and pro-inflammatory cytokine production in human monocyte/macrophages. Cytokine 2004, 28, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Abordo, E.A.; Thornalley, P.J. Synthesis and secretion of tumour necrosis factor-α by human monocytic THP-1 cells and chemotaxis induced by human serum albumin derivatives modified with methylglyoxal and glucose-derived advanced glycation endproducts. Immunol. Lett. 1997, 58, 139–147. [Google Scholar] [CrossRef]
- Bai, W.; Zhou, J.; Zhou, N.; Liu, Q.; Cui, J.; Zou, W.; Zhang, W. Hypoxia-increased RAGE expression regulates chemotaxis and pro-inflammatory cytokines release through nuclear translocation of NF-κ B and HIF1α in THP-1 cells. Biochem. Biophys. Res. Commun. 2018, 495, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Figarola, J.L.; Shanmugam, N.; Natarajan, R.; Rahbar, S. Anti-inflammatory effects of the advanced glycation end product inhibitor LR-90 in human monocytes. Diabetes 2007, 56, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Bai, W.; Liu, Q.; Cui, J.; Zhang, W. Intermittent Hypoxia Enhances THP-1 Monocyte Adhesion and Chemotaxis and Promotes M1 Macrophage Polarization via RAGE. Biomed Res. Int. 2018, 2018, 1650456. [Google Scholar] [CrossRef]
- Xu, Y.; Toure, F.; Qu, W.; Lin, L.; Song, F.; Shen, X.; Rosario, R.; Garcia, J.; Schmidt, A.M.; Yan, S.-F. Advanced glycation end product (AGE)-receptor for age (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 2010, 285, 23233–23240. [Google Scholar] [CrossRef] [Green Version]
- Bezold, V.; Rosenstock, P.; Scheffler, J.; Geyer, H.; Horstkorte, R.; Bork, K. Glycation of macrophages induces expression of pro-inflammatory cytokines and reduces phagocytic efficiency. Aging 2019, 11, 5258–5275. [Google Scholar] [CrossRef]
- Dorenkamp, M.; Müller, J.P.; Shanmuganathan, K.S.; Schulten, H.; Müller, N.; Löffler, I.; Müller, U.A.; Wolf, G.; Böhmer, F.D.; Godfrey, R.; et al. Hyperglycaemia-induced methylglyoxal accumulation potentiates VEGF resistance of diabetic monocytes through the aberrant activation of tyrosine phosphatase SHP-2/SRC kinase signalling axis. Sci. Rep. 2018, 8, 14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, N.; Kim, Y.S.; Lanting, L.; Natarajan, R. Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J. Biol. Chem. 2003, 278, 34834–34844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abordo, E.A.; Westwood, M.E.; Thornalley, P.J. Synthesis and secretion of macrophage colony stimulating factor by mature human monocytes and human monocytic THP-1 cells induced by human serum albumin derivatives modified with methylglyoxal and glucose-derived advanced glycation endproducts. Immunol. Lett. 1996, 53, 7–13. [Google Scholar] [CrossRef]
- Iida, Y.; Miyata, T.; Inagi, R.; Sugiyama, S.; Maeda, K. β2-Microglobulin modified with advanced glycation end products induces interleukin-6 from human macrophages: Role in the pathogenesis of hemodialysis-associated amyloidosis. Biochem. Biophys. Res. Commun. 1994, 201, 1235–1241. [Google Scholar] [CrossRef]
- Kirstein, M.; Brett, J.; Radoff, S.; Ogawa, S.; Stern, D.; Vlassara, H. Advanced protein glycosylation induces transendothelial human monocyte chemotaxis and secretion of platelet-derived growth factor: Role in vascular disease of diabetes and aging. Proc. Natl. Acad. Sci. USA 1990, 87, 9010–9014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, T.; Inagi, R.; Iida, Y.; Sato, M.; Yamada, N.; Oda, O.; Maeda, K.; Seo, H. Involvement of beta 2-microglobulin modified with advanced glycation end products in the pathogenesis of hemodialysis-associated amyloidosis. Induction of human monocyte chemotaxis and macrophage secretion of tumor necrosis factor-alpha and interleukin-1. J. Clin. Investig. 1994, 93, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Wu, H. Protective effects of curcumin on methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Acta Pharmacol. Sin. 2006, 27, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.V.; Mone, M.; Koehne, C.; Rediske, J.; Hughes, T.E. Binding of receptor for advanced glycation end products (RAGE) ligands is not sufficient to induce inflammatory signals: Lack of activity of endotoxin-free albumin-derived advanced glycation end products. Diabetologia 2004, 47, 844–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Liu, L.; Zhang, Y.; Xiang, Y.; Yin, G.; Lu, Y.; Shi, L.; Dong, J.; Shen, C. Advanced glycation end products enhance murine monocyte proliferation in bone marrow and prime them into an inflammatory phenotype through MAPK signaling. J. Diabetes Res. 2018, 2018, 2527406. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Di Gao, Q.; Zhu, L.; Peppa, M.; He, C.; Vlassara, H. Oxidative stress-inducing carbonyl compounds from common foods: Novel mediators of cellular dysfunction. Mol. Med. 2002, 8, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yin, L.; Xu, Z.; An, F.-M.; Liu, A.-R.; Wang, Y.; Yao, W.-B.; Gao, X.-D. Inhibiting receptor for advanced glycation end product (AGE) and oxidative stress involved in the protective effect mediated by glucagon-like peptide-1 receptor on AGE induced neuronal apoptosis. Neurosci. Lett. 2016, 612, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Loske, C.; Neumann, A.; Cunningham, A.M.; Nichol, K.; Schinzel, R.; Riederer, P.; Münch, G. Cytotoxicity of advanced glycation endproducts is mediated by oxidative stress. J. Neural Transm. 1998, 105, 1005–1015. [Google Scholar] [CrossRef]
