
Determination of Glycerophospholipids in Biological Material Using High-Performance Liquid Chromatography with Charged Aerosol Detector HPLC-CAD—A New Approach for Isolation and Quantification


Abstract
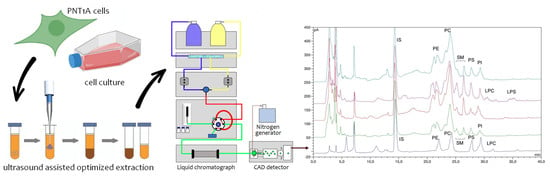
:
1. Introduction
2. Results and Discussion
2.1. Method Development
2.1.1. Optimization of Phospholipid Extraction Conditions
2.1.2. Optimization of Chromatographic Conditions
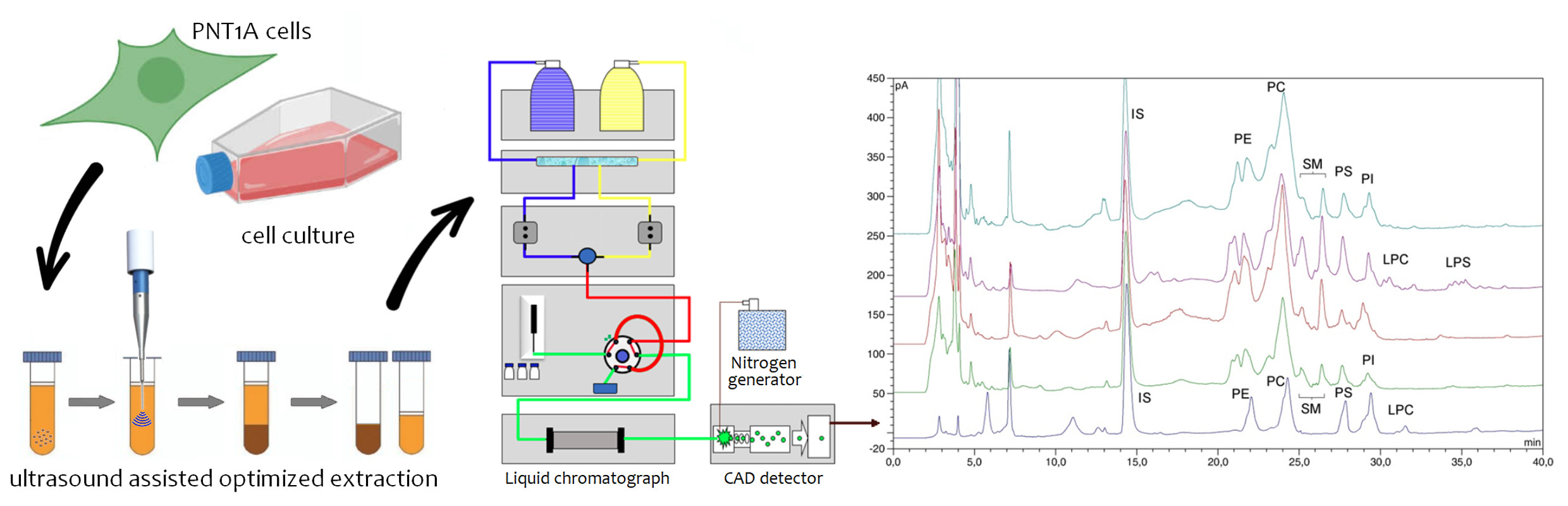
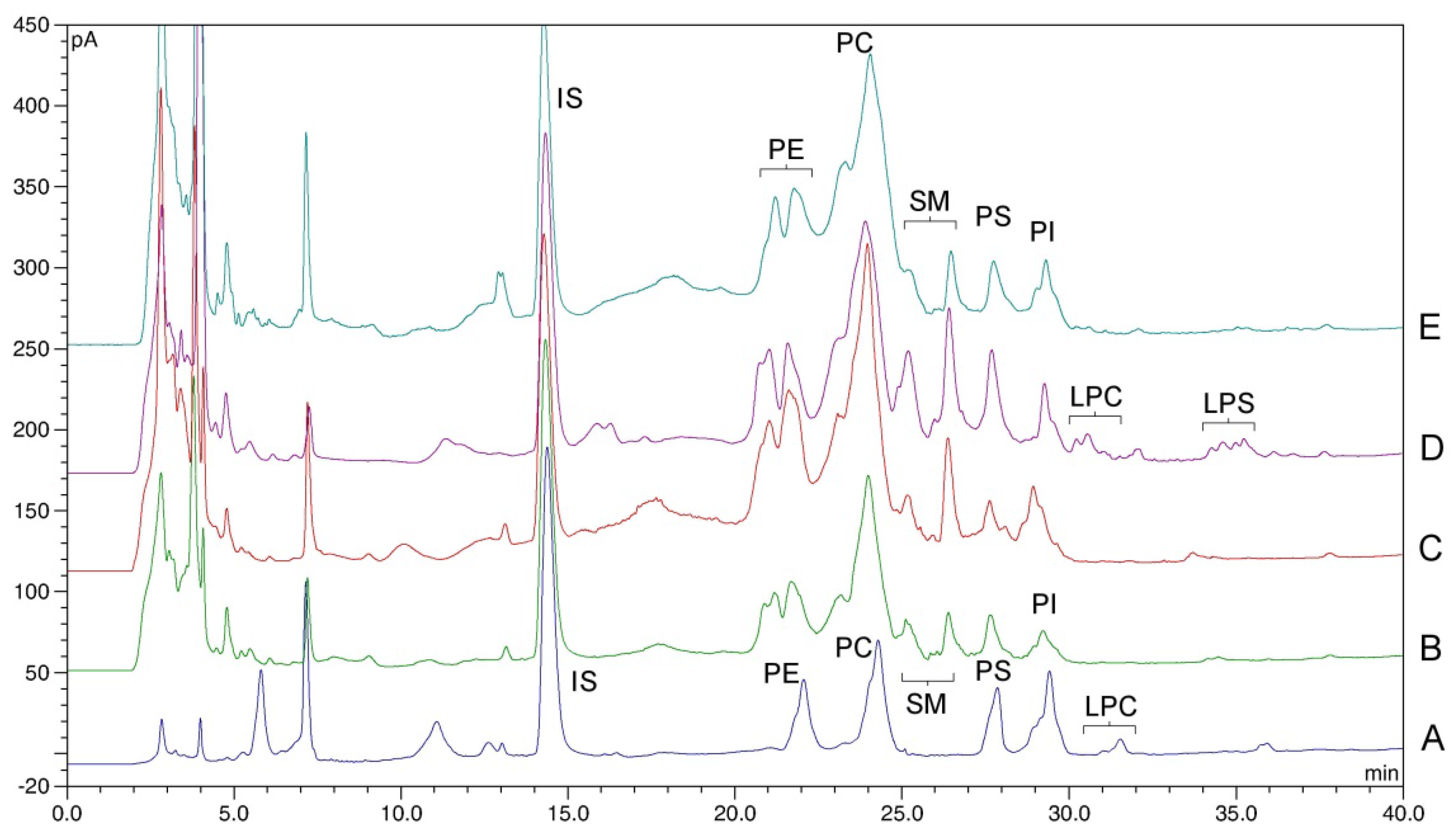
2.2. Identification of Phospholipid Peaks
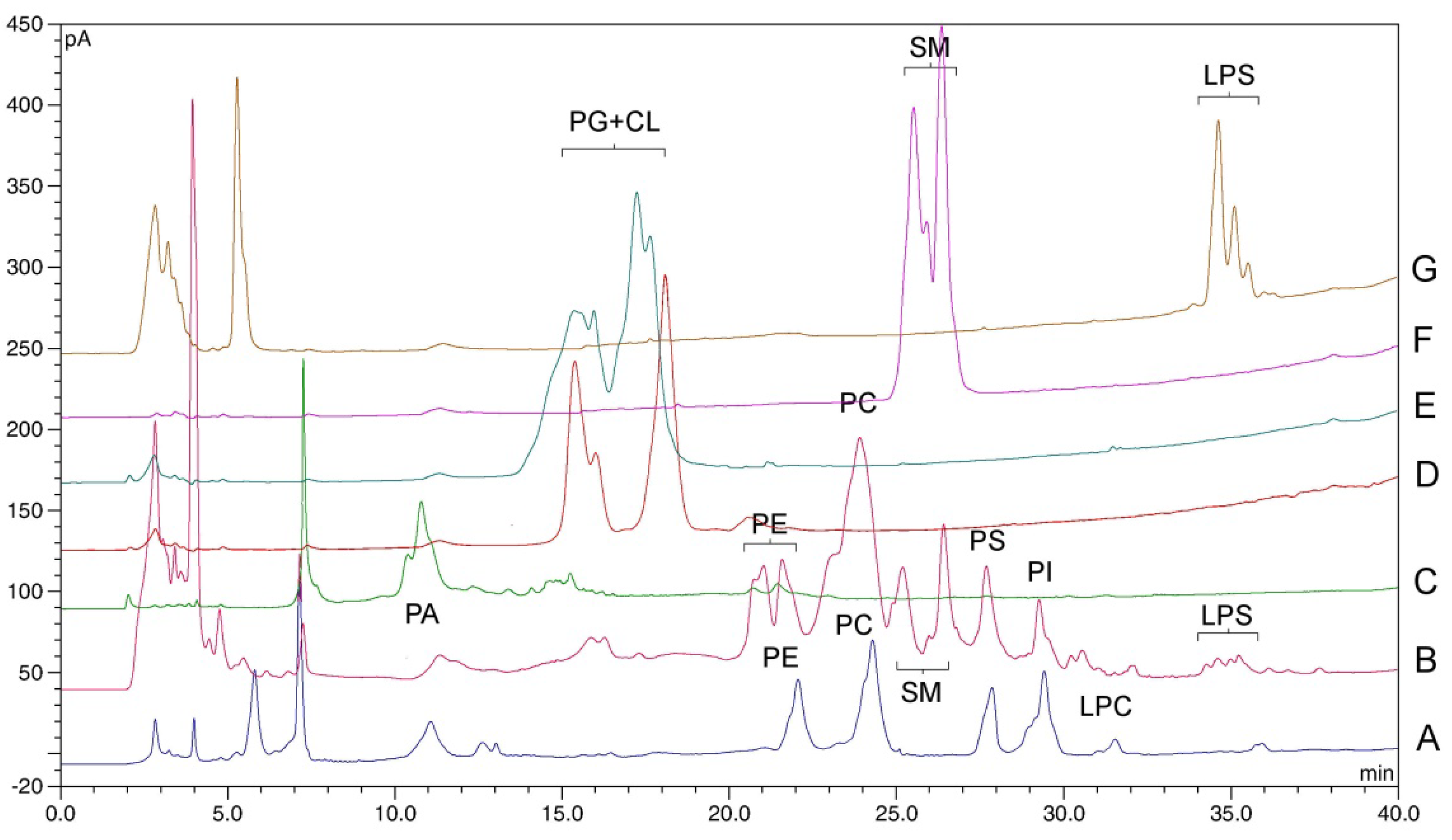
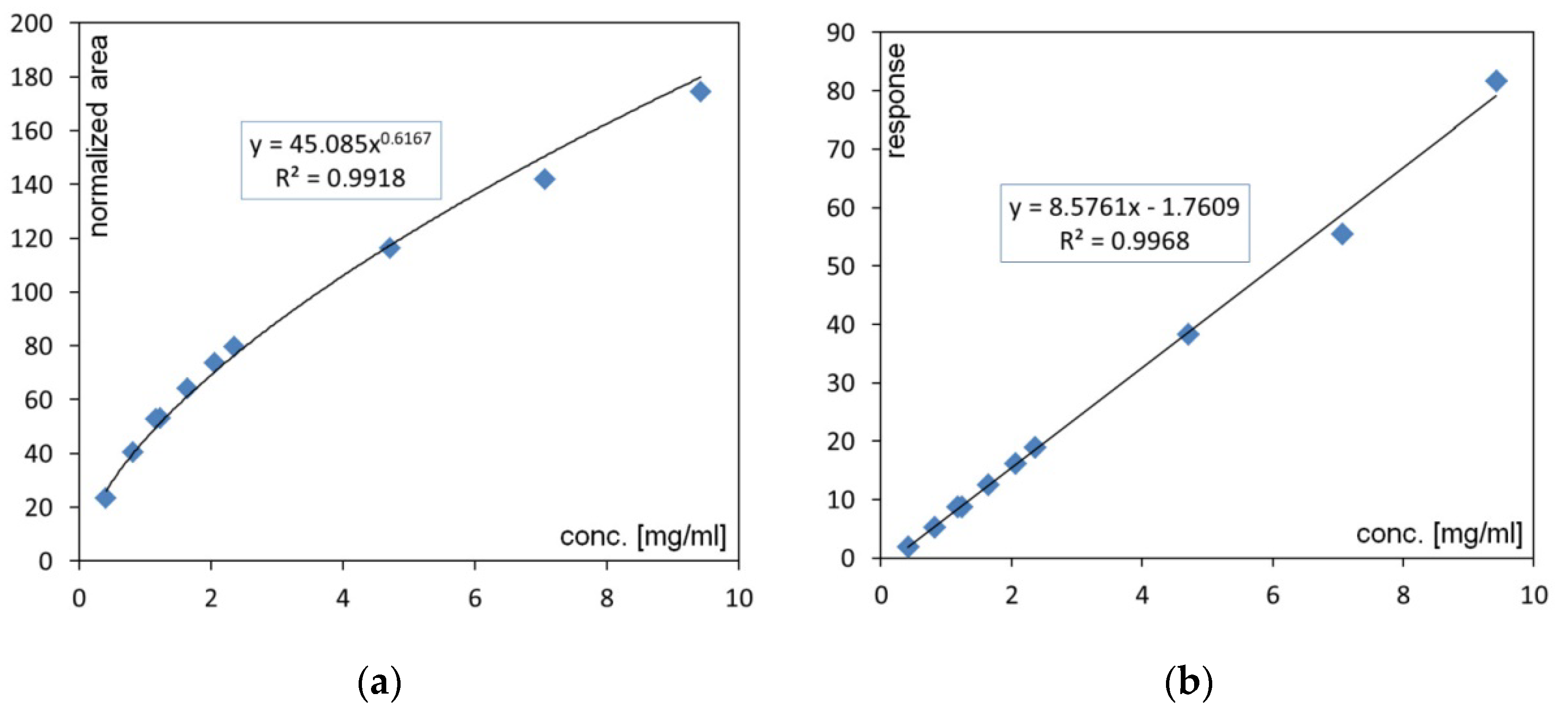
2.3. Methodology for Phospholipid Quantification
- A—normalized peak area;
- X—power function value;
- scale—full-scale range of the detector (e.g., 500 pA);
- Alin—response value eligible for linear fit calibration.
