
Synthesis of Novel Tritopic Hydrazone Ligands: Spectroscopy, Biological Activity, DFT, and Molecular Docking Studies


 , and
, and
Abstract
:
1. Introduction
2. Experimental Data
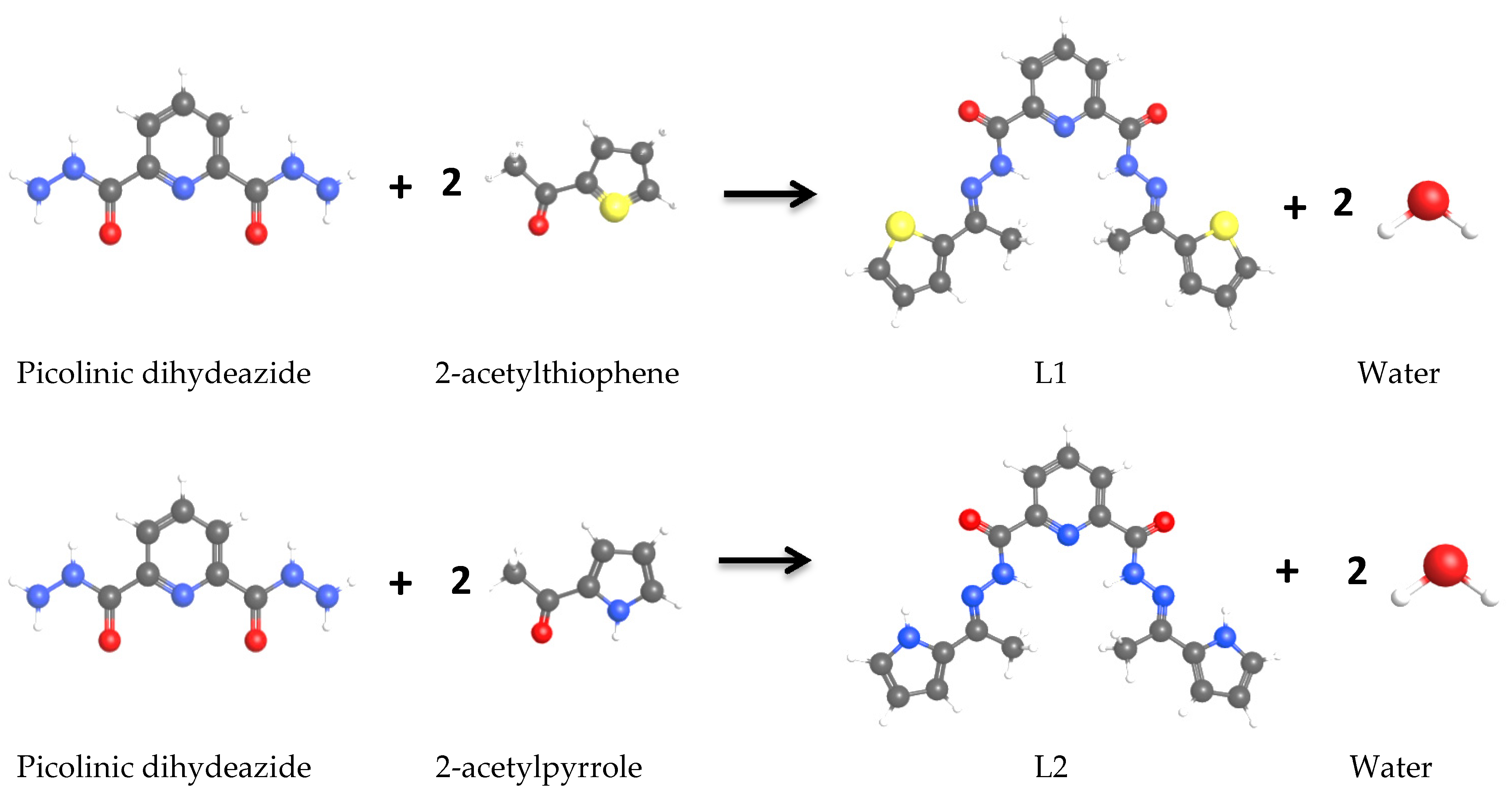
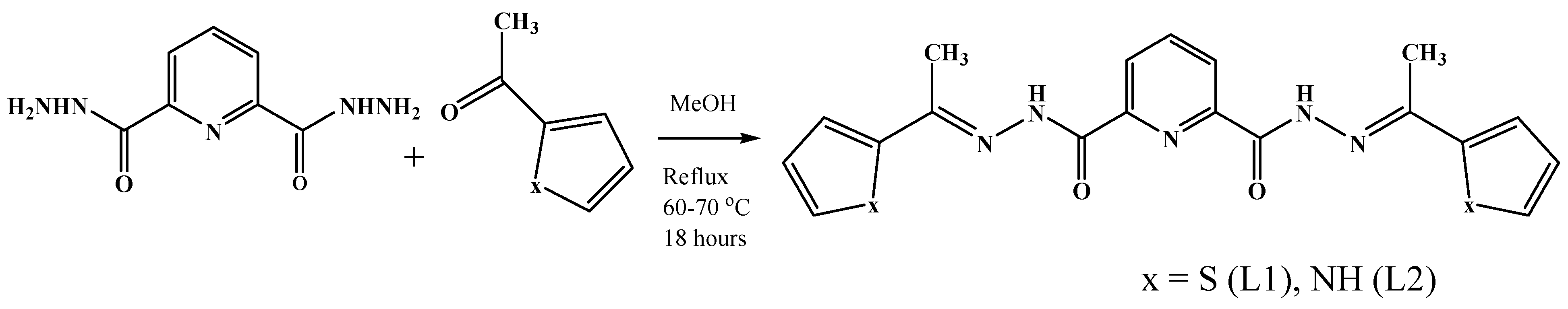
2.1. Synthesis of Ligands
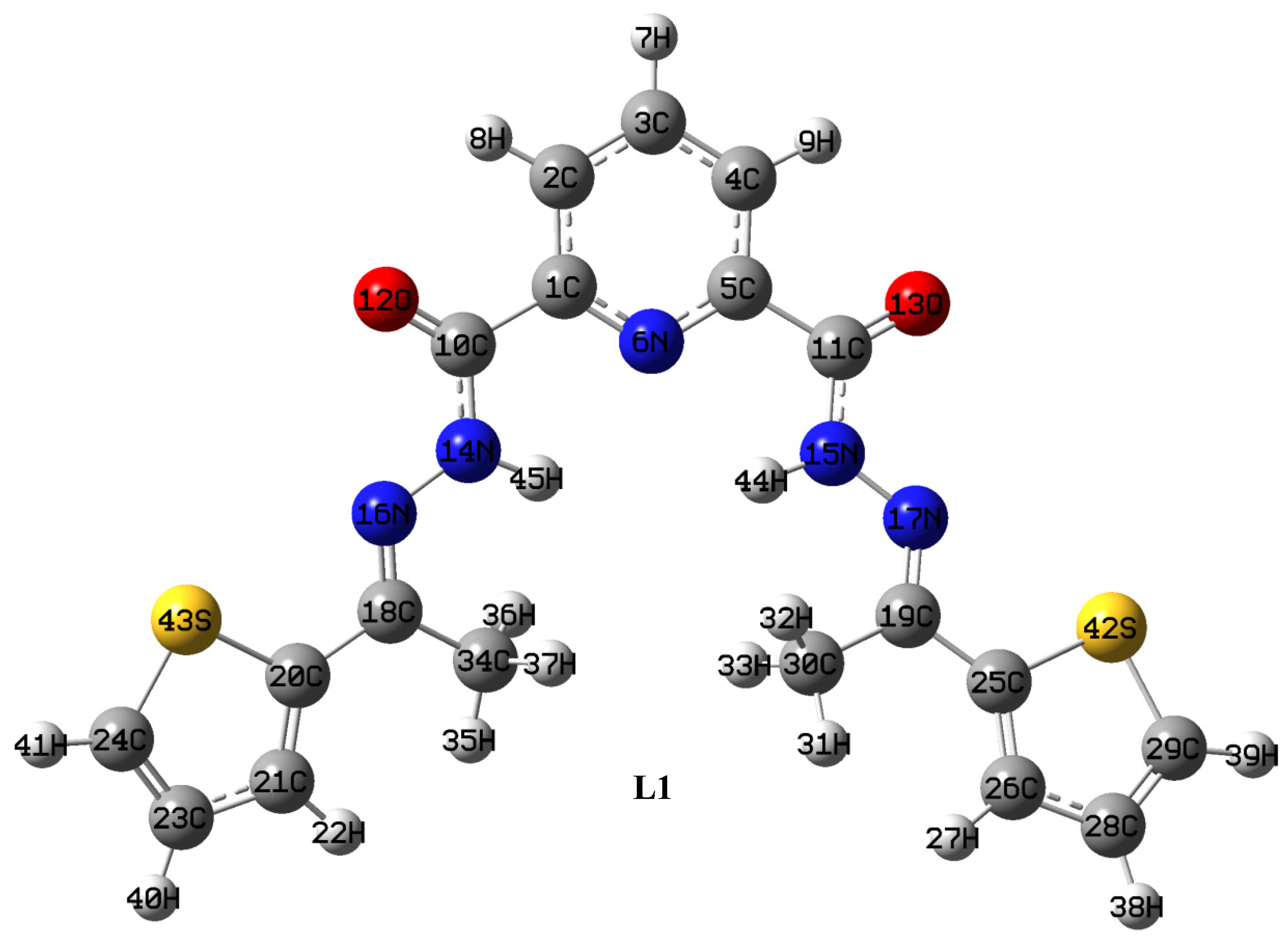
2.1.1. Synthesis of Ligand (L1)
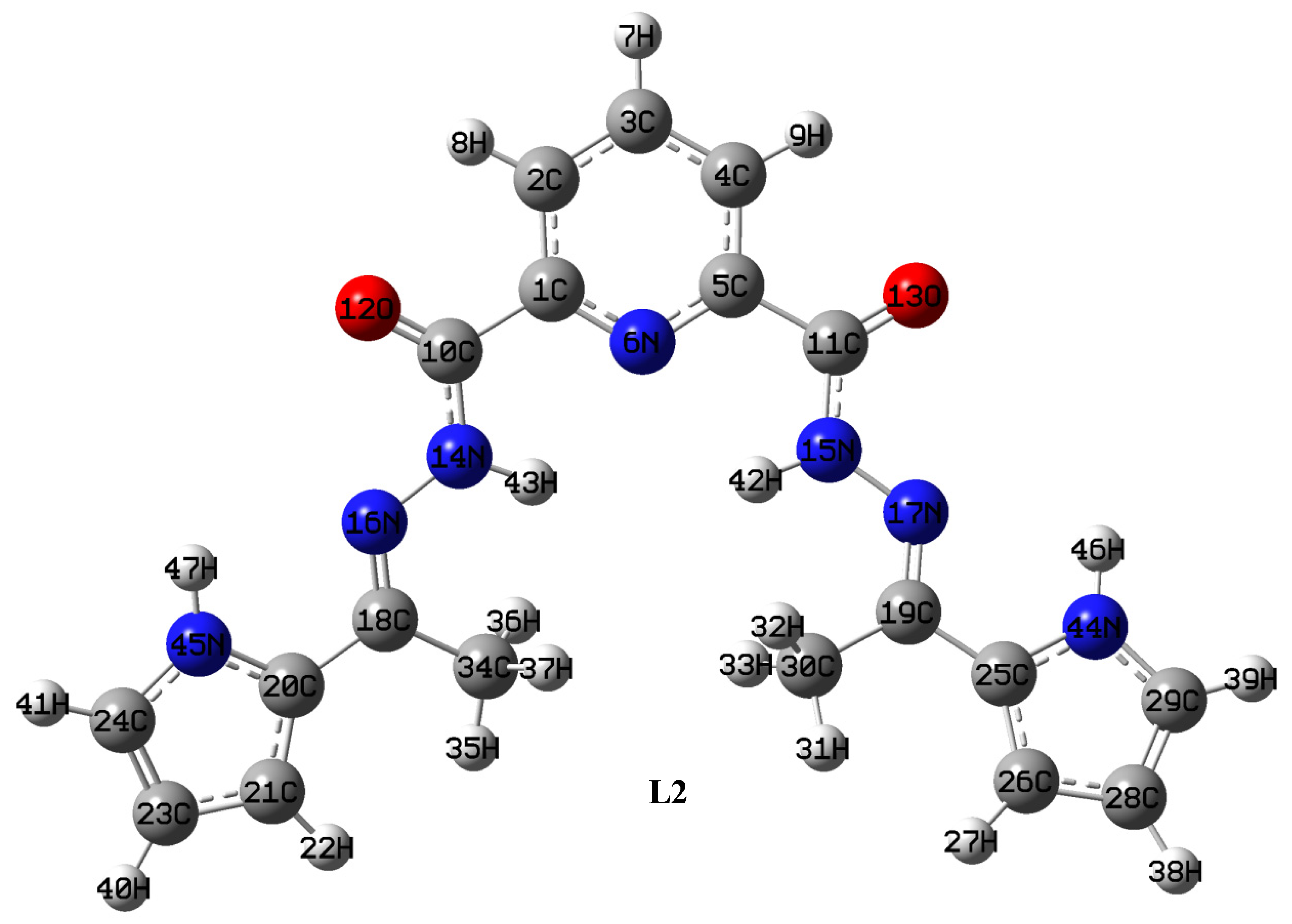
2.1.2. Synthesis of Ligand (L2)
2.1.3. Antimicrobial Activity Assay
3. Computational Details
3.1. Geometry Optimization
3.2. Protein-Ligand Docking
3.2.1. Ligand and Protein Preparation
3.2.2. Molecular Docking Analysis
4. Results and Discussion
4.1. Optimized Geometries
4.2. Mulliken Population Analysis
4.3. Vibrational Frequencies
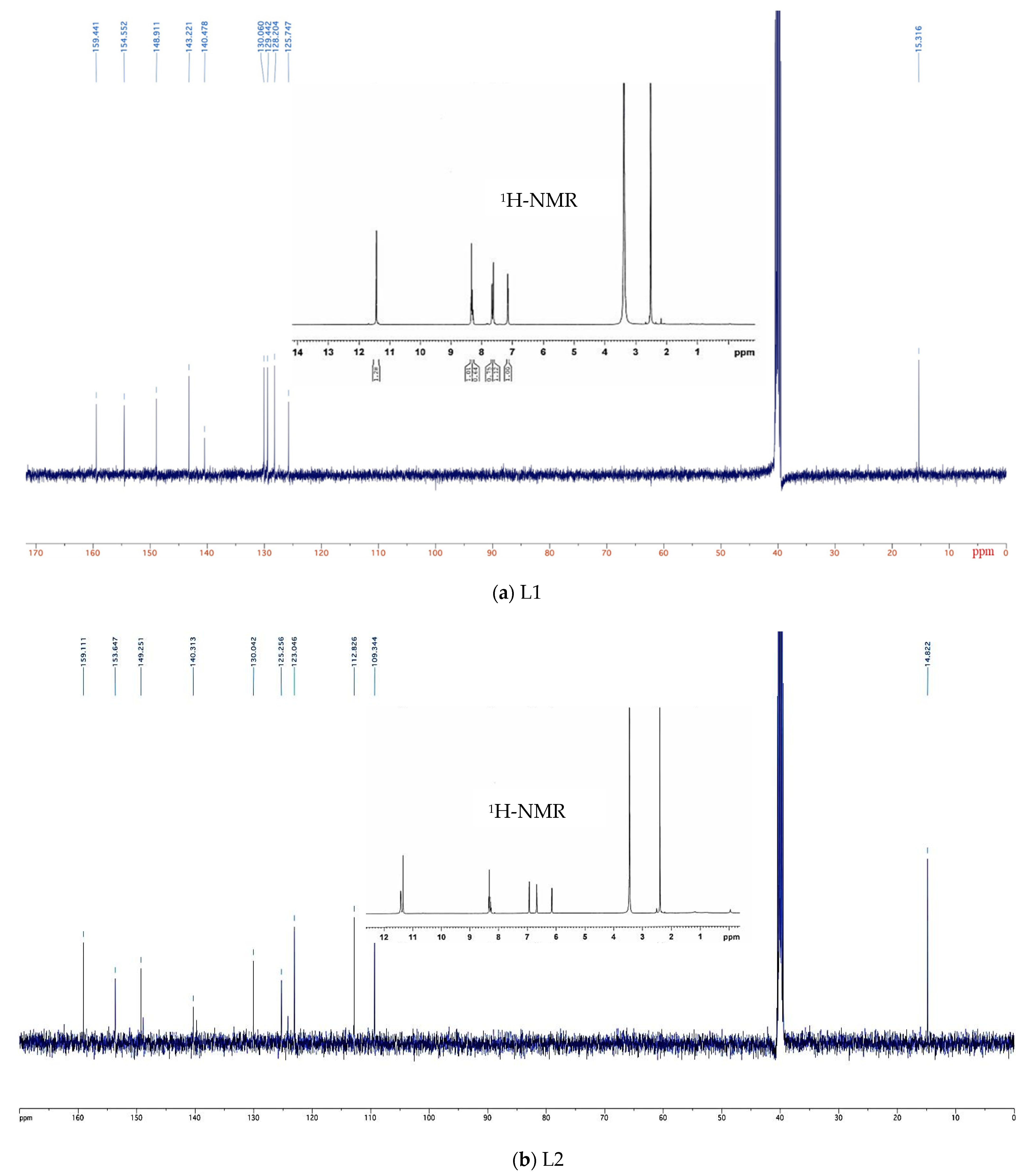
4.4. Nuclear Magnetic Resonance (NMR)
4.4.1. 1H-NMR
4.4.2. 13C-NMR
4.5. Thermodynamic Properties of the Reactions
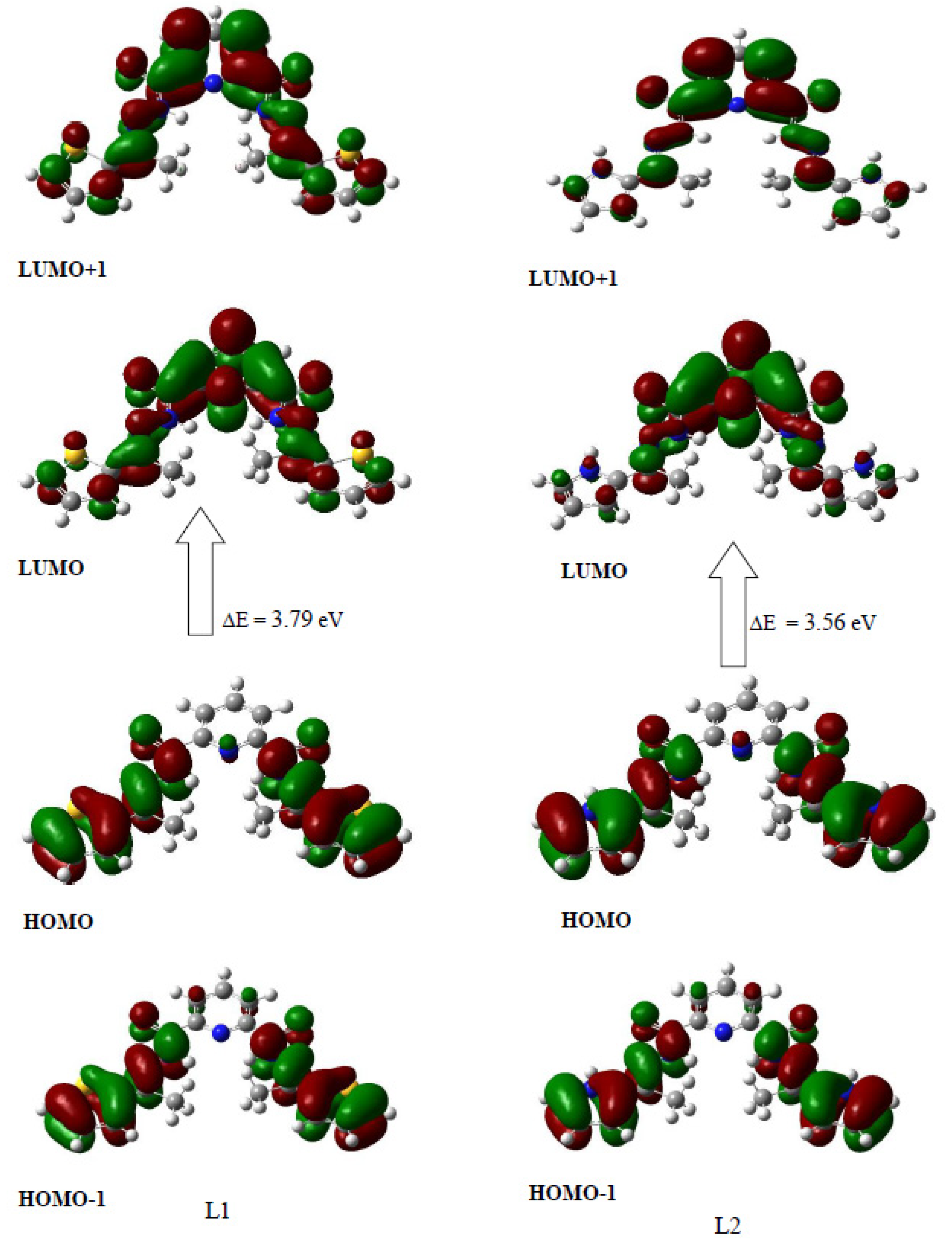
4.6. Frontier Molecular Orbital (FMO)
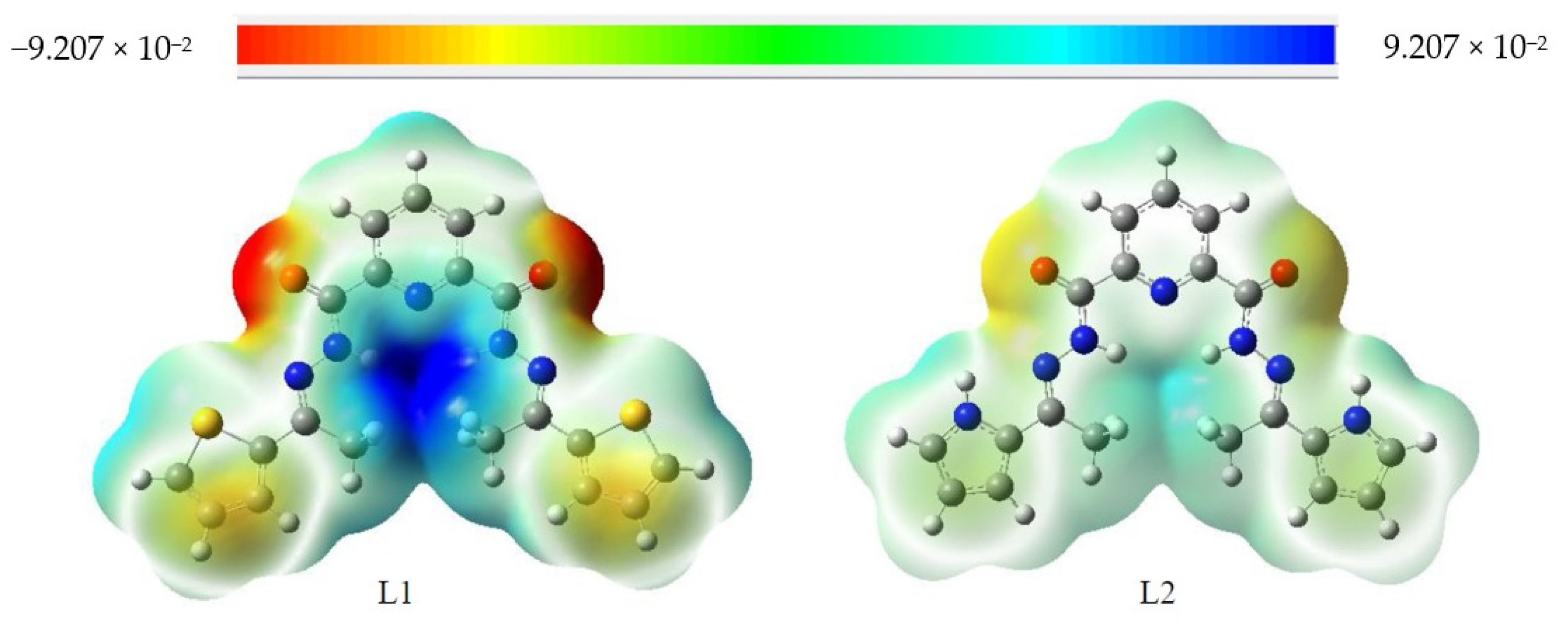
4.7. Molecular Electrostatic Potential (MEP)
4.8. Antimicrobial Activity Using the Agar Disc Diffusion Method
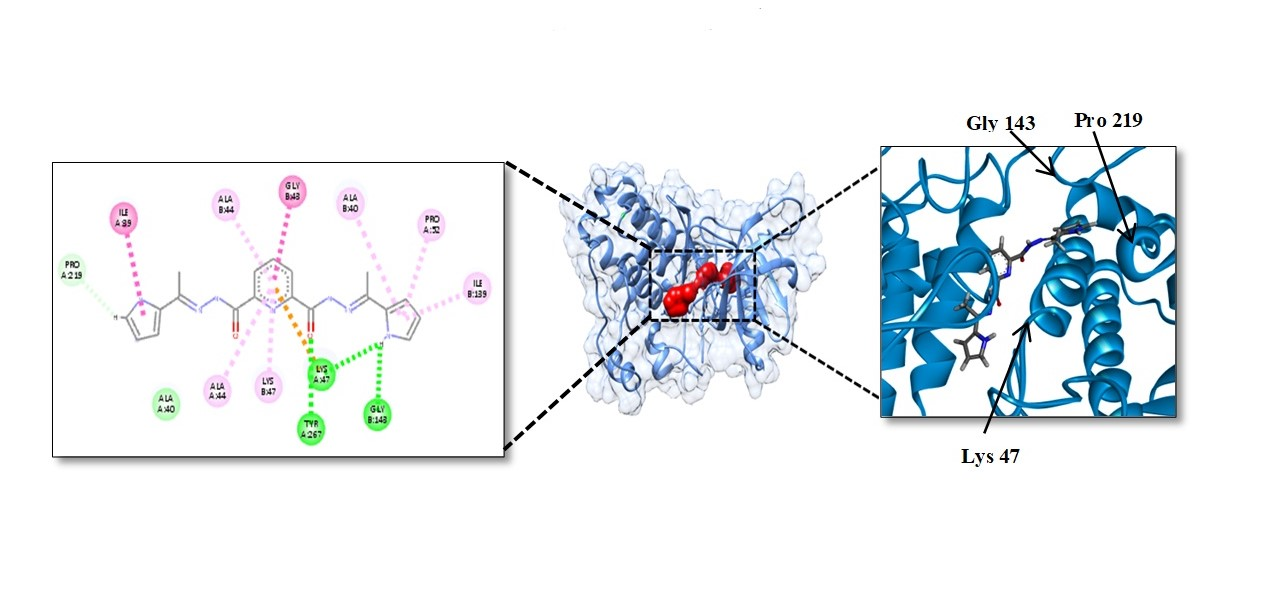
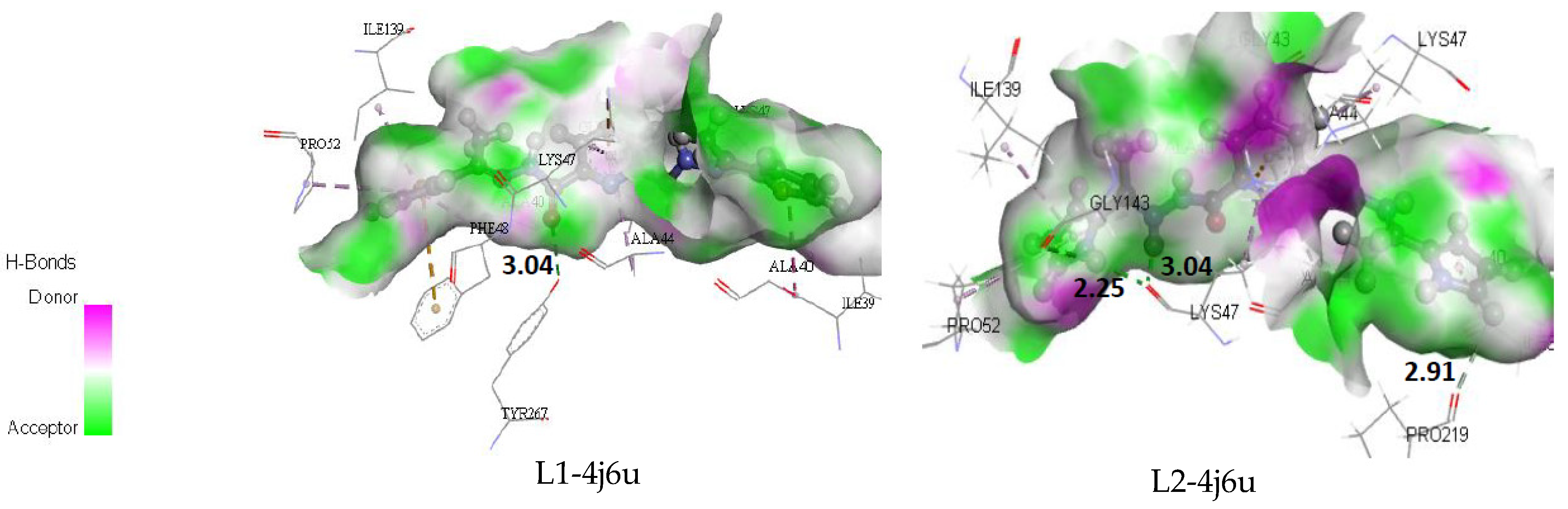
4.9. Molecular Docking Study
Binding Affinity of L1 and L2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kaan, K.; Halise, I.; Parham, T.; Ilhami, G.; Claudiu, T. Investigation of inhibitory properties of some hydrazone compounds on hCA I, hCA II and AChE enzymes. Bioorg. Chem. 2019, 86, 316–321. [Google Scholar] [CrossRef]
- Naseem, S.; Khalid, M.; Tahir, M.N.; Halim, M.A.; Braga, A.A.C.; Naseer, M.M.; Shafiq, Z. Synthesis, structural, DFT studies, docking and antibacterial activity of a xanthene-based hydrazone ligand. J. Mol. Struct. 2017, 1143, 235–244. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, T.; Xu, Z.Q.; Gu, X.N.; Wu, W.N.; Chen, H.; Wang, Y.; Jia, L.; Zhu, T.F.; Chen, R.H. Synthesis, crystal structures and antitumor activities of copper(II) complexes with a 2-acetylpyrazine isonicotinoyl hydrazone ligand. J. Mol. Struct. 2017, 1128, 448–454. [Google Scholar] [CrossRef]
- Bingul, M.; Ercan, S.; Boga, M. The design of novel 4,6-dimethoxyindole based hydrazide- hydrazones: Molecular modeling, synthesis and anticholinesterase activity. J. Mol. Struct. 2020, 1213, 128202. [Google Scholar] [CrossRef]
- El-Medani, S.M.; Makhlouf, A.A.; Moustafa, H.; Afifi, M.A.; Haukka, M.; Ramadan, R.M. Spectroscopic, crystal structural, theoretical and biological studies of phenylacetohydrazide Schiff base derivatives and their copper complexes. J. Mol. Struct. 2020, 1208, 127860. [Google Scholar] [CrossRef]
- Shebl, M. Coordination behavior of new bis(tridentate ONO, ONS and ONN) donor hydrazones towards some transition metal ions: Synthesis, spectral, thermal, antimicrobial and antitumor studies. J. Mol. Struct. 2017, 1128, 79–93. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Youns, M. Anticancer activity of new coumarin substituted hydrazide–hydrazone derivatives. Eur. J. Med. Chem. 2014, 76, 539–548. [Google Scholar] [CrossRef]
- Zhao, L.; Xu, Z.; Thompson, L.K.; Heath, S.L.; Miller, D.O.; Ohba, M. Synthesis, Structure, and Magnetism of a Novel Alkoxide Bridged Nonacopper(II) (Cu9O12) [3 × 3] Square Grid Generated by a Strict Self-Assembly Process. Angew. Chem. Int. Ed. 2000, 112, 3244–3247. [Google Scholar] [CrossRef]
- Pavan, F.R.; Maia, P.I.S.; Leite, S.R.A.; Deflon, V.M.; Batista, A.A.; Sato, D.N.; Franzblau, S.G.; Leite, C.Q.F. Ruthenium (II) phosphine/picolinate complexes as antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 598–601. [Google Scholar] [CrossRef]