- Nan, F.; Sun, G.; Xie, W.; Ye, T.; Sun, X.; Zhou, P.; Dong, X.; Sun, J.; Sun, X.; Zhang, M. Ginsenoside Rb1 mitigates oxidative stress and apoptosis induced by methylglyoxal in SH-SY5Y cells via the PI3K/Akt pathway. Mol. Cell. Probes 2019, 48, 101469. [Google Scholar] [CrossRef]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The effect of glyceraldehyde-derived advanced glycation end products on β-tubulin-inhibited neurite outgrowth in sh-sy5y human neuroblastoma cells. Nutrients 2020, 12, 2958. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z.; Amazaki, M.A.Y.; Oike, T.A.K.; et al. Neurotoxicity of Advanced Glycation End-Products for Cultured Cortical Neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, L.; Harris, G.; Delp, J.; Valadares, M.; Pamies, D.; Hogberg, H.T.; Waldmann, T.; Leist, M.; Hartung, T. A LUHMES 3D dopaminergic neuronal model for neurotoxicity testing allowing long-term exposure and cellular resilience analysis. Arch. Toxicol. Arch. Für Toxikol. 2016, 90, 2725–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malagelada, C.; Greene, L.A. Chapter 29-PC12 Cells as a model for parkinson’s disease research. In Parkinson’s Disease Molecular and Therapeutic Insights from Model Systems; Nass, R., Przedborski, S.B.T.-P.D., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 375–387. ISBN 978-0-12-374028-1. [Google Scholar]
- Grau, C.M.; Greene, L.A. Use of PC12 cells and rat superior cervical ganglion sympathetic neurons as models for neuroprotective assays relevant to Parkinson’s disease. Methods Mol. Biol. 2012, 846, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.-C.; Chen, Y.-Y.; Kan, D.; Chien, C.-L. A neuronal death model: Overexpression of neuronal intermediate filament protein peripherin in PC12 cells. J. Biomed. Sci. 2012, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.-Y.; Yang, N.-C.; Chen, C.-L.; Thi, T.L.V. Protective Effects and Possible Mechanisms of Ergothioneine and Hispidin against Methylglyoxal-Induced Injuries in Rat Pheochromocytoma Cells. Oxid. Med. Cell. Longev. 2017, 2017, 4824371. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.-Y.; Huang, C.-L.; Yang, J.-M.; Huang, W.-J.; Huang, N.-K.; Chen, Y.-W.; Lin, R.-J.; Yang, Y.-C. The role of ribosylated-BSA in regulating PC12 cell viability. Cell Biol. Toxicol. 2012, 28, 255–267. [Google Scholar] [CrossRef]
- Yan, F.; Han, G.; Wu, G. Cytotoxic role of advanced glycation end-products in PC12 cells treated with β-amyloid peptide. Mol. Med. Rep. 2013, 8, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zang, D.; Xu, Y.; Meng, J.; Qian, S. Protective effect of ISO-1 against advanced glycation end product aggravation of PC12 cell injury induced by Aβ1-40. Mol. Med. Rep. 2019, 20, 2135–2142. [Google Scholar] [CrossRef]
- Huang, S.-M.; Hsu, C.-L.; Chuang, H.-C.; Shih, P.-H.; Wu, C.-H.; Yen, G.-C. Inhibitory effect of vanillic acid on methylglyoxal-mediated glycation in apoptotic Neuro-2A cells. Neurotoxicology 2008, 29, 1016–1022. [Google Scholar] [CrossRef]
- Huang, S.-M.; Chuang, H.-C.; Wu, C.-H.; Yen, G.-C. Cytoprotective effects of phenolic acids on methylglyoxal-induced apoptosis in Neuro-2A cells. Mol. Nutr. Food Res. 2008, 52, 940–949. [Google Scholar] [CrossRef]
- Lee, C.-C.; Lee, B.-H.; Wu, S.-C. Actinidia callosa peel (kiwi fruit) ethanol extracts protected neural cells apoptosis induced by methylglyoxal through Nrf2 activation. Pharm. Biol. 2014, 52, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, N.; Girón, M.D.; Salido, M.; Vargas, A.M.; Vilches, J.; Salto, R. Internalization of the receptor for advanced glycation end products (RAGE) is required to mediate intracellular responses. J. Biochem. 2009, 145, 21–30. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, H.; Liu, Y.; Chen, Y.; Liu, Y.; Yin, X. The Antidepressant-Like Effects of Hesperidin in Streptozotocin-Induced Diabetic Rats by Activating Nrf2/ARE/Glyoxalase 1 Pathway. Front. Pharmacol. 2020, 11, 1325. [Google Scholar] [CrossRef] [PubMed]