2.4. Method Validation
2.5. Applicability of the Method
- Human normal (PNT1A) and neoplastic (LNCaP) prostate cells;
- Human normal fibroblasts (BJ) and melanoma cells (HTB 140).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | PNT1A Normal Prostate Cells | LNCaP Neoplastic Prostate Cells | BJ Human Normal Fibroblasts | HTB 140 Human Melanoma Cells | |
|---|---|---|---|---|---|
| Phospholipids | |||||
| Content | n = 5 | n = 5 | n = 4 | n = 5 | |
| PE | Mean ± SD (nmol/mln cells) | 12.7 ± 1.0 | 40.5 ± 5.0 *** | 17.1 ± 1.8 | 23.6 ± 3.1 ** |
| mean (mol%) 1 | 48.2% | 56.7% ** | 35.7% | 33.9% | |
| PC | Mean ± SD (nmol/mln cells) | 10.8 ± 0.8 | 24.5 ± 1.8 *** | 24.1 ± 1.0 | 40.4 ± 6.2 ** |
| mean (mol%) 1 | 41.1% | 34.4% ** | 50.3% | 57.6% * | |
| PS | Mean ± SD (nmol/mln cells) | 1.1 ± 0.1 | 1.9 ± 0.2 *** | 3.3 ± 0.3 | 2.1 ± 0.2 *** |
| mean (mol%) 1 | 4.3% | 2.6% *** | 6.9% | 3.0% *** | |
| PI | Mean ± SD (nmol/mln cells) | 1.7 ± 0.2 | 4.5 ± 0.3 *** | 3.4 ± 0.3 | 3.8 ± 0.6 |
| mean (mol%) 1 | 6.4% | 6.3% | 7.1% | 5.5% * | |
3. Materials and Methods
3.1. Reagents and Reference Materials
3.2. Cell Lines and Cultivation Conditions
3.3. Sample Preparation for Phospholipids Assay
3.4. Chromatographic Conditions
3.5. Calculations and Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Guo, Z.; Vikbjerg, A.F.; Xu, X. Enzymatic modification of phospholipids for functional applications and human nutrition. Biotechnol. Adv. 2005, 23, 203–259. [Google Scholar] [CrossRef] [PubMed]
- Shewan, A.; Eastburn, D.J.; Mostov, K. Phosphoinositides in Cell Architecture. Cold Spring Harb. Perspect. Biol. 2011, 3, a004796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, R.G.; Libong, D.; Rakotomanga, M. Comparioson between charge aerosol detection and light scattering detection for the analysis of Leishmania membrane phospholipids. J. Chromatogr. A 2008, 1209, 88–94. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Micek, P.; Wawrzeńczyk, C. A new liquid chromatography method with charge aerosol detector (CAD) for determination of phospholipid classes. Application to milk phospholipids. Talanta 2013, 105, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Masayama, A.; Takahashi, T.; Tsukada, K.; Nishikawa, S.; Takahashi, R. Streptomyces Phospholipase D Mutants with Altered Substrate Specifity Capable of Phosphatydylinositol Synthesis. ChemBioChem 2008, 9, 974–981. [Google Scholar] [CrossRef]
- Seri, H.I.; Naim, H.Y.; Hassan, T. A high performance liquid chromatography method with evaporative light scattering detector for quantification of major phospholipids classes of donkey serum. Vet. Arh. 2010, 80, 365–373. [Google Scholar]
- Buré, C.; Ayciriex, S.; Testet, E.; Schmitter, J.M. A single run LC-MS/MS method for phospholipidomics. Anal. Bioanal. Chem. 2013, 405, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, D.; Wehtje, E. A simple HPLC method for the simultaneous analysis of phosphatidylcholine and its partial hydrolysis products 1- and 2-acyl lysophosphatidylcholine. JAOCS J. Am. Oil Chem. Soc. 2001, 78, 1007–1011. [Google Scholar] [CrossRef]
- Dixon, R.W.; Peterson, D.S. Development and testing of a new detector for liquid chromatography based on aerosol charging. Anal. Chem. 2002, 74, 2930–2937. [Google Scholar] [CrossRef]
- Vehoveca, T.; Obreza, A. Review of operating principle and applications of the charged aerosol detector. J. Chromatogr. A 2010, 1217, 1549–1556. [Google Scholar] [CrossRef]
- Almeling, S.; Ilkob, D.; Holzgrabe, U. Charged aerosol detection in pharmaceutical analysis. J. Pharm. Biomed. Anal. 2012, 69, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Plante, M.; Bailey, B.; Acworth, I.N. Analysis of Phospholipids in Natural Samples by Normal Phase HPLC and Corona Charged Aerosol Detection. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/posters/PN-71006-Phosphoplipids-Natural-Samples-HPLC-CAD-PN71006-EN.pdf (accessed on 9 May 2022).