- Zhao, L.; Niel, V.; Thompson, L.K.; Xu, Z.; Milway, V.A.; Harvey, R.G.; Miller, D.O.; Wilson, C.; Leech, M.; Howard, J.A.K.; et al. Self-assembled polynuclear clusters derived from some flexible polydentate dihydrazide ligands. Dalton Trans. 2004, 9, 1446–1455. [Google Scholar] [CrossRef]
- Dawe, L.N.; Shuvaev, K.V.; Thompson, L.K. Magnetic [n × n] (n = 2 − 5) Grids by Directed Self-Assembly. Inorg. Chem. 2009, 48, 3323–3341. [Google Scholar] [CrossRef] [PubMed]
- Ayme, J.F.; Lehn, J.M.; Bailly, C.; Karmazin, L. Simultaneous Generation of a [2 × 2] Grid-Like Complex and a Linear Double Helicate: A Three-Level Self-Sorting Process. J. Am. Chem. Soc. 2020, 142, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.E.; Caffrey, D.F.; Gunnlaugsson, T. Lanthanide-directed synthesis of luminescent self-assembly supramolecular structures and mechanically bonded systems from acyclic coordinating organic ligands. Chem. Soc. Rev. 2016, 45, 3244–3274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xu, Z.; Thompson, L.K.; Heath, S.L.; Miller, D.O.; Ohba, M. Synthesis, Structure, and Magnetism of a Novel Alkoxide Bridged Nonacopper(II) (Cu(9)O(12)). Angew. Chem. Int. Ed. 2000, 39, 3114–3117. [Google Scholar] [CrossRef]
- Mandal, T.N.; Roy, S.; Konar, S.; Jana, A.; Ray, S.; Das, K.; Saha, R.; Fallah, M.S.E.; Butcher, R.J.; Chaterjeee, S.; et al. Self-assembled tetranuclear Cu4(II), Ni4(II)[2 × 2] square grids and a dicopper(II) complex of heterocycle based polytopic ligands—Magnetic studies. Dalton Trans. 2011, 40, 11866–11875. [Google Scholar] [CrossRef] [Green Version]
- Dawe, L.N.; Abedin, T.S.M.; Thompson, L.K. Ligand directed self-assembly of polymetallic [n × n] grids: Rational routes to large functional molecular subunits? Dalton Trans. 2008, 13, 1661–1675. [Google Scholar] [CrossRef]
- Dawe, L.N.; Abedin, T.S.M.; Kelly, T.L.; Thompson, L.K.; Miller, D.O.; Zhao, L.; Wilson, C.; Leech, M.A.; Howard, J.A.K. Self-assembled polymetallic square grids ([2 × 2] M4, [3 × 3] M9) and trigonal bipyramidal clusters (M5)—Structural and magnetic properties. J. Mater. Chem. 2006, 16, 2645–2659. [Google Scholar] [CrossRef]
- Abedin, T.S.M.; Thompson, L.K.; Miller, D.O. An octanuclear [Co(II)2–Co(III)2]2 interlocked grid example of an inorganic [2] catenane. Chem. Commun. 2005, 44, 5512–5514. [Google Scholar] [CrossRef]
- Abedin, T.S.M.; Thompson, L.K.; Miller, D.O.; Krupicka, E. Structural and magnetic properties of a self-assembled spheroidal triakonta-hexanuclear Cu36 cluster. Chem. Commun. 2003, 9, 708–709. [Google Scholar] [CrossRef]
- Tikhomirov, A.S.; Litvinova, V.A.; Andreeva, D.V.; Tsvetkov, V.B.; Dezhenkova, L.G.; Volodina, Y.L.; Kaluzhny, D.N.; Treshalin, I.D.; Schols, D.; Ramonova, A.A.; et al. Amides of pyrrole- and thiophene-fused anthraquinone derivatives: A role of the heterocyclic core in antitumor properties. Eur. J. Med. Chem. 2020, 199, 112294. [Google Scholar] [CrossRef]
- Kundu, T.; Pramanik, A. Expeditious and eco-friendly synthesis of new multifunctionalized pyrrole derivatives and evaluation of their antioxidant property. Bioorg. Chem. 2020, 98, 103734. [Google Scholar] [CrossRef] [PubMed]
- Cherif, O.; Agrebi, A.; Alves, S.; Baleizão, C.; Farinha, J.P.; Allouche, F. Synthesis and fluorescence properties of aminocyanopyrrole and aminocyanothiophene esthers for biomedical and bioimaging applications. J. Mol. Struct. 2020, 1209, 127974. [Google Scholar] [CrossRef]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An overview of benzo[b]thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef] [PubMed]
