
Ultrafast Excited-State Decay Mechanisms of 6-Thioguanine Followed by Sub-20 fs UV Transient Absorption Spectroscopy

 , , and
, , and
Abstract
:
1. Introduction
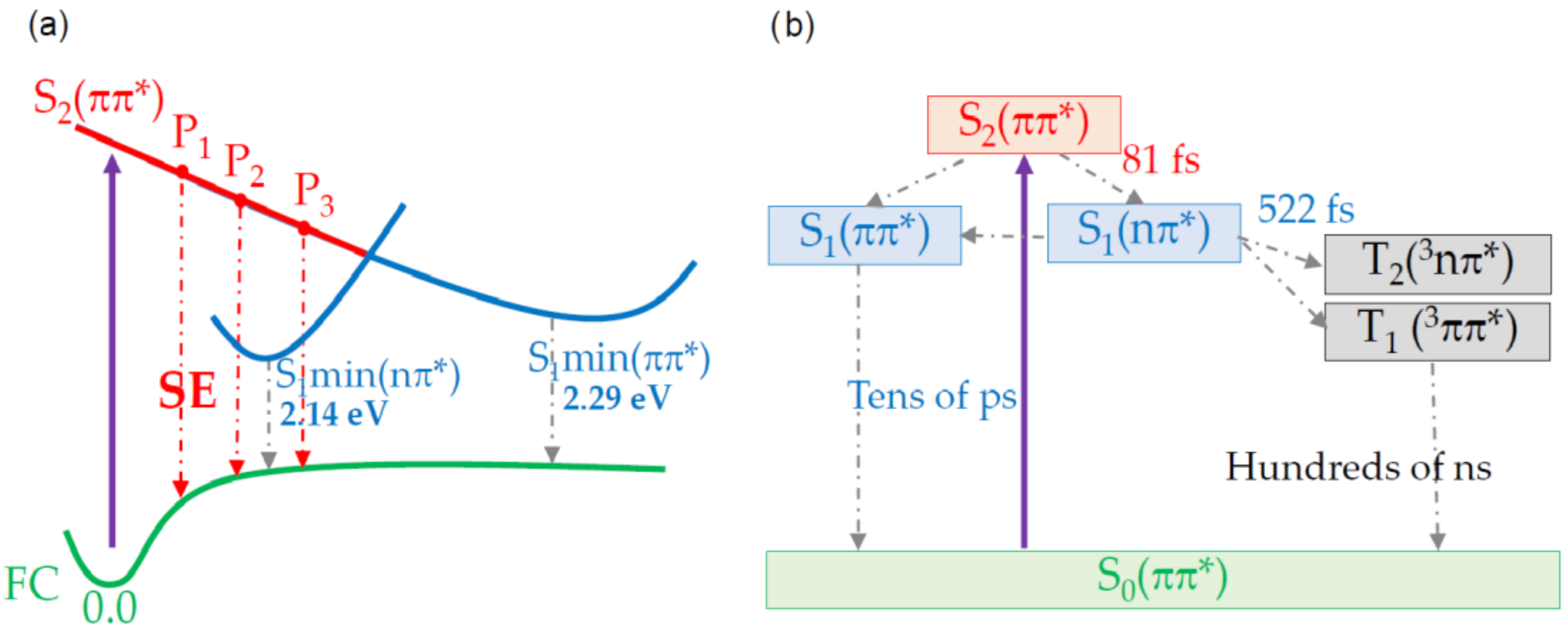
2. Results and Discussion
3. Materials and Methods
3.1. Sample Preparation
3.2. Linear Absorption
3.3. Steady-State Photoluminescence
3.4. Transient Absorption Spectroscopy
3.5. Global Analysis and Data Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hitchings, G.H.; Elion, G.B.; Falco, E.A.; Russell, P.B.; Sherwood, M.B.; VanderWerff, H. Antagonists of nucleic acid derivatives. J. Biol. Chem. 1950, 183, 1–9. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Attard, N.R.; Karran, P. UVA photosensitization of thiopurines and skin cancer in organ transplant recipients. Photochem. Photobiol. Sci. 2012, 11, 62–68. [Google Scholar] [CrossRef]
- Pridgeon, S.W.; Heer, R.; Taylor, G.A.; Newell, D.R.; O’Toole, K.; Robinson, M.; Xu, Y.Z.; Karran, P.; Boddy, A.V. Thiothymidine combined with UVA as a potential novel therapy for bladder cancer. Br. J. Cancer 2011, 104, 1869–1876. [Google Scholar] [CrossRef]
- Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and photochemistry of canonical nucleobases’ thioanalogs: From quantum mechanical studies to time resolved experiments. Molecules 2017, 22, 998. [Google Scholar] [CrossRef]
- Ashwood, B.; Pollum, M.; Crespo-Hernández, C.E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [Green Version]
- Nenov, A.; Conti, I.; Borrego-Varillas, R.; Cerullo, G.; Garavelli, M. Linear absorption spectra of solvated thiouracils resolved at the hybrid RASPT2/MM level. Chem. Phys. 2018, 515, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA Excited-State Dynamics: From Single Bases to the Double Helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Pecourt, J.M.L.; Peon, J.; Kohler, B. Ultrafast internal conversion of electronically excited RNA and DNA nucleosides in water. J. Am. Chem. Soc. 2000, 122, 9348–9349. [Google Scholar] [CrossRef]
- Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.; Nishi, N.; Xu, Y.Z. Ultrafast intersystem crossing of 4-thiothymidine in aqueous solution. J. Phys. Chem. Lett. 2010, 1, 480–484. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, J.A.; Mohamadzade, A.; Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; González, L.; Crespo-Hernández, C.E.; Ullrich, S. 2-Thiouracil intersystem crossing photodynamics studied by wavelength-dependent photoelectron and transient absorption spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhang, T.-S.; Xue, J.-D.; Zheng, X.; Cui, G.; Fang, W.-H. Short-time dynamics of 2-thiouracil in the light absorbing S2(ππ*) state. J. Chem. Phys. 2015, 143, 175103. [Google Scholar] [CrossRef]
- Koyama, D.; Milner, M.J.; Orr-Ewing, A.J. Evidence for a Double Well in the First Triplet Excited State of 2-Thiouracil. J. Phys. Chem. B 2017, 121, 9274–9280. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Barbatti, M. Why Replacing Different Oxygens of Thymine with Sulfur Causes Distinct Absorption and Intersystem Crossing. J. Phys. Chem. A 2016, 120, 6342–6350. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Dai, X.; Liu, K.; Zhao, H.; Song, D.; Su, H. Photophysical and Photochemical Properties of 4-Thiouracil: Time-Resolved IR Spectroscopy and DFT Studies. J. Phys. Chem. B 2014, 118, 5864–5872. [Google Scholar] [CrossRef]
- Gobbo, J.P.; Borin, A.C. 2-Thiouracil deactivation pathways and triplet states population. Comput. Theor. Chem. 2014, 1040, 195–201. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; Corral, I.; Granucci, G.; Persico, M. Competing ultrafast intersystem crossing and internal conversion: A time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 2014, 5, 1336. [Google Scholar] [CrossRef]