- Sajithlal, G.; Huttunen, H.; Rauvala, H.; Munch, G. Receptor for Advanced Glycation End Products Plays a More Important Role in Cellular Survival than in Neurite Outgrowth during Retinoic Acid-induced Differentiation of Neuroblastoma Cells. J. Biol. Chem. 2002, 277, 6888–6897. [Google Scholar] [CrossRef] [Green Version]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Whiteley, T.R.; Le Maitre, C.L.; Duce, J.A.; Dalton, C.F.; Smith, D.P. Recapitulating Parkinson’s disease pathology in a three-dimensional human neural cell culture model. Dis. Model. Mech. 2019, 12, dmm038042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.-T.; Tsai, Y.-H.; Fülöp, F.; Chang, F.-R.; Lo, Y.-C. 2-Iodo-4′-methoxychalcone attenuates methylglyoxal-induced neurotoxicity by activation of GLP-1 receptor and enhancement of neurotrophic signal, antioxidant defense and glyoxalase pathway. Molecules 2019, 24, 2249. [Google Scholar] [CrossRef] [Green Version]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective Effect of Sulforaphane against Methylglyoxal Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, J.; Kaur, H.; Collier, T.; Chang, K.; Brooks, A.E.S.; Allison, J.R.; Brimble, M.A.; Hickey, A.; Birch, N.P. Site-specific glycation of A1- 42 affects fibril formation and is neurotoxic. J. Biol. Chem. 2019, 294, 8806–8818. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Liu, W.Y.; Liou, S.-S.; Liu, I.-M. A bibenzyl component moscatilin mitigates glycation-mediated damages in an SH-SY5Y cell model of neurodegenerative diseases through AMPK activation and RAGE/NF-κB Pathway Suppression. Molecules 2020, 25, 4574. [Google Scholar] [CrossRef] [PubMed]
- Kuhla, B.; Loske, C.; Garcia de Arriba, S.; Schinzel, R.; Huber, J.; Münch, G. Differential effects of “Advanced glycation endproducts” and β-amyloid peptide on glucose utilization and ATP levels in the neuronal cell line SH-SY5Y. J. Neural Transm. 2004, 111, 427–439. [Google Scholar] [CrossRef] [PubMed]
- De Arriba, S.G.; Loske, C.; Meiners, I.; Fleischer, G.; Lobisch, M.; Wessel, K.; Tritschler, H.; Schinzel, R.; Münch, G. Advanced Glycation Endproducts Induce Changes in Glucose Consumption, Lactate Production, and ATP Levels in SH-SY5Y Neuroblastoma Cells by a Redox-Sensitive Mechanism. J. Cereb. Blood Flow Metab. 2003, 23, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, S.; Wang, C.-Y.; Wang, Y.; Liu, H.-X.; Cui, Y.; Zhang, L.-D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. Vitr. Cell. Dev. Biol. -Anim. 2014, 51, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Guo, J.; Tian, Y.; Zhao, K.; Li, J.; Xiao, Q. α-lipoic acid can greatly alleviate the toxic effect of AGES on SH-SY5Y cells. Int. J. Mol. Med. 2018, 41, 2855–2864. [Google Scholar] [CrossRef]
- Chen, L.; Wei, Y.; Wang, X.; He, R. Ribosylation Rapidly Induces α-Synuclein to Form Highly Cytotoxic Molten Globules of Advanced Glycation End Products. PLoS ONE 2010, 5, e9052. [Google Scholar] [CrossRef] [Green Version]
- Buttiglione, M.; Vitiello, F.; Sardella, E.; Petrone, L.; Nardulli, M.; Favia, P.; d’Agostino, R.; Gristina, R. Behaviour of SH-SY5Y neuroblastoma cell line grown in different media and on different chemically modified substrates. Biomaterials 2007, 28, 2932–2945. [Google Scholar] [CrossRef]
- Korecka, J.A.; van Kesteren, R.E.; Blaas, E.; Spitzer, S.O.; Kamstra, J.H.; Smit, A.B.; Swaab, D.F.; Verhaagen, J.; Bossers, K. Phenotypic Characterization of Retinoic Acid Differentiated SH-SY5Y Cells by Transcriptional Profiling. PLoS ONE 2013, 8, e63862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Agholme, L.; Lindström, T.; Kgedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, R.; Constantinescu, A.; Reichmann, H.; Janetzky, B. Neuronal differentiation and long-term culture of the human neuroblastoma line SH-SY5Y. J. Neural Transm. Suppl. 2007, 2007, 17–28. [Google Scholar] [CrossRef]
- Cai, C.; Dai, X.; Zhu, Y.; Lian, M.; Xiao, F.; Dong, F.; Zhang, Q.; Huang, Y.; Zheng, Q. A specific RAGE-binding peptide biopanning from phage display random peptide library that ameliorates symptoms in amyloid β peptide-mediated neuronal disorder. Appl. Microbiol. Biotechnol. 2016, 100, 825–835. [Google Scholar] [CrossRef]
- Mazzola, J.L.; Sirover, M.A. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J. Neurochem. 2001, 76, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, J.L.; Sirover, M.A. Subcellular alteration of glyceraldehyde-3-phosphate dehydrogenase in Alzheimer’s disease fibroblasts. J. Neurosci. Res. 2003, 71, 279–285. [Google Scholar] [CrossRef]