- Barry, K.M.; Dinan, T.G.; Murray, B.A.; Kelly, P.M. Comparison of dairy phospholipid preparative extraction protocols in combination with analysis by high performance liquid chromatography coupled to a charged aerosol detector. Int. Dairy J. 2016, 56, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Cascone, A.; Eerola, S.; Ritieni, A.; Rizzo, A. Development of analytical procedures to study changes in the composition of meat phospholipids caused by induced oxidation. J. Chromatogr. A 2006, 1120, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Moreau, R.A. The Analysis of Lipids via HPLC with a Charged Aerosol Detector. Lipids 2006, 41, 27–734. [Google Scholar] [CrossRef] [PubMed]
- Kiełbowicz, G.; Trziszka, T.; Wawrzeńczyk, C. Separation and Quantification of Phospholipid and Neutral Lipid Classes by HPLC–CAD: Application to Egg Yolk Lipids. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 898–903. [Google Scholar] [CrossRef]
- Plante, M.; Bailey, B.; Acworth, I.N. Analysis of Lipids by RP-HPLC Using the Dionex Corona Ultra. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-71759-HPLC-Lipids-Corona-ultra-AN71759-EN.pdf (accessed on 9 May 2022).
- Weber, F.; Rahnfeld, L.; Luciani, P. Analytical profiling and stability evaluation of liposomal drug delivery systems: A rapid UHPLC-CAD-based approach for phospholipids in research and quality control. Talanta 2020, 220, 121320. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.M.; Deprêtre, N.; Sibille, E.; Cabaret, S.; Grégoire, S.; Soubeyre, V.; Masson, E.; Acar, N.; Bretillon, L.; Grosmaitre, X.; et al. Comprehensive study of rodent olfactory tissue lipid composition. Prostaglandins Leukot. Essent. Fatty Acids 2018, 131, 32–43. [Google Scholar] [CrossRef]
- Nair, L.M.; Werling, J.O. Aerosol based detectors for the investigation of phospholipid hydrolysis in a pharmaceutical suspension formulation. J. Pharm. Biomed. Anal. 2009, 49, 95–99. [Google Scholar] [CrossRef]
- Garoma, T.; Janda, D. Investigation of the effects of microalgal cell concentration and electroporation, microwave and ultrasonication on lipid extraction efficiency. Renew. Energy 2016, 86, 117–123. [Google Scholar] [CrossRef]
- Hadrich, B.; Akremi, I.; Dammak, M.; Barkallah, M.; Fendri, I.; Abdelkafi, S. Optimization of lipids’ ultrasonic extraction and production from Chlorella sp. using response-surface methodology. Lipids Health Dis. 2018, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Carreira-Casais, A.; Otero, P.; Garcia-Perez, P.; Garcia-Oliveira, P.; Pereira, A.G.; Carpena, M.; Soria-Lopez, A.; Simal-Gandara, J.; Prieto, M.A. Benefits and Drawbacks of Ultrasound-Assisted Extraction for the Recovery of Bioactive Compounds from Marine Algae. Int. J. Environ. Res. Public Health 2021, 18, 9153. [Google Scholar] [CrossRef] [PubMed]
- Branson Ultrasonic Corporation. Model 250/450 Sonifier. Instruction Manual. 100-413-016 Rev. A. In Appendix B: The Effects of Ultrasonic Irradiation on Various Biological Material; Branson Ultrasonic Corporation: Danbury, CT, USA, 1998. [Google Scholar]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Garrett, T.J.; Bowden, J.A. Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [Google Scholar] [CrossRef]
- Tsuji, T.; Morita, S.; Ikeda, Y.; Terada, T. Enzymatic fluorometric assays for quantifying all major phospholipid classes in cells and intracellular organelles. Sci. Rep. 2019, 9, 8607. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “Proteomic Ruler” for Protein Copy Number and Concentration Estimation without Spike-in Standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Klock, J.C.; Pieprzyk, J.K. Cholesterol, phospholipids, and fatty acids of normal immature neutrophils: Comparison with acute myeloblastic leukemia cells and normal neutrophils. J. Lipid Res. 1979, 20, 908–911. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Casares, D.; Escrib, P.V.; Rossell, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [Green Version]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef] [Green Version]
- Górecki, T.; Lynen, F.; Szucs, R.; Sandra, P. Universal Response in Liquid Chromatography Using Charged Aerosol Detection. Anal. Chem. 2006, 78, 3186–3192. [Google Scholar] [CrossRef] [PubMed]