- Rackham, M.D.; Brannigan, J.A.; Moss, D.K.; Yu, Z.; Wilkinson, A.J.; Holder, A.A.; Tate, E.W.; Leatherbarrow, R.J. Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferase. J. Med. Chem. 2013, 56, 371–375. [Google Scholar] [CrossRef]
- Jagtap, V.A.; Agasimundin, Y.S. Synthesis and preliminary evaluation of some 2-amino-N′-[substituted]-4,5,6,7-tetrahydro-1-benzothiophene-3-carbohydrazide as antimicrobial agents. J. Pharm. Res. 2015, 9, 10–14. [Google Scholar]
- Berrade, L.; Aisa, B.; Ramirez, M.J.; Galiano, S.; Guccione, S.; Moltzau, L.R.; Levy, F.O.; Nicoletti, F.; Battaglia, G.; Molinaro, G.; et al. Novel Benzo[b]thiophene Derivatives as New Potential Antidepressants with Rapid Onset of Action. J. Med. Chem. 2011, 54, 3086–3090. [Google Scholar] [CrossRef]
- Mourey, R.J.; Burnette, B.L.; Brustkern, S.J.; Daniels, J.S.; Hirsch, J.L.; Hood, W.F.; Meyers, M.J.; Mnich, S.J.; Pierce, B.S.; Saabye, M.J.; et al. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor alpha production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010, 333, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Rao, G.K.; Subramaniam, R. Synthesis, Antitubercular and Antibacterial Activities of Some Quinazolinone Analogs Substituted with Benzothiophene. Chem. Sci. J. 2015, 6, 92–96. [Google Scholar] [CrossRef]
- Zaher, A.F.; Khalil, N.A.; Ahmed, E.M. Synthesis and anticonvulsant activity of new 3′-aryl-7-bromo-spiro[[1]benzothiophene-3,2′-[1,3] thiazolidine]-2,4′-dione derivatives. Ori. J. Chem. 2010, 26, 1241–1248. [Google Scholar]
- Gündüzalp, A.B.; Özsen, E.; Alyar, H.; Alyar, S.; Özbek, N. Biologically active Schiff bases containing thiophene/furan ring and their copper (II) complexes: Synthesis, spectral, nonlinear optical and density functional studies. J. Mol. Struct. 2016, 1120, 259–266. [Google Scholar] [CrossRef]
- Ermiş, E. Synthesis, spectroscopic characterization and DFT calculations of novel Schiff base containing thiophene ring. J. Mol. Struct. 2018, 1156, 91–104. [Google Scholar] [CrossRef]
- Balachandran, V.; Santhi, G.; Karpagam, V.; Lakshmi, A. Molecular structure, spectroscopic (FT-IR, FT-Raman), NBO and HOMO–LUMO analyses, computation of thermodynamic functions for various temperatures of 2,6-dichloro-3-nitrobenzoic acid. Spectrochim. Acta Part A Mol. Biomol. Spect. 2013, 110, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Aljahdali, M.S.; Abdou El-Sherif, A.; Hilal, R.H.; Abdel-Karim, A.T. Mixed bivalent transition metal complexes of 1,10-phenanthroline and 2-aminomethylthiophenyl-4-bromo salicyl aldehyde Schiff base: Spectroscopic, molecular modeling and biological activities. Chem. Eur. J. 2013, 4, 370–378. [Google Scholar] [CrossRef]
- Balachandran, V.; Santhi, G.; Karpagam, V.; Lakshmi, A. DFT computation and spectroscopic analysis of N-(p-methoxybenzylidene)aniline, a potentially useful NLO material. J. Mol. Struct. 2013, 1047, 249–261. [Google Scholar] [CrossRef]
- Naveen, R.K.; Tittal, V.D.; Ghule, N.; Kumar, L.; Lal, K.; Kumar, A. Design, synthesis, biological activity, molecular docking and computational studies on novel 1,4-disubstituted-1,2,3-Triazole-Thiosemicarbazone hybrid molecules. J. Mol. Struct. 2020, 1209, 127951. [Google Scholar] [CrossRef]
- Palafox, M.A. DFT computations on vibrational spectra: Scaling procedures to improve the wavenumbers. Phy. Sci. Rev. 2018, 3, 20170184–20170214. [Google Scholar] [CrossRef]
- James, C.; Raj, A.A.; Reghunathan, R.; Jayakumar, V.S.; Joe, I.H. Structural conformation and vibrational spectroscopic studies of 2,6-bis(p-N,N-dimethyl benzylidene)cyclohexanone using density functional theory. J. Raman Spectrosc. 2006, 37, 1381–1392. [Google Scholar] [CrossRef]
