PEG-Poly(1-Methyl-l-Tryptophan)-Based Polymeric Micelles as Enzymatically Activated Inhibitors of Indoleamine 2,3-Dioxygenase

 and
and
Abstract
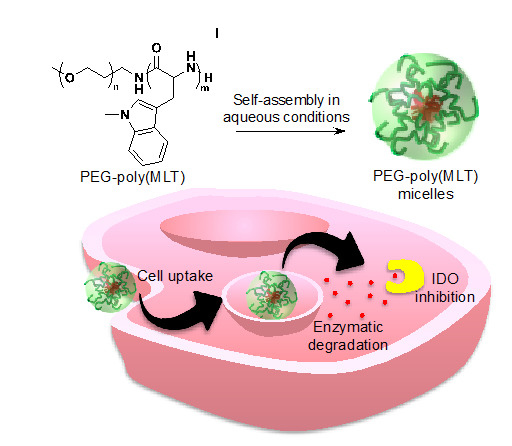
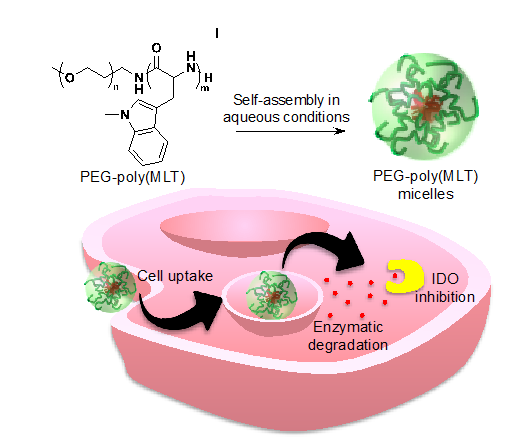
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
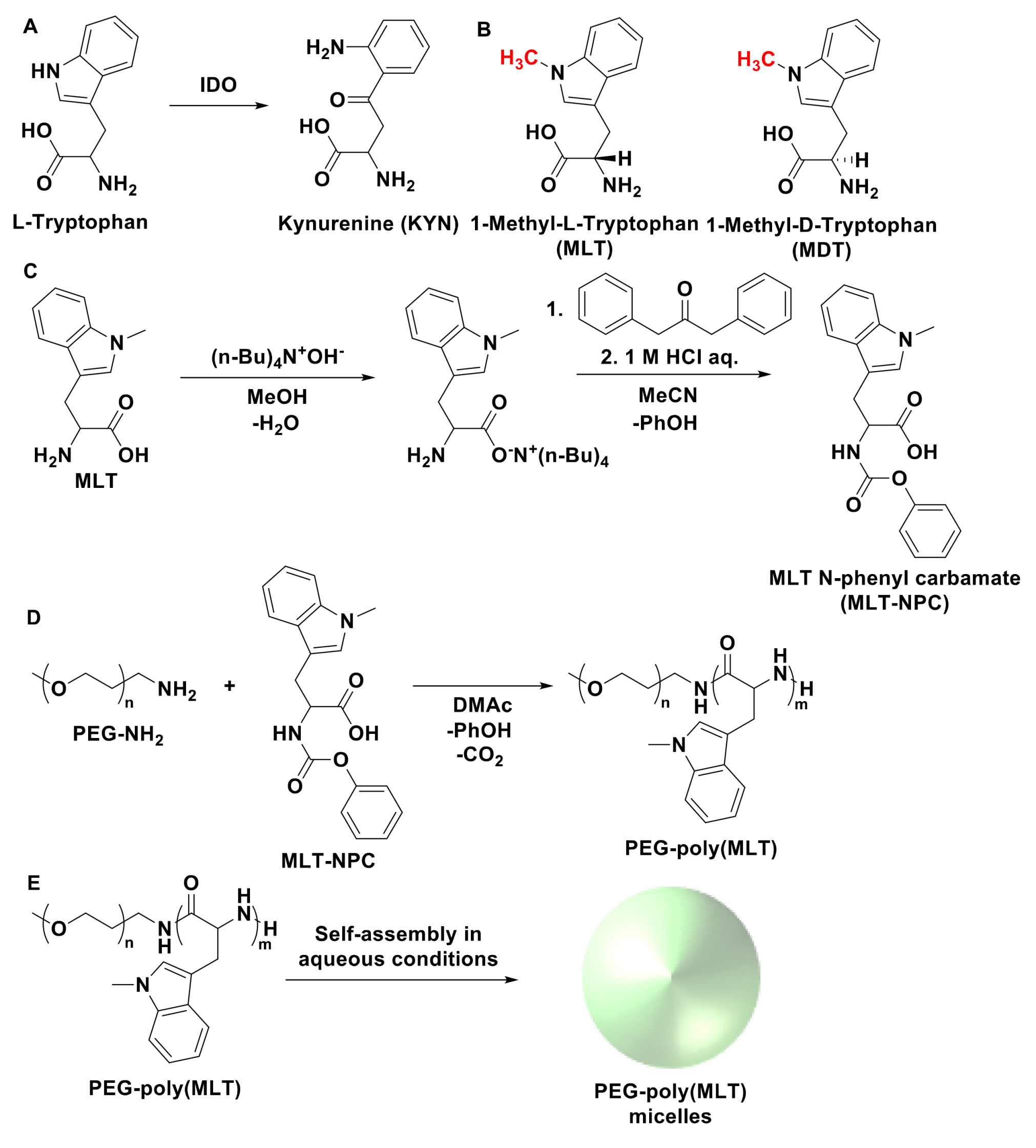
2.3. Synthesis of P(MLT) and P(MDT) by Condensation Reaction
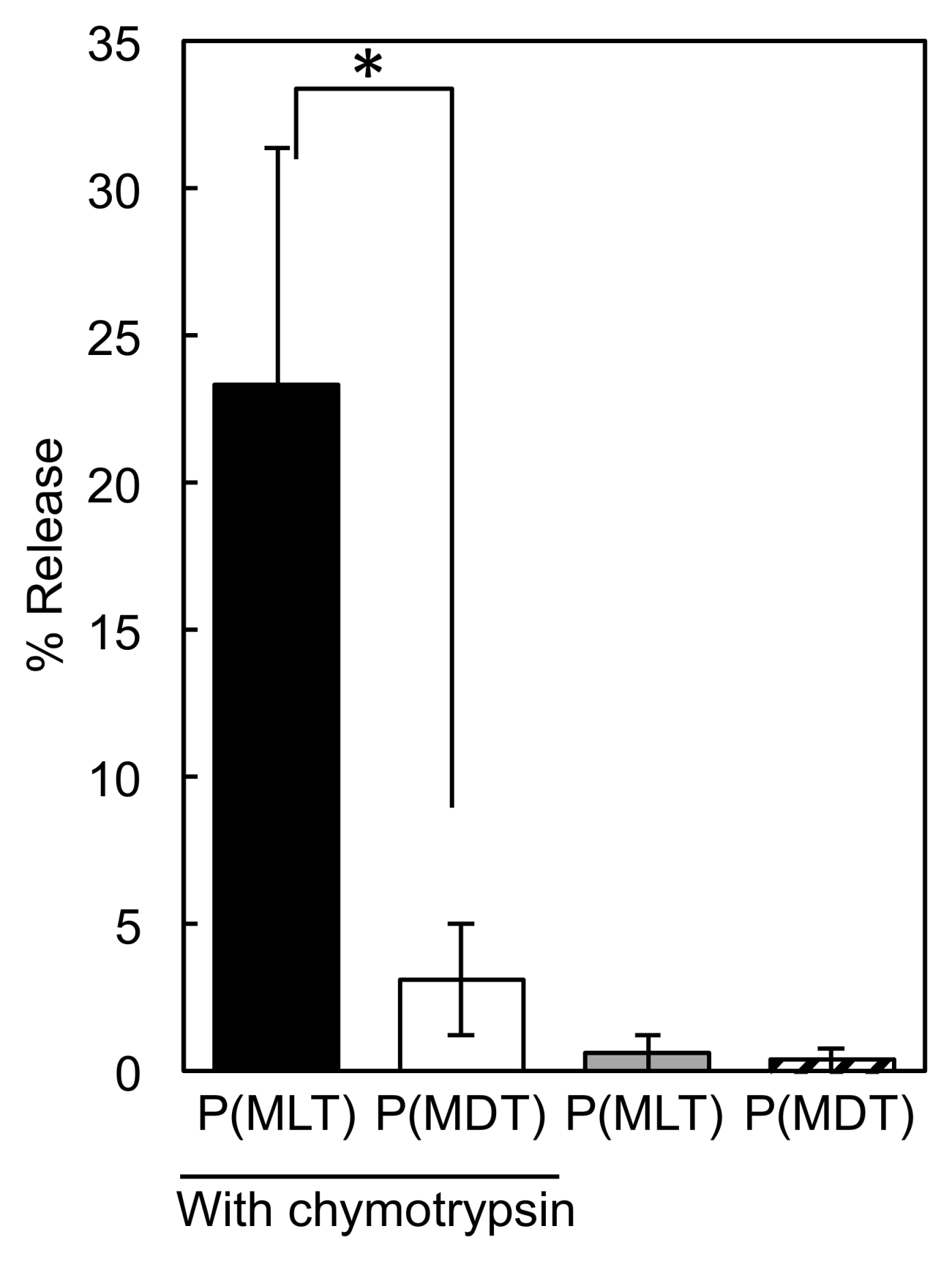
2.4. Enzymatic Digestion of P(MLT) and P(MDT)
2.5. Synthesis of MLT-NPC
2.6. Synthesis of PEG-P(MLT) Block Copolymer
2.7. Enzymatic Digestion of PEG-P(MLT)
2.8. Cytotoxicity of PEG-P(MLT)
2.9. Self-Assembly of Polymeric Micelles
2.10. Cell Uptake Assay
2.11. KYN Inhibition Assay
2.12. Quantification of TNF-α Expression by THP-1 by Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Quantification of NF-κB Expression in RAW 264.7 by Secreted Alkaline Phosphatase (SEAP) Reporter Gene Assay
2.14. HPLC Detection
3. Results
3.1. Enzymatic Degradation of P(MLT) and P(MDT)
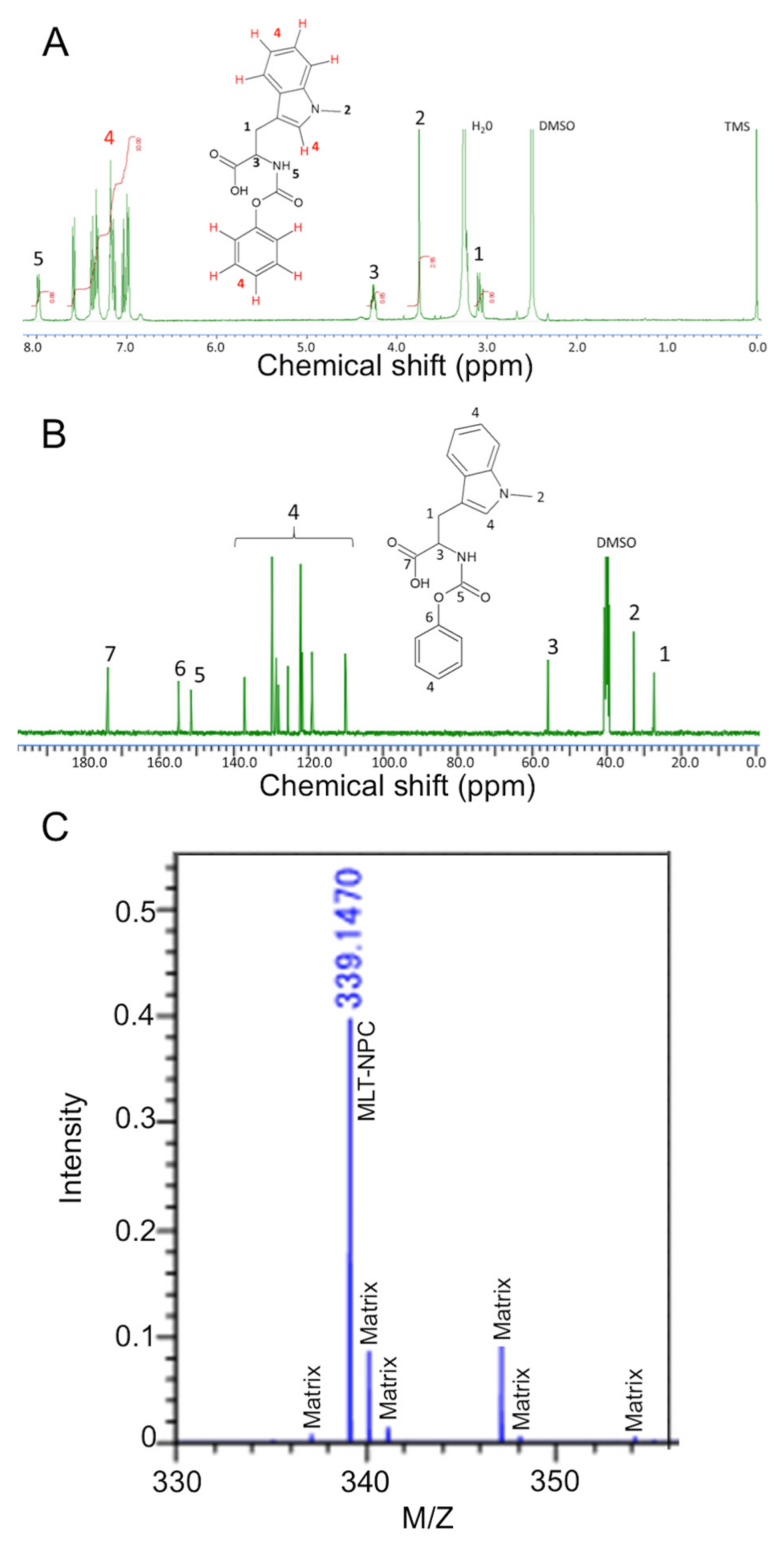
3.2. Synthesis and Characterization of NPC-MLT
3.3. Synthesis of PEG-P(MLT) and Self-Assembly of Micelles
3.4. Release of MLT from PEG-P(MLT) after Enzymatic Cleavage