- Lisa, M.; Lynen, F.; Holčapek, M.; Sandra, P. Quantitation of triacylglycerols from plant oils using charged aerosol detection with gradient compensation. J. Chromatogr. A 2007, 1176, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Patton, G.M.; Robins, S.J. Separation and quantitation of phospholipid classes by HPLC. Methods Mol. Biol. 1998, 110, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.Y.; Terada, T. Enzymatic measurement of phosphatidylglycerol and cardiolipin in cultured cells and mitochondria. Sci. Rep. 2015, 5, 11737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, V.R. Pitfalls and Errors of HPLC in Pictures, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–42. [Google Scholar] [CrossRef]
- Vervoort, N.; Daemen, D.; Török, G. Performance evaluation of evaporative light scattering detection and charged aerosol detection in reversed phase liquid chromatography. J. Chromatogr. A 2008, 1189, 92–100. [Google Scholar] [CrossRef]
- Gamache, P.H.; Kaufman, S.L. Fundamentals of Charged Aerosol Detection. In Charged Aerosol Detection for Liquid Chromatography and Related Separation Techniques; Gamache, P.H., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 3–59. [Google Scholar]
- Joseph, A.; Patel, S.; Rustum, A. Development and Validation of a RP-HPLC Method for the Estimation of Netilmicin Sulfate and its Related Substances using Charged Aerosol Detection. J. Chromatogr. Sci. 2010, 48, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Hartley, D.; Emanuele, M.; Awad, A.; Swartz, M. Charged Aerosol Detection in Pharmaceutical Analysis: An Overview. LCGC Suppl. 2009, 27, 40–48. [Google Scholar]
- Gamache, P.; Muellner, T.; Eggart, B.; Lovejoy, K.; Acworth, I. Charged Aerosol Detection-Use of the Power Function and Robust Calibration Practices to Achieve the Best Quantitative Results. Thermo Fisher Scientific Technical Note 73299. Available online: https://assets.thermofisher.com/TFSAssets/CMD/Technical-Notes/tn-73299-lc-cad-calibration-tn73299-en.pdf (accessed on 12 April 2022).
- ICH. ICH Harmonised Tripartite Guideline “Validation of analytical procedures: Text and methodology Q2 (R1)”. In Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 1–13 November 2005.
- Sorvina, A.; Bader, C.A.; Caporale, C.; Carter, E.A.; Johnson, I.R.D.; Parkinson-Lawrence, E.J.; Simpson, P.V.; Wright, P.J.; Stagni, S.; Lay, P.A.; et al. Lipid profiles of prostate cancer cells. Oncotarget 2018, 9, 35541–35552. [Google Scholar] [CrossRef] [Green Version]
- Young, R.S.E.; Bowman, A.P.; Williams, E.D.; Tousignant, K.D.; Bidgood, C.L.; Narreddula, V.R.; Gupta, R.; Marshall, D.L.; Poad, B.L.J.; Nelson, C.C.; et al. Apocryphal FADS2 activity promotes fatty acid diversification in cancer. Cell Rep. 2021, 34, 108738. [Google Scholar] [CrossRef]
- Yi, X.; Li, Y.; Hu, X.; Wang, F.; Liu, T. Changes in phospholipid metabolism in exosomes of hormone-sensitive and hormone-resistant prostate cancer cells. J. Cancer 2021, 12, 2893–2902. [Google Scholar] [CrossRef]
- Podo, F. Tumour phospholipid metabolism. NMR Biomed. 1999, 12, 413–439. [Google Scholar] [CrossRef]
- Preetha, A.; Banerjee, R.; Huilgol, N. Surface activity, lipid profiles and their implications in cervical cancer. J. Cancer Res. Ther. 2005, 1, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.L.; Sherr, D.; Degani, H. Variations in energy and phospholipid metabolism in normal and cancer human mammary epithelial cells. Anticancer Res. 1996, 16, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, M.; Iwamoto, N.; Kawaguchi-Sakita, N.; Sugimoto, M.; Ueno, T.; Mikami, Y.; Terasawa, K.; Sato, T.A.; Tanaka, K.; Shimizu, K.; et al. High-resolution imaging mass spectrometry reveals detailed spatial distribution of phosphatidylinositols in human breast cancer. Cancer Sci. 2013, 104, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Aboagye, E.O.; Bhujwalla, Z.M. Malignant Transformation Alters Membrane Choline Phospholipid Metabolism of Human Mammary Epithelial Cells. Cancer Res. 1999, 59, 80–84. [Google Scholar]
- Ahmad, I.A.H.; Blasko, A.; Tam, J.; Variankaval, N.; Halsey, H.M.; Hartman, R.; Regalado, E.L. Revealing the inner workings of the power function algorithm in Charged Aerosol Detection: A simple and effective approach to optimizing power function value for quantitative analysis. J. Chromatogr. A 2019, 1603, 1–7. [Google Scholar] [CrossRef]
- Avanti Polar Lipids. Available online: https://avantilipids.com (accessed on 12 April 2022).