- Harischandra, D.S.; Rokad, D.; Ghaisas, S.; Verma, S.; Robertson, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Enhanced differentiation of human dopaminergic neuronal cell model for preclinical translational research in Parkinson’s disease. Biochim. Biophys. Acta -Mol. Basis Dis. 2020, 1866, 165533. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Yin, M.; Zhang, M.-H. Cell-based assays for Parkinson’s disease using differentiated human LUHMES cells. Acta Pharmacol. Sin. 2014, 35, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Park, T.I.-H.; Schweder, P.; Lee, K.; Dieriks, B.V.; Jung, Y.; Smyth, L.; Rustenhoven, J.; Mee, E.; Heppner, P.; Turner, C.; et al. Isolation and culture of functional adult human neurons from neurosurgical brain specimens. Brain Commun. 2020, 2, fcaa171. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Watai, T.; Sasaki, N.; Choei, H.; Iwaki, M.; Ashizawa, T.; Inagaki, Y.; Yamagishi, S.I.; Kikuchi, S.; Riederer, P.; et al. Neurotoxicity of acetaldehyde-derived advanced glycation end products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2003, 62, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Roux, F.; Couraud, P.-O. Rat Brain Endothelial Cell Lines for the Study of Blood-Brain Barrier Permeability and Transport Functions. Cell. Mol. Neurobiol. 2005, 25, 41–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, K.; Weksler, B.; Romero, I.; Couraud, P.-O.; Gelli, A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryot. Cell 2009, 8, 1803–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, A.; Pestana, D.; Teixeira, D.; Azevedo, J.; De Freitas, V.; Mateus, N.; Calhau, C. Flavonoid transport across RBE4 cells: A blood-brain barrier model. Cell. Mol. Biol. Lett. 2010, 15, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Sbai, O.; Nawroth, P.P.; Bierhaus, A. The Complexity of Sporadic Alzheimer’s Disease Pathogenesis: The Role of RAGE as Therapeutic Target to Promote Neuroprotection by Inhibiting Neurovascular Dysfunction. Int. J. Alzheimers Dis. 2012, 2012, 734956. [Google Scholar] [CrossRef] [Green Version]
- Montesano, R.; Pepper, M.S.; Möhle-Steinlein, U.; Risau, W.; Wagner, E.F.; Orci, L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell 1990, 62, 435–445. [Google Scholar] [CrossRef]
- Omidi, Y.; Campbell, L.; Barar, J.; Connell, D.; Akhtar, S.; Gumbleton, M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b. End3, as an in vitro blood–brain barrier model for drug uptake and transport studies. Brain Res. 2003, 990, 95–112. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Abuasal, B.; Romero, I.; Weksler, B.; Couraud, P.-O.; Keller, J.; Kaddoumi, A. Differences in amyloid-β clearance across mouse and human blood–brain barrier models: Kinetic analysis and mechanistic modeling. Neuropharmacology 2014, 79, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Chan, Y.; Wan, W.; Li, Y.; Zhang, C. Aβ1-42 induces cell damage via RAGE-dependent endoplasmic reticulum stress in bEnd.3 cells. Exp. Cell Res. 2018, 362, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jin, C.; Cui, L.; Xing, H.; Liu, J.; Liao, W.; Liao, H.; Yu, Y. HMGB1 contributes to the irradiation-induced endothelial barrier injury through receptor for advanced glycation endproducts (RAGE). J. Cell. Physiol. 2018, 233, 6714–6721. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ1-42 oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef]
- Park, R.; Kook, S.-Y.; Park, J.-C.; Mook-Jung, I. Aβ1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef]
- Liu, C.; Chen, K.; Lu, Y.; Fang, Z.; Yu, G. Catalpol provides a protective effect on fibrillary Aβ1–42-induced barrier disruption in an in vitro model of the blood–brain barrier. Phyther. Res. 2018, 32, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Youdim, K.A.; Dobbie, M.S.; Kuhnle, G.; Proteggente, A.R.; Abbott, N.J.; Rice-Evans, C. Interaction between flavonoids and the blood-brain barrier: In vitro studies. J. Neurochem. 2003, 85, 180–192. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]
- Dauchy, S.; Miller, F.; Couraud, P.-O.; Weaver, R.J.; Weksler, B.; Romero, I.-A.; Scherrmann, J.-M.; De Waziers, I.; Declèves, X. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem. Pharmacol. 2009, 77, 897–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okura, T.; Kato, S.; Deguchi, Y. Functional expression of organic cation/carnitine transporter 2 (OCTN2/SLC22A5) in human brain capillary endothelial cell line hCMEC/D3, a human blood-brain barrier model. Drug Metab. Pharmacokinet. 2014, 29, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Potin, S.; Chapy, H.; Crete, D.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O.; Scherrmann, J.-M.; Declèves, X. Aryl hydrocarbon receptor regulates CYP1B1 but not ABCB1 and ABCG2 in hCMEC/D3 human cerebral microvascular endothelial cells after TCDD exposure. Brain Res. 2015, 1613, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Guyot, L.; Garcia, J.; Vilchez, G.; Bardel, C.; Chenel, M.; Tod, M.; Payen, L. Impact of interleukin-6 on drug transporters and permeability in the hCMEC/D3 blood–brain barrier model. Fundam. Clin. Pharmacol. 2021, 35, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.-P.; Fenart, L. Modelling of the blood-Brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Ram, M.S.; Krishna, K.V.; Saha, R.N.; Singhvi, G.; Agrawal, M.; Ajazuddin; Saraf, S.; Saraf, S.; Alexander, A. Recent Expansions on Cellular Models to Uncover the Scientific Barriers Towards Drug Development for Alzheimer’s Disease. Cell. Mol. Neurobiol. 2019, 39, 181–209. [Google Scholar] [CrossRef]