- Furic, K.; Duric, J.R. Proton-pair disorder in dimers of aromatic carboxylic acids: Vibrational spectra of benzoic acid at low temperatures. Chem. Phys. Lett. 1986, 126, 92–97. [Google Scholar] [CrossRef]
- Thompson, L.K.; Dawe, L.N. Magnetic properties of transition metal (Mn(II), Mn(III), Ni(II), Cu(II)) and lanthanide (Gd(III), Dy(III), Tb(III), Eu(III), Ho(III), Yb(III)) clusters and [nxn] grids: Isotropic exchange and SMM behaviour. Coord. Chem. Rev. 2015, 289–290, 13–31. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1993, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; Delano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Com. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 4.0; Accelrys Software Inc.: San Diego, CA, USA, 2013. [Google Scholar]
- Grove, H.; Kelly, T.L.; Thompson, L.K.; Zhao, L.; Xu, Z.; Abedin, T.S.M.; Miller, D.O.; Goeta, A.E.; Wilson, C.; Howard, J.A.K. Copper(II) Complexes of a Series of Alkoxy Diazine Ligands: Mononuclear, Dinuclear, and Tetranuclear Examples with Structural, Magnetic, and DFT Studies. Inorg. Chem. 2004, 43, 4278–4288. [Google Scholar] [CrossRef]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P.; Verma, D. Synthesis Molecular Structure and Spectral Analysis of Ethyl 4-Formyl-3, 5-Dimethyl 1H-Pyrrole-2-Carboxylate Thiosemicarbazone: A Combined DFT and AIM Approach. J. Mol. Struct. 2012, 1016, 97–108. [Google Scholar] [CrossRef]
- Pramodh, B.; Lokanath, N.K.; Naveen, S.; Naresh, P.; Ganguly, S.; Panda, J. Molecular structure, Hirshfeld surface analysis, theoretical investigations and nonlinear optical properties of a novel crystalline chalcone derivative: (E)-1-(5-bromothiophen-2-yl)-3-(p-tolyl)prop-2-en-1-one. J. Mol. Struct. 2018, 1161, 9–17. [Google Scholar] [CrossRef]
- Jouad, E.M.; Riou, A.; Allain, M.; Khan, M.A.; Boue, G.M. Synthesis, structural and spectral studies of 5-methyl 2-furaldehyde thiosemicarbazone and its Co, Ni, Cu and Cd complexes. Polyhedron 2001, 20, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Guerraoui, A.; Djedouani, A.; Jeanneau, E.; Boumaza, A.; Alsalme, A.; Zarrouk, A.; Salih, K.S.M.; Warad, I. Crystal structure and spectral of new hydrazine-pyran-dione derivative: DFT enol↔hydrazone tautomerization via zwitterionic intermediate, hirshfeld analysis and optical activity studies. J. Mol. Struct. 2020, 1220, 128728. [Google Scholar] [CrossRef]
- Anderson, B.J.; Jasinski, J.P.; Freedman, M.B.; Millikan, S.P.; O’Rourke, K.A.; Smolenski, V.A. Synthesis, Crystal Structural Investigations, and DFT Calculations of Novel Thiosemicarbazones. Crystals 2016, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Uzun, S.; Demircioglu, Z.; Taşdogan, M.; Agar, E. Quantum chemical and X-ray diffraction studies of (E)-3-(((3,4-dimethoxybenzyl)imino)methyl)benzene-1,2-diol. J. Mol. Struct. 2020, 1206, 127749. [Google Scholar] [CrossRef]
- Mahmoudi, F.; Farhadi, S.; Dusek, M.; Poupon, M. Synthesis, Spectroscopy and X-ray Crystallography Structure of Pyridine 4-Carbaldehyde Semicarbazone Schiff Base Ligand. Adv. J. Chem. Sec. A 2020, 3, 534–541. [Google Scholar] [CrossRef]
- Balachandran, V.; Karpagam, V.; Revathi, B.; Kavimani, M.; Ilango, G. Conformational stability, spectroscopic and computational studies, HOMO–LUMO, NBO, ESP analysis, thermodynamic parameters of natural bioactive compound with anticancer potential of 2-(hydroxymethyl)anthraquinone. Spectrochim. Acta Part A Mol. Biomol. Spect. 2015, 150, 631–640. [Google Scholar] [CrossRef]