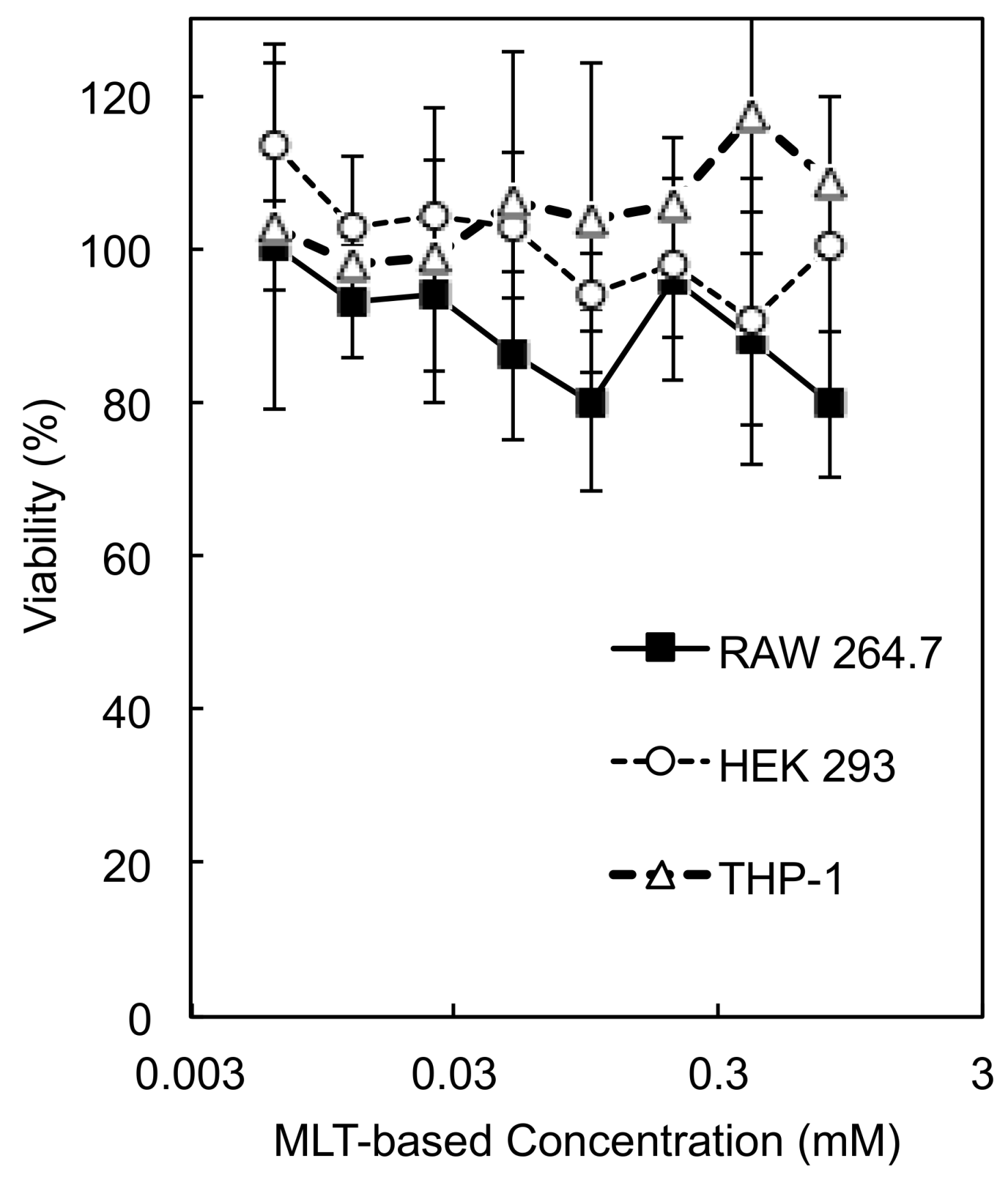
3.5. Cytotoxicity of PEG-P(MLT)
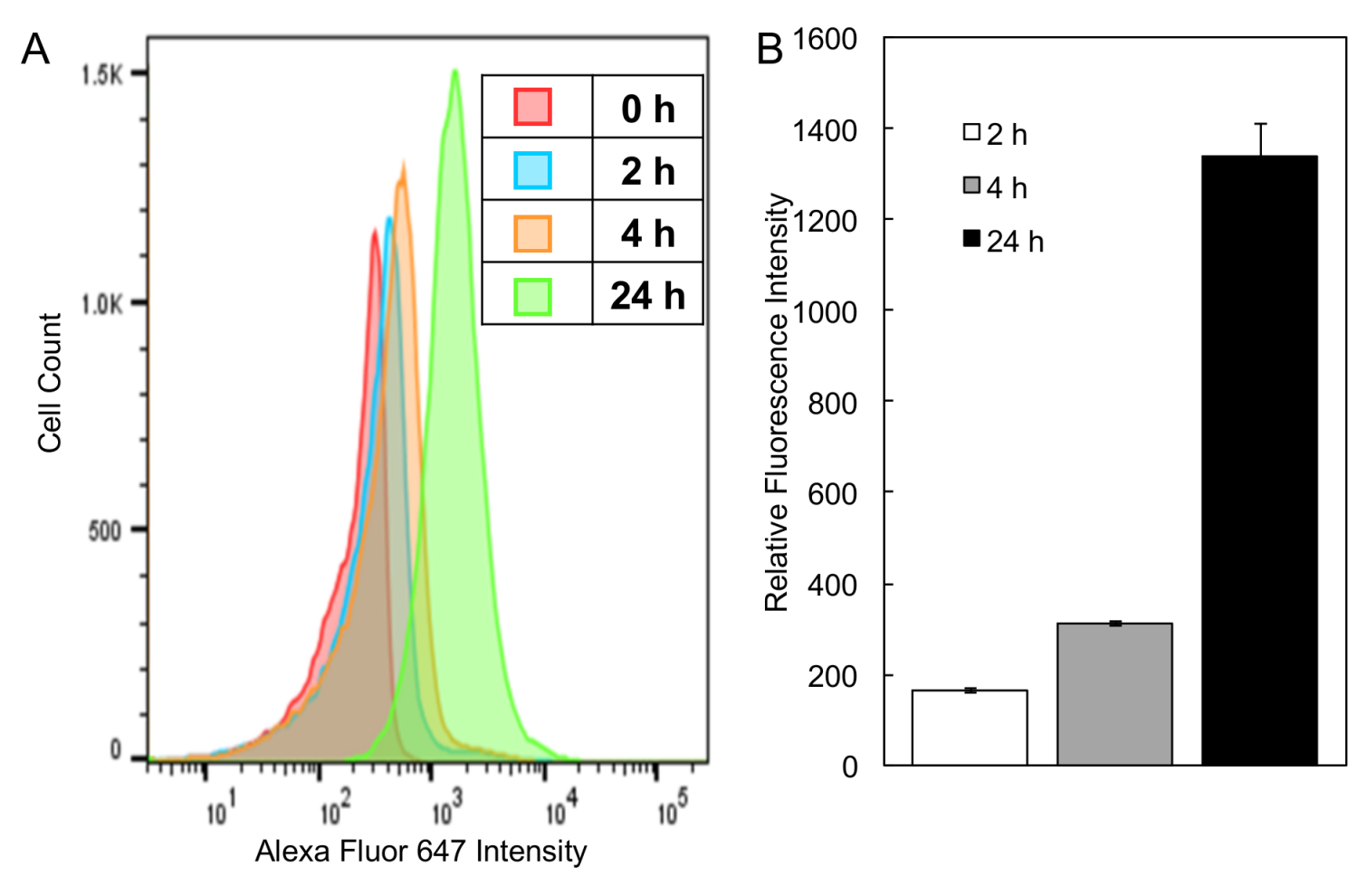
3.6. Cellular Uptake of PEG-P(MLT) Micelles
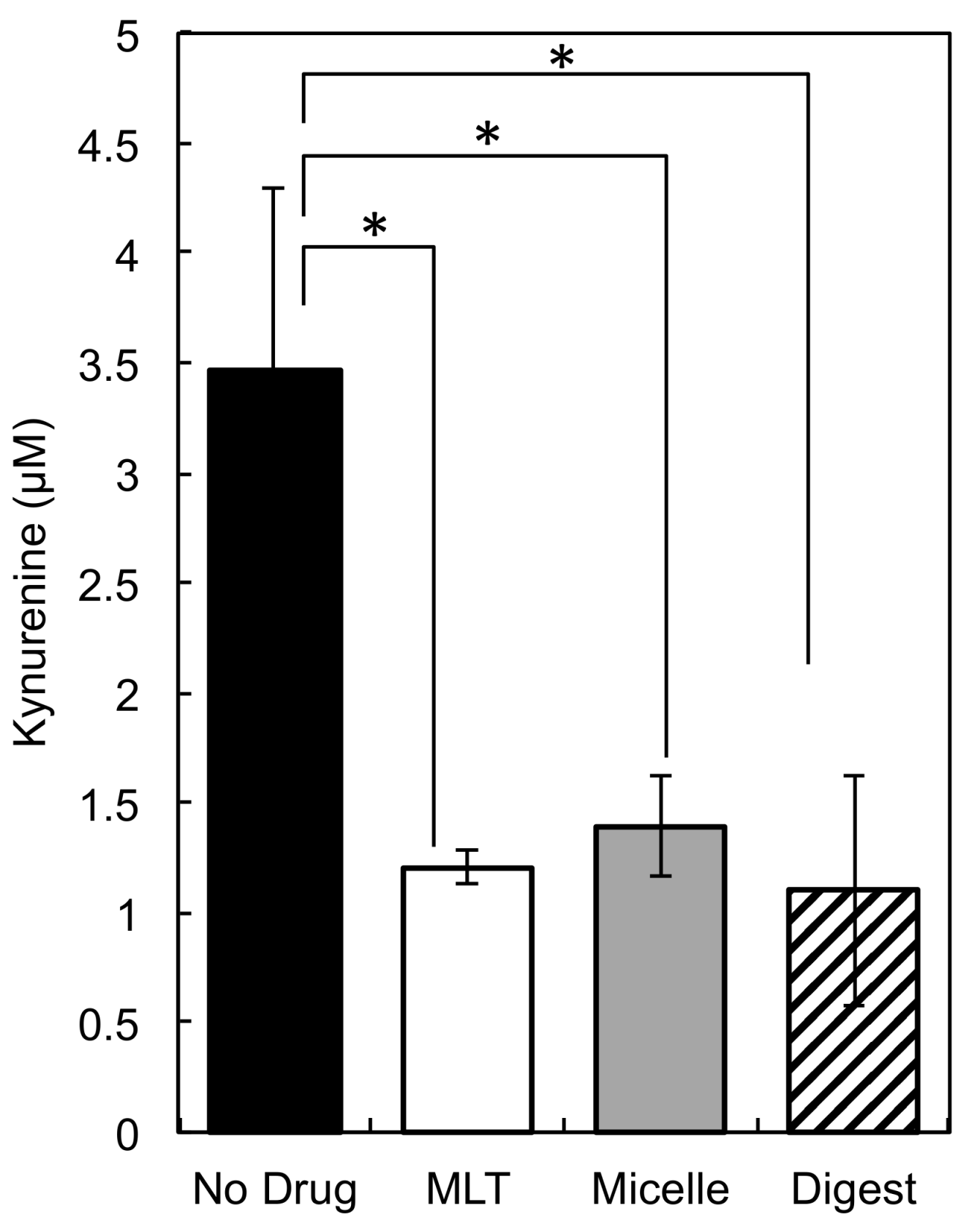
3.7. Inhibition of KYN by PEG-P(MLT) Micelles
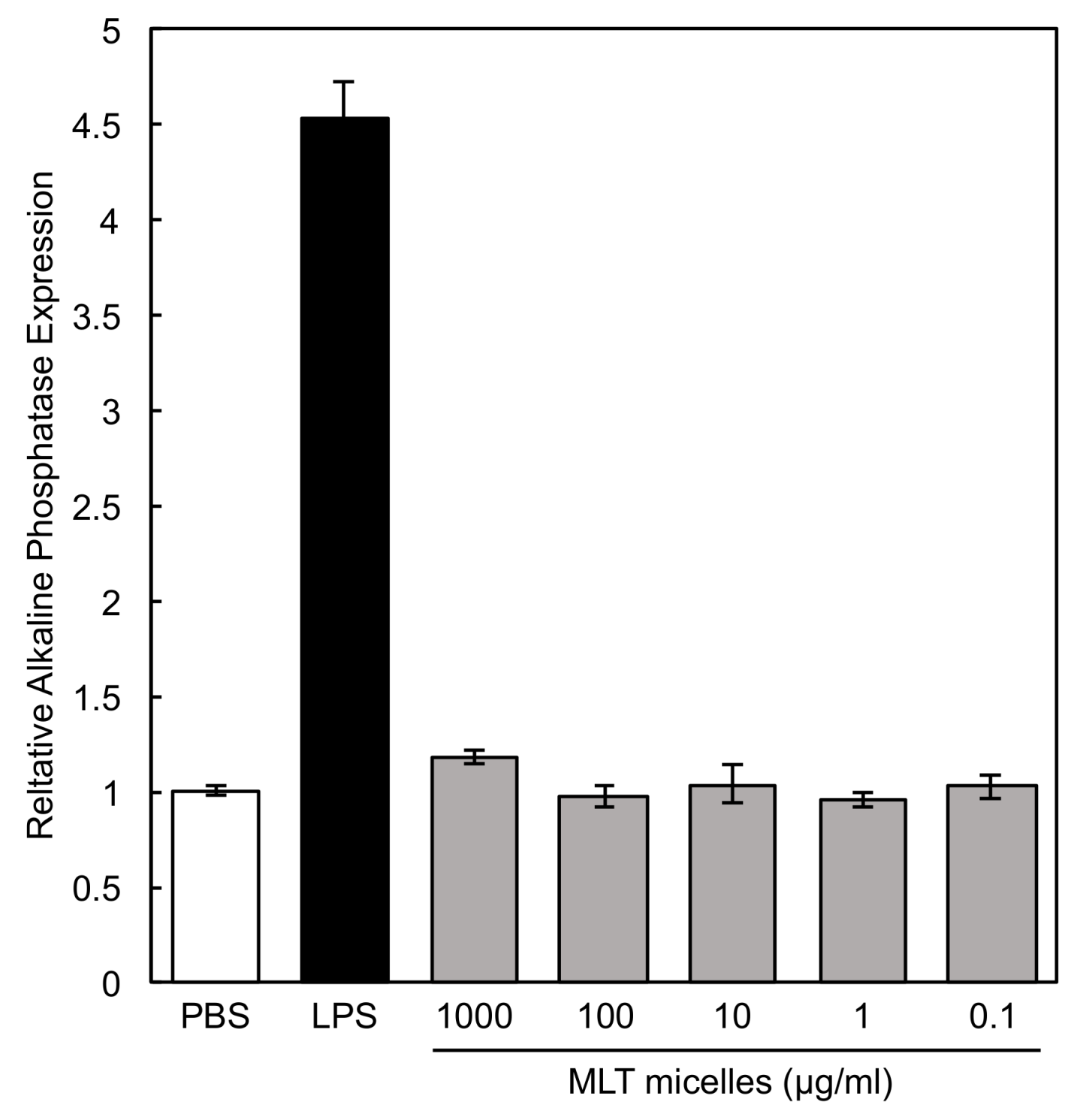
3.8. Macrophage Activity in the Presence of PEG-P(MLT) Micelles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tang, J.; Pearce, L.; Donnell-tormey, J.O.; Hubbard-lucey, V.M. FROM THE ANALYST ’ S COUCH Trends in the global immuno-oncology landscape. Nat. Publ. Gr. 2018, 17, 783–784. [Google Scholar]
- Prendergast, G.C.; Jaffee, E.M. Cancer Immunologists and Cancer Biologists: Why We Didn’t Talk Then but Need to Now. Cancer Res. 2007, 3500–3505. [Google Scholar] [CrossRef] [PubMed]
- Rieth, J.; Subramanian, S. Mechanisms of intrinsic tumor resistance to immunotherapy. Int. J. Mol. Sci. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D.; et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928–22938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ino, K.; Yoshida, N.; Kajiyama, H.; Shibata, K.; Yamamoto, E.; Kidokoro, K.; Takahashi, N.; Terauchi, M.; Nawa, A.; Nomura, S.; et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 2006, 95, 1555–1561. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Hou, D.Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D.H. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef]
- Cabral, H.; Kataoka, K. Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M. Comparison of the Proteolytic Susceptibilities of Homologous L-Amino Acid, D-Amino Acid, and N-Substituted Clycine Peptide and Peptoid Oligomers. Drug Dev. Res. 1995, 32, 20–32. [Google Scholar] [CrossRef]
- Yamada, S.; Atsushi, S.; Goto, M.; Endo, T. Facile Synthesis of Poly ( L -tryptophan ) through Polycondensation of Activated Urethane Derivatives. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4565–4571. [Google Scholar] [CrossRef]
- Wu, H.; Tao, A.; Martin, J.D.; Quader, S.; Liu, X.; Takahashi, K.; Hespel, L.; Miura, Y.; Hayakawa, Y.; Irimura, T.; et al. Proteasome Inhibitor e Loaded Micelles Enhance Antitumor Activity Through Macrophage Reprogramming by NF- k B Inhibition. J. Pharm. Sci. 2017, 106, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, S.; Saito, K.; Sekikawa, K.; Tone, S.; Takikawa, O.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-γ-independent mechanism. Eur. J. Immunol. 2001, 31, 2313–2318. [Google Scholar] [CrossRef] [Green Version]
- Laich, A.; Neurauter, G.; Widner, B.; Fuchs, D. More Rapid Method for Simultaneous Measurement of Tryptophan and Kynurenine by HPLC. Clin. Chem. 2002, 48, 579–581. [Google Scholar] [PubMed]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- HO, C.H.; Odermatt, E.; Berndt, I.; Tiller, J.C. Ways of Selective Polycondensation of L-Lysine Towards Linear a- and e-Poly-L-Lysine. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 5053–5063. [Google Scholar] [CrossRef]
- Okamura, A.; Hirai, T.; Tanihara, M.; Yamaoka, T. Synthesis and properties of novel biodegradable polyamides containing α-amino acids. Polymer (Guildf) 2002, 43, 3549–3554. [Google Scholar] [CrossRef]
- Cheng, J.; Deming, T.J. Synthesis of Polypeptides by Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides BT - Peptide-Based Materials. In Peptide-Based Materials; Deming, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–26. ISBN 978-3-642-27139-7. [Google Scholar]
- Doriti, A.; Brosnan, S.M.; Weidner, S.M.; Schlaad, H. Synthesis of polysarcosine from air and moisture stable N-phenoxycarbonyl-N-methylglycine assisted by tertiary amine base. Polym. Chem. 2016, 7, 3067–3070. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Koga, K.; Endo, T. Useful synthetic method of polypeptides with well-defined structure by polymerization of activated urethane derivatives of α-amino acids. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Sudo, A.; Goto, M.; Endo, T. Phosgene-free synthesis of polypeptides using activated urethane derivatives of α-amino acids: An efficient synthetic approach to hydrophilic polypeptides. RSC Adv. 2014, 4, 29890–29896. [Google Scholar] [CrossRef]
- Yamada, S.; Koga, K.; Sudo, A.; Goto, M.; Endo, T. Phosgene-free synthesis of polypeptides: Useful synthesis for hydrophobic polypeptides through polycondensation of activated urethane derivatives of α-amino acids. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3726–3731. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Baqui, A.A.M.A.; Meiller, T.F.; Turng, B.F.; Kelley, J.I.; Falkler, W.A. Functional changes in THP-1 human monocytic cells after stimulation with lipopolysacchar1de of oral microorganisms and granulocyte macrophage colony stimulating factor. Immunopharmacol. Immunotoxicol. 1998, 20, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 171–176. [Google Scholar] [CrossRef]
- Trevejo, J.M.; Marino, M.W.; Philpott, N.; Josien, R.; Richards, E.C.; Elkon, K.B.; Falck-Pedersen, E. TNF-α-dependent maturation of local dendritic cells is critical for activating the adaptive immune response to virus infection. Proc. Natl. Acad. Sci. USA 2002, 98, 12162–12167. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Mak, T.W.; Ohashi, P.S. TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Ikkyu, K.; Iso, K.; Goto, M.; Endo, T. Facile synthesis of polymethionine oxides through polycondensation of activated urethane derivative of α-amino acid and their application to antifouling polymer against proteins and cells. Polym. Chem. 2015, 6, 1838–1845. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Review Hallmarks of Cancer The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fruton, J.S.; Bergmann, M. The Multiple Specificity of Chymotrypsin. J. Biol. Chem. 1942, 145, 253–265. [Google Scholar]
- Kondakova, I.V.; Spirina, L.V.; Koval, V.D.; Shashova, E.E.; Choinzonov, E.L.; Ivanova, E.V.; Kolomiets, L.A.; Chernyshova, A.L.; Slonimskaya, E.M.; Usynin, E.A.; et al. BIOLOGY Chymotrypsin Like Activity and Subunit Composition of Proteasomes in Human Cancers. Mol. Biol. 2014, 48, 384–389. [Google Scholar] [CrossRef]