- Yarong, H.; Yao, Y.; Tsirka, S.E.; Yu, C. Cell-Culture Models of the Blood–Brain Barrier. Stroke 2014, 45, 2514–2526. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Lee, J.; Lim, J.; Cho, S.; An, S.; Lee, M.; Yoon, N.; Seo, M.; Lim, S.; Park, S. Soluble RAGE attenuates AngII-induced endothelial hyperpermeability by disrupting HMGB1-mediated crosstalk between AT1R and RAGE. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.-Y.; Liu, C.-L.; Chen, J.-Y.; Hu, M.-L. Curcumin ameliorates methylglyoxal-induced alterations of cellular morphology and hyperpermeability in human umbilical vein endothelial cells. J. Funct. Foods 2013, 5, 745–754. [Google Scholar] [CrossRef]
- Cao, Y.; Gong, Y.; Liu, L.; Zhou, Y.; Fang, X.; Zhang, C.; Li, Y.; Li, J. The use of human umbilical vein endothelial cells (HUVECs) as an in vitro model to assess the toxicity of nanoparticles to endothelium: A review. J. Appl. Toxicol. 2017, 37, 1359–1369. [Google Scholar] [CrossRef]
- Boerma, M.; Burton, G.R.; Wang, J.; Fink, L.M.; McGehee, R.E., Jr.; Hauer-Jensen, M. Comparative expression profiling in primary and immortalized endothelial cells: Changes in gene expression in response to hydroxy methylglutaryl-coenzyme A reductase inhibition. Blood Coagul. Fibrinolysis 2006, 17, 173–180. [Google Scholar] [CrossRef]
- Yuan, J.; Zhu, C.; Hong, Y.; Sun, Z.; Fang, X.; Wu, B.; Li, S. The role of cPLA2 in Methylglyoxal-induced cell apoptosis of HUVECs. Toxicol. Appl. Pharmacol. 2017, 323, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, J.H.; Ahn, J.; Hong, M.J.; Kim, J.; Kim, S.Y. Isosamidin from Peucedanum japonicum Roots Prevents Methylglyoxal-Induced Glucotoxicity in Human Umbilical Vein Endothelial Cells via Suppression of ROS-Mediated Bax/Bcl-2. Antioxidants 2020, 9, 531. [Google Scholar] [CrossRef]
- Münch, G.; Apelt, J.; Kientsch-Engel, R.; Stahl, P.; Lüth, H.J.; Schliebs, R. Advanced glycation endproducts and pro-inflammatory cytokines in transgenic Tg2576 mice with amyloid plaque pathology. J. Neurochem. 2003, 86, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Nakamura, N.; Sotokawauchi, A.; Higashimoto, Y.; Yamagishi, S.-I. Methylglyoxal-derived hydroimidazolone-1 evokes inflammatory reactions in endothelial cells via an interaction with receptor for advanced glycation end products. Diabetes Vasc. Dis. Res. 2017, 14, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Yamamoto, Y.; Munesue, S.; Harashima, A.; Watanabe, T.; Yonekura, H.; Yamamoto, H.; Tsuchiya, H. Low molecular weight heparin suppresses receptor for advanced glycation end products-mediated expression of malignant phenotype in human fibrosarcoma cells. Cancer Sci. 2013, 104, 740–749. [Google Scholar] [CrossRef] [Green Version]
- Myint, K.-M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K.; et al. RAGE Control of Diabetic Nephropathy in a Mouse Model. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, S.; Rüster, C.; Pester, J.; Hofmann, G.; Oelzner, P.; Wolf, G. Advanced glycation end products affect growth and function of osteoblasts. Clin. Exp. Rheumatol. 2011, 29, 650–660. [Google Scholar]
- Simard, E.; Söllradl, T.; Maltais, J.-S.; Boucher, J.; D’Orléans-Juste, P.; Grandbois, M. Receptor for Advanced Glycation End-Products Signaling Interferes with the Vascular Smooth Muscle Cell Contractile Phenotype and Function. PLoS ONE 2015, 10, e0128881. [Google Scholar] [CrossRef] [Green Version]
- Talalay, P.; Dinkova-Kostova, A.T.; Holtzclaw, W.D. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv. Enzym. Regul. 2003, 43, 121–134. [Google Scholar] [CrossRef]