- Srikanth, K.E.; Veeraiah, A.; Pooventhiran, T.; Thomas, R.; Solomon, K.A.; Raju, C.J.S.; Naveena, J.; Latha, L. Detailed molecular structure (XRD), conformational search, spectroscopic characterization (IR, Raman, UV, fluorescence), quantum mechanical properties and bioactivity prediction of a pyrrole analogue. Heliyon 2020, 6, e04106–e04117. [Google Scholar] [CrossRef]
- Karpagakalyaani, G.; Daisy Magdaline, J.; Chithambarathanu, T.; Aruldhas, D.; Ronaldo Anuf, A. Spectroscopic (FT-IR, FT-Raman, NBO) Investigation and Molecular Docking study of a Herbicide compound Bifenox. Chem. Data Collect. 2020, 27, 100393. [Google Scholar] [CrossRef]
- Sundaram, M.S.S.; Karthick, S.; Sailaja, K.; Karkuzhali, R.; Gopu, G. Theoretical study on cyclophane amide molecular receptors and its complexation behavior with TCNQ. J. Photochem. Photobio. B Biol. 2020, 203, 111735. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Karabacak, M.; Bilgili, S.; Atac, A. Theoretical study on molecular structure and vibrational analysis included FT-IR, FT-Raman and UV techniques of 2,4,5-trimethylbenzoic acid (monomer and dimer structures). Spectrochim. Acta Part A Mol. Biomol. Spect. 2015, 134, 598–607. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X. Spectroscopic Identification of Organic Compound, 6th ed.; John Willey and Sons: New York, NY, USA, 1998. [Google Scholar]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications; John Wiley and Sons: Oxford, UK, 2004. [Google Scholar]
- El-Gammal, O.A.; Abu El-Reash, G.M.; Bedier, R.A. Synthesis, spectroscopic, DFT, biological studies and molecular docking of oxovanadium (IV), copper (II) and iron (III) complexes of a new hydrazone derived from heterocyclic hydrazide. Appl. Organometal. Chem. 2019, 33, e5141. [Google Scholar] [CrossRef]
- Bharati, N.; Sharma, S.; Naqvi, F.; Azam, A. New palladium(II) complexes of 5-nitrothiophene-2-carboxaldehyde thiosemicarbazones, synthesis, spectral studies and in vitro anti-amoebic activity. Bioinorg. Med. Chem. 2003, 11, 2923–2929. [Google Scholar] [CrossRef]
- Varsanyi, G. Vibrational Spectra of 700 Benzene Derivatives; Academic Kiego: Budapest, Hungary, 1974; Volume I–II. [Google Scholar]
- Dubis, A.T.; Grabowski, S.J.; Romanowska, D.B.; Misiaszek, T.; Leszczynski, J. Pyrrole-2-carboxylic Acid and Its Dimers: Molecular Structures and Vibrational Spectrum. J. Phys. Chem. A 2002, 106, 10613–10621. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Niu, H.; Wang, C.; Qin, C.; Bai, X.; Wang, W. Schiff bases containing triphenylamine and pyrrole units: Synthesis and electrochromic, acidochromic properties. New J. Chem. 2016, 40, 5245–5254. [Google Scholar] [CrossRef]
- Tanak, H.; Ağar, A.A.; Büyükgüngör, O. Experimental (XRD, FT-IR and UV–Vis) and theoretical modeling studies of Schiff base (E)-N’-((5-nitrothiophen-2-yl)methylene)-2-phenoxyaniline. Spectrochim. Acta Part A Mol. Biomol. Spect. 2014, 118, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Maryam, K.; Safar Ali, B.; Azar, G. Novel Schiff Bases of Pyrrole: Synthesis, Experimental and Theoretical Characterizations, Fluorescent Properties and Molecular Docking. Iran. J. Chem. Chem. Eng. 2018, 37, 59–72. [Google Scholar]
- Azad, I.; Akhter, Y.; Khan, T.; Azad, M.I.; Chandra, S.; Singh, P.; Kumar, D.; Nasibullah, M. Synthesis, quantum chemical study, AIM simulation, in silico ADMET profile analysis, molecular docking and antioxidant activity assessment of aminofuran derivatives. J. Mol. Struct. 2020, 1203, 127285. [Google Scholar] [CrossRef]