| Column | Resolution between | S/N forPS | ||
|---|---|---|---|---|
| No. | Description, Dimensions | PE/PC | PI/LPC | |
| 1 | Lichrospher Diol 100 250 × 4 mm, 5 μm, batch A | 2.4 | 3.8 | 99.1 |
| 2 | Lichrospher Diol 100 250 × 4 mm, 5 μm, batch B | 3.7 | 4.5 | 86.0 |
| 3 | Lichrosorb Diol 250 × 4 mm, 5 μm | 2.4 | 3.2 | 89.3 |
| 4 | Supelcosil LC-Fuji-Diol 250 × 4.6 mm, 5 μm | 3.2 | 4.8 | 9.8 |
| Phospholipid | Analytical Range (mg/mL) | Determination Coefficient R2 for Linearized Data | S/N for the Lowest Concentration of Analytical Range | Concentration at Precision Test (mg/mL) | Precision (n = 6), RSD (%) | Intermediate Precision (n = 12), RSD (%) | Recovery (%) |
|---|---|---|---|---|---|---|---|
| PE | 0.40–9.40 | 0.9968 (n = 10) | 220 | 2.4 | 9.03% | 8.10% | 104.5% |
| PC | 0.20–10.0 | 0.9963 (n = 12) | 620 | 3.1 | 3.52% | 5.90% | 98.9% |
| PS | 0.04–1.70 | 0.9969 (n = 12) | 250 | 0.3 | 4.32% | 5.18% | 108.0% |
| PI | 0.07–1.75 | 0.9978 (n = 10) | 90 | 0.5 | 5.01% | 8.46% | 96.3% |
| Time (min) | Solvent A (%) | Solvent B (%) |
|---|---|---|
| 0 | 95 | 5 |
| 3 | 95 | 5 |
| 35 | 50 | 50 |
| 37 | 20 | 80 |
| 41 | 20 | 80 |
| 43 | 95 | 5 |
| 61 | stop | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosłon, M.; Jaworska, M.; Anuszewska, E.L. Determination of Glycerophospholipids in Biological Material Using High-Performance Liquid Chromatography with Charged Aerosol Detector HPLC-CAD—A New Approach for Isolation and Quantification. Molecules 2022, 27, 3356. https://doi.org/10.3390/molecules27103356
Rosłon M, Jaworska M, Anuszewska EL. Determination of Glycerophospholipids in Biological Material Using High-Performance Liquid Chromatography with Charged Aerosol Detector HPLC-CAD—A New Approach for Isolation and Quantification. Molecules. 2022; 27(10):3356. https://doi.org/10.3390/molecules27103356
Chicago/Turabian StyleRosłon, Magdalena, Małgorzata Jaworska, and Elżbieta L. Anuszewska. 2022. "Determination of Glycerophospholipids in Biological Material Using High-Performance Liquid Chromatography with Charged Aerosol Detector HPLC-CAD—A New Approach for Isolation and Quantification" Molecules 27, no. 10: 3356. https://doi.org/10.3390/molecules27103356