- Ruckenbauer, M.; Mai, S.; Marquetand, P.; González, L. Photoelectron spectra of 2-thiouracil, 4-thiouracil, and 2,4-dithiouracil. J. Chem. Phys. 2016, 144, 074303. [Google Scholar] [CrossRef] [Green Version]
- Cerullo, G.; Garavelli, M. A novel spectroscopic window on conical intersections in biomolecules. Proc. Natl. Acad. Sci. USA 2020, 117, 26553–26555. [Google Scholar] [CrossRef]
- Borrego-Varillas, R.; Nenov, A.; Kabacinski, P.; Conti, I.; Ganzer, L.; Oriana, A.; Delno, I.; Weingart, O.; Manzoni, C.; Rivalta, I.; et al. Tracking excited state decay mechanisms of pyrimidine nucleosides in real time. Nat. Commun. 2021, 12, 7285. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.-B.; Wang, Q.; Guo, W.-W.; Cui, G. The excited-state decay mechanism of 2,4-dithiothymine in the gas phase, microsolvated surroundings, and aqueous solution. Phys. Chem. Chem. Phys. 2017, 19, 7689–7698. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Guo, C.; Crespo-Hernández, C.E. Excited-state dynamics in 6-thioguanosine from the femtosecond to microsecond time scale. J. Phys. Chem. B 2011, 115, 3263–3270. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Crespo-Hernaíndez, C.E. Room-temperature phosphorescence of the DNA monomer analogue 4-thiothymidine in aqueous solutions after UVA excitation. J. Phys. Chem. Lett. 2010, 1, 2239–2243. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-hernández, C.E. 2,4-Dithiothymine as a Potent UVA Chemotherapeutic Agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef] [PubMed]
- Sismour, A.M. The use of thymidine analogs to improve the replication of an extra DNA base pair: A synthetic biological system. Nucleic Acids Res. 2005, 33, 5640–5646. [Google Scholar] [CrossRef]
- Heuberger, B.D.; Pal, A.; Del Frate, F.; Topkar, V.V.; Szostak, J.W. Replacing Uridine with 2-Thiouridine Enhances the Rate and Fidelity of Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2015, 137, 2769–2775. [Google Scholar] [CrossRef] [Green Version]