- Cuenú, F.; Londoño-Salazar, J.; Torres, J.E.; Abonia, R.; D’Vries, R.F. Synthesis, structural characterization and theoretical studies of a new Schiff base 4-(((3-(tert-Butyl)-(1-phenyl) pyrazol-5-yl) imino) methyl) phenol. J. Mol. Struct. 2018, 1152, 163–176. [Google Scholar] [CrossRef]
- Singh, R.N.; Rawat, P.; Verma, D.; Bharti, S.K. Experimental and DFT study on pyrrole tosylhydrazones. J. Mol. Struct. 2015, 1081, 543–554. [Google Scholar] [CrossRef]
- Anđelković, K.; Sladić, D. Complexes of iron(II), iron(III) and zinc(II) with condensation derivatives of 2-acetylpyridine and oxalic or malonic dihydrazide. Crystal structure of tris[(1-(2-pyridyl)ethylidene)hydrazine]iron(II) perchlorate. Transit. Met. Chem. 2005, 30, 243–250. [Google Scholar] [CrossRef]
- Cuenú, F.; Restrepo-Acevedo, A.; Murillo, M.I.; Torres, J.E.; Moreno-Fuquen, R.; Abonia, R.; Kennedy, A.R.; Tenorio, J.C.; Lehmann, C.W. Synthesis, structural characterization, and theoretical studies of new pyrazole (E)-2-{[(5-(tert-butyl)-1H-pyrazol-3- yl)imino]methyl}phenol and (E)-2-{[(1-(4-bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl] imino]methyl}phenol. J. Mol. Struct. 2019, 1184, 59–71. [Google Scholar] [CrossRef]
- Gökce, H.; Öztürk, N.; Kazıcı, M.; Yörür Göreci, Ç.; Güneş, S. Structural, spectroscopic, electronic, nonlinear optical and thermodynamic properties of a synthesized Schiff base compound: A combined experimental and theoretical approach. J. Mol. Struct. 2017, 1136, 288–302. [Google Scholar] [CrossRef]
- Lien, E.J.; Guo, Z.R.; Li, R.; Su, C.T. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J. Pharm. Sci. 1982, 71, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Pramodh, B.; Chethan Prathap, K.N.; Hema, M.K.; Warad, I.; Lokanath, N.K. Synthesis, structure, quantum computational and biological studies of novel thiophene derivatives. J. Mol. Struct. 2021, 1229, 129587. [Google Scholar] [CrossRef]
- Snyder, S.H.; Merril, C.R. A relationship between the hallucinogenic activity of drugs and their electronic configuration. Proc. Natl. Acad. Sci. USA 1965, 54, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Aihara, J. Reduced HOMO−LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Li, J.; Cramer, C.J.; Truhlar, D.G. MIDI! basis set for silicon, bromine, and iodine. Theoret. Chem. Acc. 1998, 99, 192–196. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Pearson, R.G. The HSAB Principle—More quantitative aspects. Inorg. Chim. Acta 1995, 240, 93–98. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef] [Green Version]
- Pavitha, P.; Prashanth, J.; Ramu, G.; Ramesh, G.; Mamatha, K.; Venkatram, R.B. Synthesis, structural, spectroscopic, anti-cancer and molecular docking studies on novel 2-[(Anthracene-9-ylmethylene)amino]-2-methylpropane-1,3-diol using XRD, FTIR, NMR, UV-Vis spectra and DFT. J. Mol. Struct. 2017, 1147, 406. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Conteras, R. Quantitative Characterization of the Local Electrophilicity of Organic Molecules. Understanding the Regioselectivity on Diels−Alder Reactions. J. Phys. Chem. A 2002, 106, 6871–6875. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quant. Chem. 1978, 11, 115–193. [Google Scholar]
- Luque, F.J.; López, J.M.; Orozco, M. Electrostatic Interactions of a Solute with a Continuum. A Direct Utilization of AB initio Molecular Potentials for the Prevision of Solvent Effects. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Shukla, R.; Bandopadhyay, P.; Sathe, M.; Chopra, D. Quantitative investigation on the intermolecular interactions present in 8-(4-ethoxyphenyl)-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione with insight from interaction energies, energy framework, electrostatic potential map and fingerprint analysis. J. Chem. Sci. 2020, 132, 19–26. [Google Scholar] [CrossRef]