- Lee, J.-M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. BMB Rep. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeuerle, P.A. Pro-inflammatory signaling: Last pieces in the NF-kappaB puzzle? Curr. Biol. 1998, 8, R19–R22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Forman, H.J. Reexamination of the electrophile response element sequences and context reveals a lack of consensus in gene function. Biochim. Biophys. Acta 2010, 1799, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerboom, A.-M.J.F.; Vermeulen, M.; van der Woude, H.; Bremer, B.I.; Lee-Hilz, Y.Y.; Kampman, E.; van Bladeren, P.J.; Rietjens, I.M.C.M.; Aarts, J.M.M.J.G. Newly constructed stable reporter cell lines for mechanistic studies on electrophile-responsive element-mediated gene expression reveal a role for flavonoid planarity. Biochem. Pharmacol. 2006, 72, 217–226. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, S.C.; von Bergh, A.R.M.; van Vught-Lussenburg, B.M.A.; Jonker, L.R.A.; Teunis, M.; Krul, C.A.M.; van der Burg, B. Development of a panel of high-throughput reporter-gene assays to detect genotoxicity and oxidative stress. Mutat. Res. Toxicol. Environ. Mutagen. 2014, 760, 23–32. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s Disease Patient-Derived Induced Pluripotent Stem Cells Free of Viral Reprogramming Factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripo.tent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef]
- Sommer, A.; Maxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial alzheimer’s disease iPSC-Derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293. [Google Scholar] [CrossRef] [Green Version]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [Green Version]
- López de Maturana, R.; Lang, V.; Zubiarrain, A.; Sousa, A.; Vázquez, N.; Gorostidi, A.; Águila, J.; López de Munain, A.; Rodríguez, M.; Sánchez-Pernaute, R. Mutations in LRRK2 impair NF-ΚB pathway in iPSC-derived neurons. J. Neuroinflamm. 2016, 13, 295. [Google Scholar] [CrossRef] [Green Version]
- Mantle, J.L.; Lee, K.H. A differentiating neural stem cell-derived astrocytic population mitigates the inflammatory effects of TNF-α and IL-6 in an iPSC-based blood-brain barrier model. Neurobiol. Dis. 2018, 119, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Mantle, J.L.; Lee, K.H. Immunoglobulin G transport increases in an in vitro blood–brain barrier model with amyloid-β and with neuroinflammatory cytokines. Biotechnol. Bioeng. 2019, 116, 1752–1761. [Google Scholar] [CrossRef]
- Trindade, P.; Loiola, E.C.; Gasparotto, J.; Ribeiro, C.T.; Cardozo, P.L.; Devalle, S.; Salerno, J.A.; Ornelas, I.M.; Ledur, P.F.; Ribeiro, F.M.; et al. Short and long TNF-alpha exposure recapitulates canonical astrogliosis events in human-induced pluripotent stem cells-derived astrocytes. Glia 2020, 68, 1396–1409. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, M.J.; Gastfriend, B.D.; Canfield, S.G.; Lee, M.-S.; Richards, D.; Faubion, M.G.; Li, W.-J.; Daneman, R.; Palecek, S.P.; Shusta, E.V. Human pluripotent stem cell–derived brain pericyte–like cells induce blood-brain barrier properties. Sci. Adv. 2019, 5, eaau7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, J.J.; Linville, R.M.; Ding, Y.Y.; Gerecht, S.; Searson, P.C. Role of iPSC-derived pericytes on barrier function of iPSC-derived brain microvascular endothelial cells in 2D and 3D. Fluids Barriers CNS 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Faal, T.; Phan, D.T.T.; Davtyan, H.; Scarfone, V.M.; Varady, E.; Blurton-Jones, M.; Hughes, C.C.W.; Inlay, M.A. Induction of Mesoderm and Neural Crest-Derived Pericytes from Human Pluripotent Stem Cells to Study Blood-Brain Barrier Interactions. Stem Cell Rep. 2019, 12, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [Green Version]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Muñoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Kale, S.; El Fatimy, R.; Rabinovsky, R.; Krichevsky, A.M. Co-cultures of glioma stem cells and primary neurons, astrocytes, microglia, and endothelial cells for investigation of intercellular communication in the brain. Front. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef]
- Pabon, M.M.; Bachstetter, A.D.; Hudson, C.E.; Gemma, C.; Bickford, P.C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.R.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Gresa-Arribas, N.; Viéitez, C.; Dentesano, G.; Serratosa, J.; Saura, J.; Solà, C. Modelling Neuroinflammation In Vitro: A Tool to Test the Potential Neuroprotective Effect of Anti-Inflammatory Agents. PLoS ONE 2012, 7, e45227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-G.; Chen, S.-D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamynina, A.V.; Esteras, N.; Koroev, D.O.; Bobkova, N.V.; Balasanyants, S.M.; Simonyan, R.A.; Avetisyan, A.V.; Abramov, A.Y.; Volpina, O.M. Synthetic Fragments of Receptor for Advanced Glycation End Products Bind Beta-Amyloid 1–40 and Protect Primary Brain Cells From Beta-Amyloid Toxicity. Front. Neurosci. 2018, 12, 681. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.M.; Liu, Z.Y.; Ai, Y.H.; Zhang, L.N.; Zou, Y.; Peng, Q.Y. Blocking the CD38/cADPR pathway plays a double-edged role in LPS stimulated microglia. Neuroscience 2017, 361, 34–42. [Google Scholar] [CrossRef]