- Favre, A.; Moreno, G.; Blondel, M.O.; Kliber, J.; Vinzens, F.; Salet, C. 4-thiouridine photosensitized RNA-protein crosslinking in mammalian cells. Biochem. Biophys. Res. Commun. 1986, 141, 847–854. [Google Scholar] [CrossRef]
- Pollum, M.; Lam, M.; Jockusch, S.; Crespo-Hernández, C.E. Dithionated Nucleobases as Effective Photodynamic Agents against Human Epidermoid Carcinoma Cells. ChemMedChem 2018, 13, 1044–1050. [Google Scholar] [CrossRef]
- Harris, M.; Cote, H.; Ochoa, C.; Allavena, C.; Negredo, E.; Cahn, P.; Zala, C.; Raffi, F. A Randomized, Open-Label Study of a Nucleoside Analogue Reverse Transcriptase Inhibitor—Sparing Regimen in Antiretroviral-Naive HIV-Infected Patients. J. Acquir. Immune Defic. Syndr. 2009, 50, 339–340. [Google Scholar] [CrossRef]
- Meisenheimer, K.M.; Koch, T.H. Photocross-Linking of Nucleic Acids to Associated Proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 101–140. [Google Scholar] [CrossRef]
- Reelfs, O.; Karran, P.; Young, A.R. 4-thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy. Photochem. Photobiol. Sci. 2012, 11, 148–154. [Google Scholar] [CrossRef]
- Massey, A.; Xu, Y.Z.; Karran, P. Photoactivation of DNA thiobases as a potential novel therapeutic option. Curr. Biol. 2001, 11, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Fernández, L.; Granucci, G.; Pollum, M.; Crespo-Hernández, C.E.; Persico, M.; Corral, I. Decoding the Molecular Basis for the Population Mechanism of the Triplet Phototoxic Precursors in UVA Light-Activated Pyrimidine Anticancer Drugs. Chem.—A Eur. J. 2017, 23, 2619–2627. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. Intersystem Crossing Pathways in the Noncanonical Nucleobase 2-Thiouracil: A Time-Dependent Picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [Green Version]