- Acharya, R.; Chacko, S.; Bose, P.; Lapenna, A.; Pattanayak, S.P. Structure Based Multitargeted Molecular Docking Analysis of Selected Furanocoumarins against Breast Cancer. Sci. Rep. 2019, 9, 15743–15756. [Google Scholar] [CrossRef]
- Ahmed, M.J.; Dhumad, A.M.; Almashal, F.A.; Alshawi, J.M. Microwave-assisted synthesis, molecular docking and anti-HIV activities of some drug-like quinolone derivatives. Med. Chem. Res. 2020, 29, 1067–1076. [Google Scholar] [CrossRef]
- Thirumurugan, C.; Vadivel, P.; Lalitha, A.; Lakshmanan, S. Synthesis, characterization of novel quinoline-2-carboxamide based chalcone derivatives and their molecular docking, photochemical studies. Synth. Commun. 2020, 50, 831–839. [Google Scholar] [CrossRef]
- Khan, E.; Tandon, P.; Maurya, R.; Kumar, P. A theoretical study on molecular structure, chemical reactivity and molecular docking studies on dalbergin and methyldalbergin. J. Mol. Struct. 2019, 1183, 100–106. [Google Scholar]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Jin, W.; Nazir, Y.; Fercher, C.; Thomas Blaskovich, M.A.; Cooper, M.A.; Barnard, R.T.; Ziora, Z.M. Tyrosinase inhibitors as potential antibacterial agents. Eur. J. Med. Chem. 2020, 187, 111892. [Google Scholar] [CrossRef]
- Kang, S.M.; Heo, S.J.; Kim, K.N.; Lee, S.H.; Yang, H.M.; Kim, A.D.; Jeon, Y.J. Design, synthesis, molecular docking and biological evaluation of imides, pyridazines, imidazoles derived from itaconic anhydride for potential antioxidant and antimicrobial activities. Bioorg. Med. Chem. 2012, 20, 311–316. [Google Scholar] [CrossRef]
- Nokinsee, D.; Shank, L.; Lee, V.S.; Nimmanpipug, P. Estimation of Inhibitory Effect against Tyrosinase Activity through Homology Modeling and Molecular Docking. Enzym. Res. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, R.; Valente, R.; Souza da Costa, C.H.; da S Gonçalves Vianez, J.L.; Santana da Costa, K.; de Molfetta, F.A.; Nahum Alves, C. Analysis of Kojic Acid Derivatives as Competitive Inhibitors of Tyrosinase: A Molecular Modeling Approach. Molecules 2021, 26, 2875. [Google Scholar] [CrossRef] [PubMed]
- Gurunanjappa, P.; Kameshwar, V.H.; Kariyappa, A.K. Bioactive formylpyrazole Analogues: Synthesis, Antimicrobial, Antioxidant and Molecular docking studies. Asian J. Chem. 2017, 29, 1549–1554. [Google Scholar] [CrossRef]
- Nayak, P.S.; Narayana, B.; Sarojini, B.K.; Sheik, S.; Shashidhara, K.S.; Chandrashekar, K.R. Design, synthesis, molecular docking and biological evaluation of imides, pyridazines, and imidazoles derived from itaconic anhydride for potential antioxidant and antimicrobial activities. J. Taibah Univ. Sci. 2014, 10, 823–838. [Google Scholar] [CrossRef] [Green Version]
- Murugavel, S.; Deepa, S.; Ravikumar, C.; Ranganathand, R.; Alagusundaram, P. Synthesis, structural, spectral and antibacterial activity of 3,3a,4,5-tetrahydro-2H-benzo[g]indazole fused carbothioamide derivatives as antibacterial agents. J. Mol. Struct. 2020, 1222, 128961. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L1 | L2 | ||
|---|---|---|---|
| Atoms | Charges (e) | Atoms | Charges (e) |
| B3LYP/6-311G+(d,p) | B3LYP/6-311G+(d,p) | ||
| C (1) & C (5) | 0.152 | C (1) & C (5) | 0.191 |
| H (44) & H (45) | 0.285 | H (42) & H (43) | 0.289 |
| N (6) | 0.026 | N (6) | 0.003 |
| C (10) & C (11) | −0.124 | C (10) & C (11) | −0.216 |
| O (12) & O (13) | −0.318 | O (12) & O (13) | −0.342 |
| N (14) & N (15) | −0.030 | N (14) & N (15) | −0.173 |
| N (16) & N (17) | −0.006 | N (16) & N (17) | −0.011 |