- Chen, J.M.; Li, Q.W.; Jiang, G.X.; Liu, J.S.; Cheng, Q. IL-18 induced IL-23/IL-17 expression impairs Aβ clearance in cultured THP-1 and BV2 cells. Cytokine 2019, 119, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APP(Swe) cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef]
- Yin, P.; Wang, S.; Wei, Y.; Wang, X.; Zhang, J.; Yin, X.; Feng, J.; Zhu, M. Maresin1 Decreased Microglial Chemotaxis and Ameliorated Inflammation Induced by Amyloid-β42 in Neuron-Microglia Co-Culture Models. J. Alzheimers Dis. 2020, 73, 503–515. [Google Scholar] [CrossRef]
- Zheng, Y.-F.; Zhou, X.; Chang, D.; Bhuyan, D.J.; Zhang, J.P.; Yu, W.-Z.; Jiang, X.-S.; Seto, S.W.; Yeon, S.Y.; Li, J.; et al. A novel tri-culture model for neuroinflammation. J. Neurochem. 2021, 156, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A primary neural cell culture model to study neuron, astrocyte, and microglia interactions in neuroinflammation. J. Neuroinflamm. 2020, 17, 115. [Google Scholar] [CrossRef]
- Appelt-Menzel, A.; Cubukova, A.; Günther, K.; Edenhofer, F.; Piontek, J.; Krause, G.; Stüber, T.; Walles, H.; Neuhaus, W.; Metzger, M. Establishment of a Human Blood-Brain Barrier Co-culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Rep. 2017, 8, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, E.S.; Weidenfeller, C.; Svendsen, C.N.; Shusta, E.V. Blood–brain barrier modeling with co-cultured neural progenitor cell-derived astrocytes and neurons. J. Neurochem. 2011, 119, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Pham, L.D.D.; Arai, K.; Lo, E.H. Reactive astrocytes promote adhesive interactions between brain endothelium and endothelial progenitor cells via HMGB1 and beta-2 integrin signaling. Stem Cell Res. 2014, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Wray, S. Modelling neurodegenerative disease using brain organoids. Semin. Cell Dev. Biol. 2021, 111, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; Kao, J.; Diamandis, P. Three-dimensional modeling of human neurodegeneration: Brain organoids coming of age. Mol. Psychiatry 2020, 25, 254–274. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Koito, H.; Li, J.; Han, A. Microfluidic compartmentalized co-culture platform for CNS axon myelination research. Biomed. Microdevices 2009, 11, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achyuta, A.K.H.; Conway, A.J.; Crouse, R.B.; Bannister, E.C.; Lee, R.N.; Katnik, C.P.; Behensky, A.A.; Cuevas, J.; Sundaram, S.S. A modular approach to create a neurovascular unit-on-a-chip. Lab Chip 2013, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. BBB on chip: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef]
- Osaki, T.; Shin, Y.; Sivathanu, V.; Campisi, M.; Kamm, R.D. In Vitro Microfluidic Models for Neurodegenerative Disorders. Adv. Healthc. Mater. 2018, 7, 1700489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsatsanis, A.; McCorkindale, A.N.; Wong, B.X.; Patrick, E.; Ryan, T.M.; Evans, R.W.; Bush, A.I.; Sutherland, G.T.; Sivaprasadarao, A.; Guennewig, B.; et al. The acute phase protein lactoferrin is a key feature of Alzheimer’s disease and predictor of Aβ burden through induction of APP amyloidogenic processing. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef] [Green Version]
- Weksler, B.; Romero, I.A.; Couraud, P.-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Reading patterns of proteome damage by glycation, oxidation and nitration: Quantitation by stable isotopic dilution analysis LC-MS/MS. Essays Biochem. 2020, 64, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Detection of oxidized and glycated proteins in clinical samples using mass spectrometry—A user’s perspective. Biochim. Biophys. Acta -Gen. Subj. 2014, 1840, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Van Der Lugt, T.; Weseler, A.R.; Gebbink, W.A.; Vrolijk, M.F.; Opperhuizen, A.; Bast, A. Dietary advanced glycation endproducts induce an inflammatory response in human macrophages in vitro. Nutrients 2018, 10, 1868. [Google Scholar] [CrossRef] [Green Version]
- Soboleva, A.; Vikhnina, M.; Grishina, T.; Frolov, A. Probing Protein Glycation by Chromatography and Mass Spectrometry: Analysis of Glycation Adducts. Int. J. Mol. Sci. 2017, 18, 2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Huang, G.; Xiao, L.; Mitchell, A.E. Determination of advanced glycation endproducts by LC-MS/MS in raw and roasted almonds (Prunus dulcis). J. Agric. Food Chem. 2011, 59, 12037–12046. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Reitano, R.; Aricò, N.C.; Seraglia, R.; Traldi, P.; Marotta, E.; Tonani, R. Enzymatic digestion and mass spectrometry in the study of advanced glycation end products/peptides. J. Am. Soc. Mass Spectrom. 2004, 15, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Reznikov, L.L.; Waksman, J.; Azam, T.; Kim, S.H.; Bufler, P.; Niwa, T.; Werman, A.; Zhang, X.; Pischetsrieder, M.; Shaldon, S.; et al. Effect of advanced glycation end products on endotoxin-induced TNF-alpha, IL-1beta and IL-8 in human peripheral blood mononuclear cells. Clin. Nephrol. 2004, 61, 324–336. [Google Scholar] [CrossRef]