- Borrego-Varillas, R.; Teles-Ferreira, D.C.; Nenov, A.; Conti, I.; Ganzer, L.; Manzoni, C.; Garavelli, M.; de Paula, A.M.; Cerullo, G. Observation of the Sub-100 Femtosecond Population of a Dark State in a Thiobase Mediating Intersystem Crossing. J. Am. Chem. Soc. 2018, 140, 16087–16093. [Google Scholar] [CrossRef]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Fang, W.-H. State-specific heavy-atom effect on intersystem crossing processes in 2-thiothymine: A potential photodynamic therapy photosensitizer. J. Chem. Phys. 2013, 138, 044315. [Google Scholar] [CrossRef]
- Teles-Ferreira, D.C.; Conti, I.; Borrego-Varillas, R.; Nenov, A.; Van Stokkum, I.H.M.; Ganzer, L.; Manzoni, C.; de Paula, A.M.; Cerullo, G.; Garavelli, M. A Unified Experimental/Theoretical Description of the Ultrafast Photophysics of Single and Double Thionated Uracils. Chem.—A Eur. J. 2020, 26, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Siouri, F.M.; Boldissar, S.; Berenbeim, J.A.; de Vries, M.S. Excited State Dynamics of 6-Thioguanine. J. Phys. Chem. A 2017, 121, 5257–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Fernandez, L.; Fahleson, T.; Norman, P.; Santoro, F.; Coriani, S.; Improta, R. Optical absorption and magnetic circular dichroism spectra of thiouracils: A quantum mechanical study in solution. Photochem. Photobiol. Sci. 2017, 16, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollum, M.; Ortiz-Rodríguez, L.A.; Jockusch, S.; Crespo-Hernández, C.E. The Triplet State of 6-thio-2′-deoxyguanosine: Intrinsic Properties and Reactivity toward Molecular Oxygen. Photochem. Photobiol. 2016, 92, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, B.; Jockusch, S.; Crespo-Hernández, C.E. Excited-state dynamics of the thiopurine prodrug 6-thioguanine: Can N9-glycosylation affect its phototoxic activity? Molecules 2017, 22, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, K.M.; Brister, M.M.; Pittelkow, M.; Sølling, T.I. Heavy-Atom-Substituted Nucleobases in Photodynamic Applications: Substitution of Sulfur with Selenium in 6-Thioguanine Induces a Remarkable Increase in the Rate of Triplet Decay in 6-Selenoguanine. J. Am. Chem. Soc. 2018, 140, 11214–11218. [Google Scholar] [CrossRef]