- Garcia, M.; Platet, N.; Liaudet, E.; Laurent, V.; Derocq, D.; Brouillet, J.; Rochefort, H. Concise Review Biological and Clinical Significance of Cathepsin D in Breast Cancer Metastasis. Stem Cells 1996, 14, 642–650. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Wickström, M.; Larsson, R.; Nygren, P.; Gullbo, J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011, 102, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR A novel IDO effector pathway targeted by D -1-methyl-tryptophan. RSC Adv. 2012, 1, 1460–1468. [Google Scholar] [CrossRef]
- Zhang, J.; Kinoh, H.; Hespel, L.; Liu, X.; Quader, S.; Martin, J.; Chida, T.; Cabral, H.; Kataoka, K. E ff ective treatment of drug resistant recurrent breast tumors harboring cancer stem-like cells by staurosporine / epirubicin co-loaded polymeric micelles. J. Control. Release 2017, 264, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Schultze, J.L. New insights into IDO biology in bacterial and viral infections. Front. Immunol. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Gautam, U.S.; Foreman, T.W.; Bucsan, A.N.; Veatch, A.V.; Alvarez, X.; Adekambi, T. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, E62–E71. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Vaccari, M.; Fuchs, D.; Hardy, A.W.; Tsai, W.-P.; Tryniszewska, E.; Shearer, G.M.; Franchini, G. Combined Effect of Antiretroviral Therapy and Blockade of IDO in SIV-Infected Rhesus Macaques. J. Immunol. 2009, 182, 4313–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.L.; Tao, A.; Miyazaki, T.; Khan, T.; Hong, T.; Nakagawa, Y.; Cabral, H. PEG-Poly(1-Methyl-l-Tryptophan)-Based Polymeric Micelles as Enzymatically Activated Inhibitors of Indoleamine 2,3-Dioxygenase. Nanomaterials 2019, 9, 719. https://doi.org/10.3390/nano9050719
Huang GL, Tao A, Miyazaki T, Khan T, Hong T, Nakagawa Y, Cabral H. PEG-Poly(1-Methyl-l-Tryptophan)-Based Polymeric Micelles as Enzymatically Activated Inhibitors of Indoleamine 2,3-Dioxygenase. Nanomaterials. 2019; 9(5):719. https://doi.org/10.3390/nano9050719
Chicago/Turabian StyleHuang, George Lo, Anqi Tao, Takuya Miyazaki, Thahomina Khan, Taehun Hong, Yasuhiro Nakagawa, and Horacio Cabral. 2019. "PEG-Poly(1-Methyl-l-Tryptophan)-Based Polymeric Micelles as Enzymatically Activated Inhibitors of Indoleamine 2,3-Dioxygenase" Nanomaterials 9, no. 5: 719. https://doi.org/10.3390/nano9050719