| S (42) & S (43) | −0.094 | N (44) & N (45) | −0.126 |
| Experimental | Calculated without Scaling (Intensity) | Calculated with Scaling a (Intensity) | Assignment b | ||
|---|---|---|---|---|---|
| 6-311G+(d,p) | cc-pvdz | 6-311G+(d,p) | cc-pvdz | ||
| L1 | |||||
| 3318 | 3533 (75) | 3514 (76) | 3422 | 3404 | ʋ (N–H hydrazide) |
| 3067 | 3240–3183 (~11) | 3246–3193 (~10) | 3139–3084 | 3145–3093 | ʋasy (=C–H thiophene, pyridine) |
| 1698 | 1760 (540) | 1787 (538) | 1705 | 1731 | ʋ (CO) + δ (N–H) |
| 1589 | 1638 (4) | 1651 (3) | 1587 | 1599 | ʋsym (C=N) |
| 1537 | 1622 (40) | 1633 (45.8) | 1570 | 1582 | ʋsym (C=C) + ω (C–H, pyridine) |
| 1504 | 1576 (243) | 1585 (103) | 1527 | 1536 | ʋsym (C=C) + ω (NH) + δ (C–H thiophene) + θ (sp3 C–H) |
| 994 | 970 (0.2) | 1041 (0.3) | 940 | 1008 | π (C–H pyridine) |
| 848 | 911 (0.02) | 913 (0.11) | 883 | 885 | π (C–H thiophene) |
| 727 | 744 (17) | 745 (14) | 721 | 722 | ʋ (C-S-C) |
| L2 | |||||
| 3469 | 3617 (127) | 3621 (129) | 3508 | 3391 | ʋ (NH pyrrole) |
| 3350 | 3524 (64) | 3520 (65) | 3414 | 3410 | ʋ (NH hydrazide) |
| 3089 | 3005 (21) | 3021 (28) | 2921 | 2926 | ʋasy (=C–H pyrrole, pyridine) |
| 1677 | 1712 (526) | 1778 (564) | 1658 | 1723 | ʋsym (CO) + ω (NH) + ω (=CH pyridine) |
| 1550 | 1619 (16) | 1657 (58) | 1568 | 1605 | ʋsym (C=N) |
| 1568 | 1607 (116) | 1633(93) | 1557 | 1582 | ʋsym (C=C) + ω (C–H pyridine) |
| 1519 | 1593 (44) | 1605 (115) | 1544 | 1555 | ʋsym (C=C pyrrole) + ω (=C–H) |
| 999 | 974 (0.2) | 981 (0.1) | 944 | 950 | π (C–H pyridine) |
| 834 | 950 (56) | 876 (0.05) | 920 | 848 | π (C–H pyrrole) |
| Name | ∆H (kcal/mol) | ∆G (kcal/mol) | ∆S (cal/mol K) | Dipole Moment (Debye) | ||||
|---|---|---|---|---|---|---|---|---|
| Gas | CH3OH | Gas | CH3OH | Gas | CH3OH | Gas | CH3OH | |
| L1 | 1.63 | 1.24 | 2.66 | 2.36 | 23.81 | 6.13 | 4.73 | 7.65 |
| L2 | 3.09 | 2.88 | 3.99 | 3.22 | 2.16 | 1.49 | 2.26 | 3.79 |
| Energies (eV) | L1 | L2 | FMOs (eV) | L1 | L2 |
|---|---|---|---|---|---|
| I | 6.17 | 5.79 | HOMO-2 | −7.21 | −7.16 |
| A | 2.38 | 2.23 | HOMO-1 | −6.18 | −5.8 |
| μ | −4.27 | −4.01 | HOMO | −6.17 | −5.79 |
| ω | 4.82 | 4.52 | LUMO | −2.38 | −2.23 |
| Property | LUMO+1 | −2.35 | −2.22 | ||
| η | 1.89 | 1.78 | LUMO+2 | −1.69 | −1.27 |
| S | 390.7 | 415.9 | Gap (∆E) | 3.79 | 3.56 |
| Compound | Gram (+) Bacteria | Gram (−) Bacteria | Fungi | |||
|---|---|---|---|---|---|---|
| S. aureus | B. megaterium | E. coli | S. typhi | T. harzianum | A. niger | |
| L1 | 10 | 10 | 11 | 9 | 6 | 11 |
| L2 | 9 | 12 | 10 | 8 | 6 | 6 |
| Ceftriaxone | 40.0 | 50.0 | 38.0 | 44.0 | -- | -- |
| Amphotericin-B | -- | -- | -- | -- | 17.0 | 8.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rupa, S.A.; Moni, M.R.; Patwary, M.A.M.; Mahmud, M.M.; Haque, M.A.; Uddin, J.; Abedin, S.M.T. Synthesis of Novel Tritopic Hydrazone Ligands: Spectroscopy, Biological Activity, DFT, and Molecular Docking Studies. Molecules 2022, 27, 1656. https://doi.org/10.3390/molecules27051656
Rupa SA, Moni MR, Patwary MAM, Mahmud MM, Haque MA, Uddin J, Abedin SMT. Synthesis of Novel Tritopic Hydrazone Ligands: Spectroscopy, Biological Activity, DFT, and Molecular Docking Studies. Molecules. 2022; 27(5):1656. https://doi.org/10.3390/molecules27051656
Chicago/Turabian StyleRupa, Sharmin Akther, Md. Rassel Moni, Md. Abdul Majed Patwary, Md. Mayez Mahmud, Md. Aminul Haque, Jamal Uddin, and S. M. Tareque Abedin. 2022. "Synthesis of Novel Tritopic Hydrazone Ligands: Spectroscopy, Biological Activity, DFT, and Molecular Docking Studies" Molecules 27, no. 5: 1656. https://doi.org/10.3390/molecules27051656