- Pirillo, J.; Mazzone, G.; Russo, N.; Bertini, L. Photophysical Properties of S, Se and Te-Substituted Deoxyguanosines: Insight into Their Ability to Act as Chemotherapeutic Agents. J. Chem. Inf. Modeling 2017, 57, 234–242. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; González, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef]
- Santhosh, C.; Mishra, P.C. Electronic structures and spectra of 6-mercaptopurine and 6-thioguanine. Spectrochim. Acta Part A Mol. Spectrosc. 1993, 49, 985–993. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Roos, B.O.; Andersson, K. Multiconfigurational perturbation theory with level shift—The Cr2 potential revisited. Chem. Phys. Lett. 1995, 245, 215–223. [Google Scholar] [CrossRef]
- Finley, J.; Malmqvist, P.Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Pou-Amérigo, R.; Merchán, M.; Nebot-Gil, I.; Widmark, P.O.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions—III. First row transition metal atoms. Theor. Chim. Acta 1995, 92, 149–181. [Google Scholar] [CrossRef]
- Fernández Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef]
- Zou, X.; Zhao, H.; Yu, Y.; Su, H. Formation of guanine-6-sulfonate from 6-thioguanine and singlet oxygen: A combined theoretical and experimental study. J. Am. Chem. Soc. 2013, 135, 4509–4515. [Google Scholar] [CrossRef]
- Borrego-Varillas, R.; Ganzer, L.; Cerullo, G.; Manzoni, C. Ultraviolet Transient Absorption Spectrometer with Sub-20-fs Time Resolution. Appl. Sci. 2018, 8, 989. [Google Scholar] [CrossRef] [Green Version]