- Buetler, T.M.; Latado, H.; Leclerc, E.; Weigle, B.; Baumeyer, A.; Heizmann, C.W.; Scholz, G. Glycolaldehyde-modified β-lactoglobulin AGEs are unable to stimulate inflammatory signaling pathways in RAGE-expressing human cell lines. Mol. Nutr. Food Res. 2011, 55, 291–299. [Google Scholar] [CrossRef]
- Buetler, T.M.; Leclerc, E.; Baumeyer, A.; Latado, H.; Newell, J.; Adolfsson, O.; Parisod, V.; Richoz, J.; Maurer, S.; Foata, F.; et al. Nε-carboxymethyllysine-modified proteins are unable to bind to RAGE and activate an inflammatory response. Mol. Nutr. Food Res. 2008, 52, 370–378. [Google Scholar] [CrossRef]
- Teodorowicz, M.; Perdijk, O.; Verhoek, I.; Govers, C.; Savelkoul, H.F.J.; Tang, Y.; Wichers, H.; Broersen, K. Optimized Triton X-114 assisted lipopolysaccharide (LPS) removal method reveals the immunomodulatory effect of food proteins. PLoS ONE 2017, 12, e0173778. [Google Scholar] [CrossRef] [Green Version]


| Relevant Endpoint | Cell Type | Line Name | Origin | Exposed to | Outcome | Reference |
|---|---|---|---|---|---|---|
| Neuroinflammation/inflammation | Microglia | BV2 | mouse | MGO-BSA | ↑ •NO, ROS, NLRP3, NF-κΒ, TNF-α, IL-6, MAPKs, RAGE, COX-2 | [89] |
| N11 | mouse | Glu-BSA | Microglial activation, ↑ •NO, IL-6, MCP-1, TNF-α | [119,120,242] | ||
| Glu-CEA | ↑ •NO, iNOS, TNF-α, IL_6 | [114,118] | ||||
| HMC3 | Human | Glu-HSA | ↑ •NO, apoptosis Anti-RAGE prevented cell death | [133] | ||
| Glu-BSA | Activated microglia (↑ Glu-5, CR3/43), ↑ RAGE, TNF-α Anti-RAGE decreased TNF-α | [132] | ||||
| HMO6 | Human | Aβ peptide | ↑ RAGE, synthesis of AGE-albumin | [96,98,122] | ||
| Monocytes/macrophages | THP-1 | Human | Glu-BSA | ↑ VEGF, IL-6, TNF-α, iNOS, RAGE, promoted M1 phenotype | [92,151,160] | |
| MG-H1 | ↑ Adhesion to HUVECs | [243] | ||||
| S100b | ↑ RAGE, MCP-1, IP-10, COX-2, NOX2, O2•, NF-κB activation. LR90 blocked the expression of pro-inflammatory cytokines, NF-κΒ and decreased the adhesion of THP-1 to endothelial cells. | [154] | ||||
| Hypoxia | ↑ AGEs, MCP-1, RAGE, NF-κΒ, Μ1 phenotype | [153,155,156] | ||||
| MGO | ↑ secretion of AGEs, glycation of cell surface proteins, unchanged ROS and RAGE, ↑ IL-1β, IL-8, TNF-α ↑ migration, Impaired chemotaxis | [157,158] | ||||
| MGO-BSA | ↑ COX2, TNF-α, M-CSF | [152,159,160] | ||||
| Neurodegeneration/neurotoxicity | Neuronal | PC12 | Rat | Ribosylated-BSA | ↑ apoptosis, iNOS, COX2, pp38 | [177,178,179] |
| Glu-BSA | ↑ apoptosis, RAGE, NF-κΒ | |||||
| Glu + MGO | ↑ protein carbonyls, ROS, RAGE, NF-κΒ | |||||
| Neuro2A | mouse | MGO | ↑ ROS, apoptosis, intracellular CML | [181,182] | ||
| SH-SY5Y | human | AGE-BSA | ↑ RAGE, ROS, apoptosis, NF-κB, AMPK | [195] | ||
| MGO | ↑ apoptosis, ROS, MDA, mitochondrial damage ↓SOD, CAT, GSH | [170] | ||||
| Ribosylated α-synuclein | ↑ ROS | [200] | ||||
| GA | ↑ AGE accumulation intracellularly, apoptosis, VEGF, TGF-β, phosphorylated tau ↓GAPDH activity | [100,171] | ||||
| LUHMES | Human | AGE-BSA | ↑ RAGE, MAPKs, Bax | [122] | ||
| BBB | Endothelial | bEnd.3 | Mouse | MGO-BSA | ↑ monolayer permeability, ROS ↓SOD2 activity, impaired respiratory metabolism | [59] |
| BMECs | Human or animal | AGE-BSA | ↓claudin-5, TEER, Unchanged occludin-1 and ZO-1, ↑ monolayer permeability due to ↑ VEGF, MMP-2 leading to thickening of the basement membrane and barrier disruption | [58] | ||
| HUVECs | human | Glu-BSA MGO-BSA MGO-OvA, CML-BSA | ↓GSH activity ↑ GPx activity | [167] | ||
| Glycated insulin | ↓viability, Bcl-2 ↑ Bax, ROS, permeability | [57] | ||||
| MGO | ↑ ROS, apoptosis, Bax, MAPKs ↓Bcl-2, GLO-1, unchanged Nrf2 ↑ monolayer permeability, actin stress fibers ↓ZO-1, Cx43, GJIC | [237,240,241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysanthou, M.; Miro Estruch, I.; Rietjens, I.M.C.M.; Wichers, H.J.; Hoppenbrouwers, T. In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration. Nutrients 2022, 14, 363. https://doi.org/10.3390/nu14020363
Chrysanthou M, Miro Estruch I, Rietjens IMCM, Wichers HJ, Hoppenbrouwers T. In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration. Nutrients. 2022; 14(2):363. https://doi.org/10.3390/nu14020363
Chicago/Turabian StyleChrysanthou, Marialena, Ignacio Miro Estruch, Ivonne M. C. M. Rietjens, Harry J. Wichers, and Tamara Hoppenbrouwers. 2022. "In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration" Nutrients 14, no. 2: 363. https://doi.org/10.3390/nu14020363