- Varillas, R.B.; Candeo, A.; Viola, D.; Garavelli, M.; De Silvestri, S.; Cerullo, G.; Manzoni, C.; De Silvestri, S.; Cerullo, G.; Manzoni, C.; et al. Microjoule-level, tunable sub-10 fs UV pulses by broadband sum-frequency generation. Opt. Lett. 2014, 39, 3849–3852. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.P.; Seger, R.; Mullen, K.M.; van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Van Stokkum, I.H.M.; Larsen, D.S.; Van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta—Bioenerg. 2004, 1657, 82–104. [Google Scholar] [CrossRef] [Green Version]




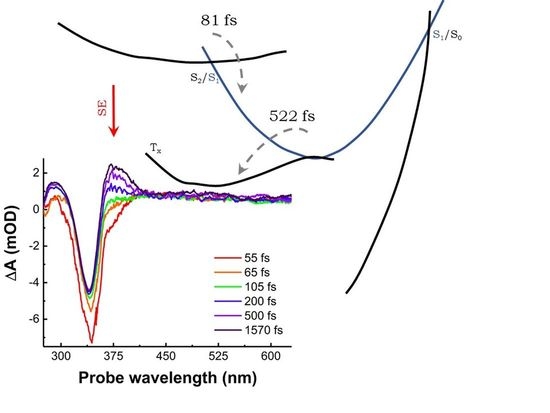
{kind=link}
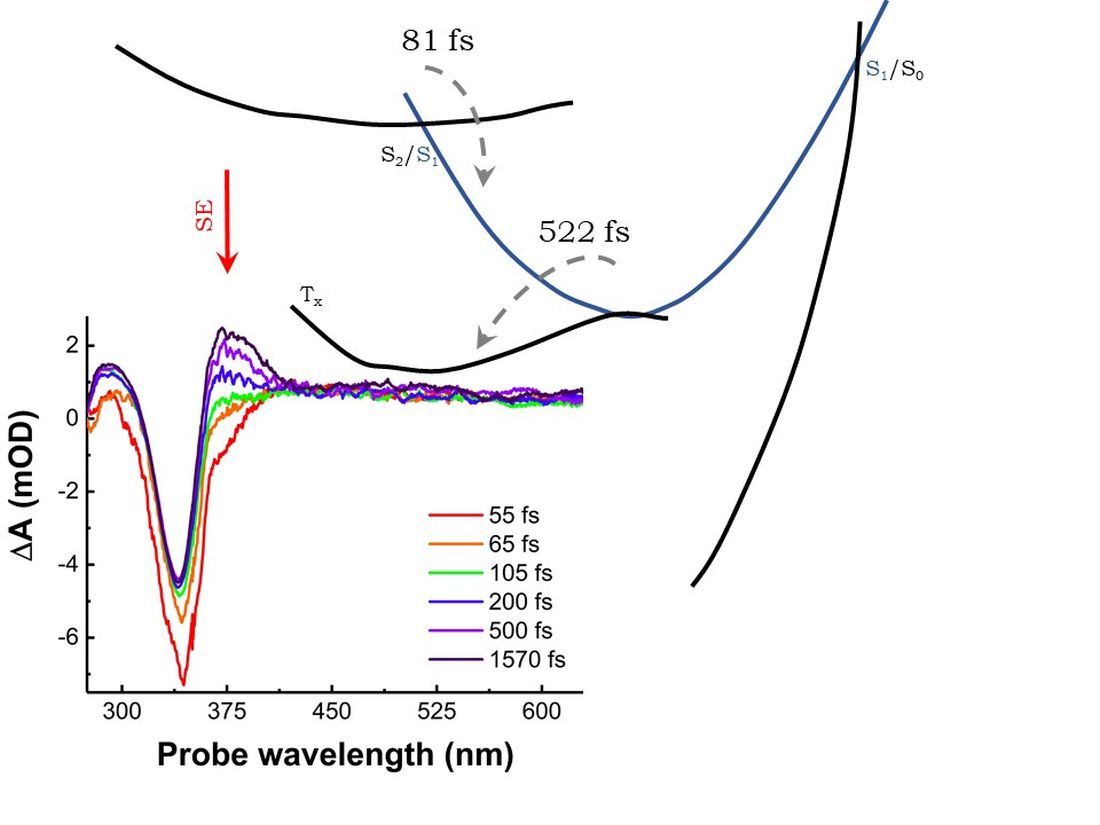
{kind=link}
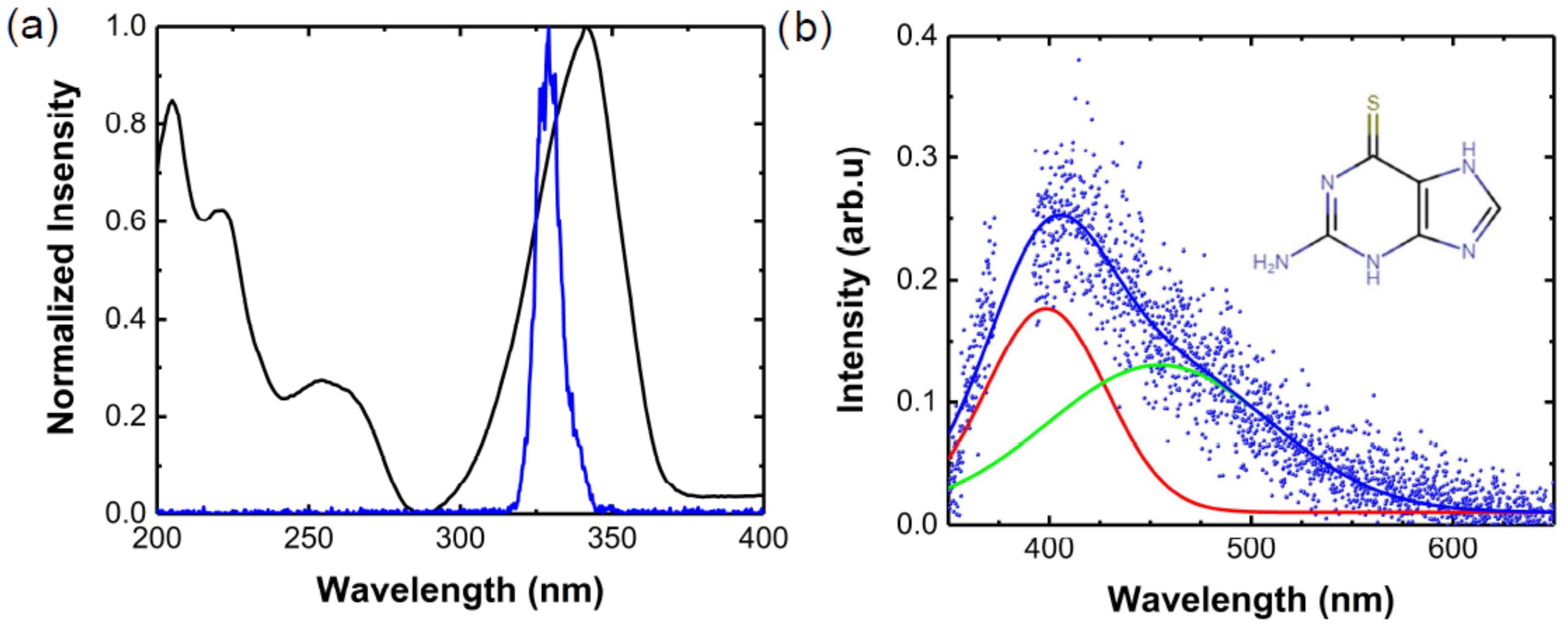
{kind=link}
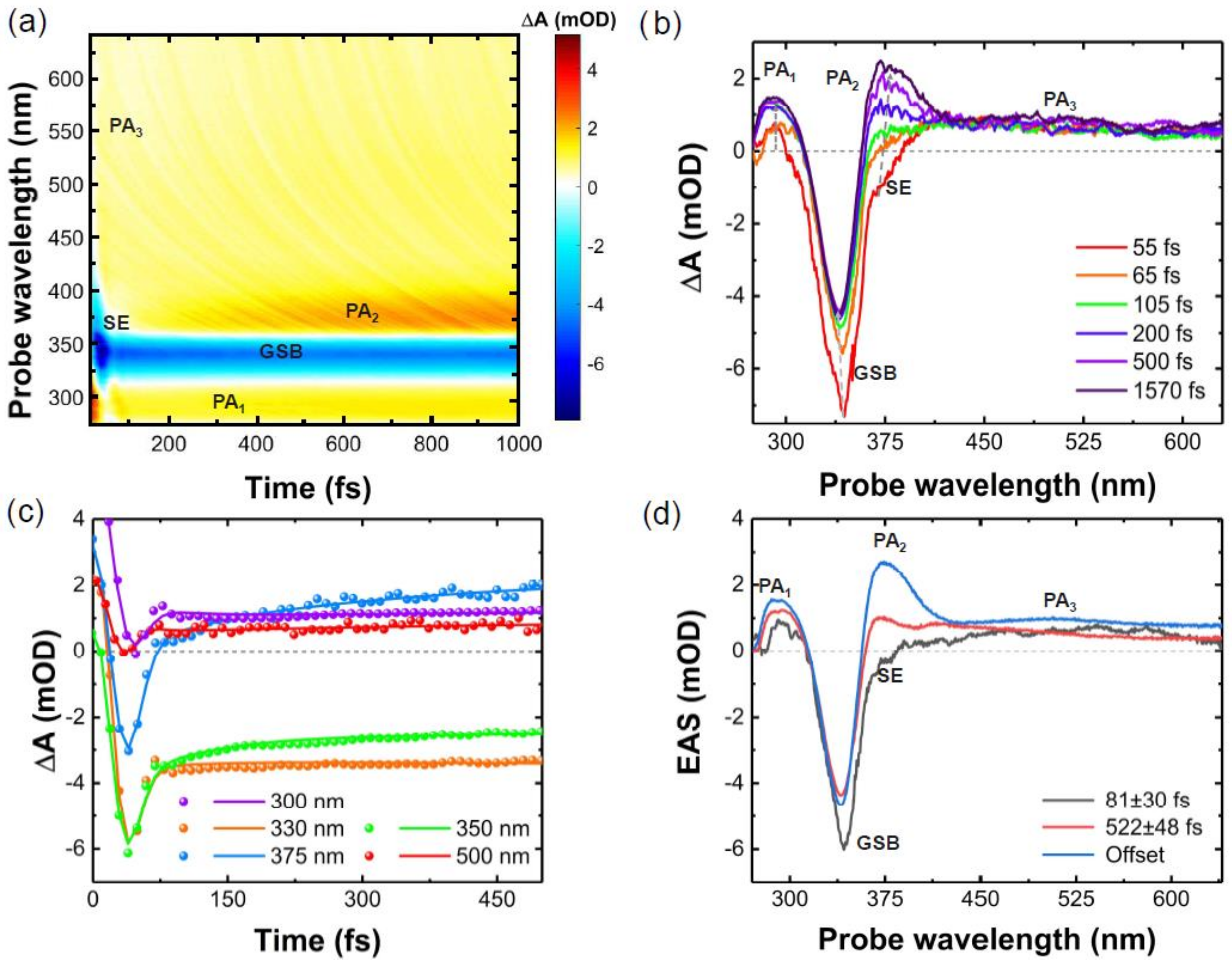
{kind=link}
| State Character | Vertical Emission Energy eV (nm) | Adiabatic Emission Energy eV (nm) | f |
|---|---|---|---|
| FC→S2(ππ*) | 4.00 (311) | 0.187 | |
| P1 S2(ππ*)→S0(ππ*) | 3.16 (393) | 3.87 (320) | 0.101 |
| P2 S2(ππ*)→S0(ππ*) | 3.04 (408) | 3.87 (320) | 0.095 |
| P3 S2(ππ*)→S0(ππ*) | 2.86 (434) | 3.78 (328) | 0.061 |
| S1(ππ*)min→S0(ππ*) | 2.29 (541) | 3.79 (328) | 0.024 |
| S1(nπ*)min→S0(ππ*) | 2.14 (580) | 3.18 (392) | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teles-Ferreira, D.C.; Manzoni, C.; Martínez-Fernández, L.; Cerullo, G.; de Paula, A.M.; Borrego-Varillas, R. Ultrafast Excited-State Decay Mechanisms of 6-Thioguanine Followed by Sub-20 fs UV Transient Absorption Spectroscopy. Molecules 2022, 27, 1200. https://doi.org/10.3390/molecules27041200
Teles-Ferreira DC, Manzoni C, Martínez-Fernández L, Cerullo G, de Paula AM, Borrego-Varillas R. Ultrafast Excited-State Decay Mechanisms of 6-Thioguanine Followed by Sub-20 fs UV Transient Absorption Spectroscopy. Molecules. 2022; 27(4):1200. https://doi.org/10.3390/molecules27041200
Chicago/Turabian StyleTeles-Ferreira, Danielle C., Cristian Manzoni, Lara Martínez-Fernández, Giulio Cerullo, Ana Maria de Paula, and Rocío Borrego-Varillas. 2022. "Ultrafast Excited-State Decay Mechanisms of 6-Thioguanine Followed by Sub-20 fs UV Transient Absorption Spectroscopy" Molecules 27, no. 4: 1200. https://doi.org/10.3390/molecules27041200