
Design, Synthesis, and Anticancer Screening for Repurposed Pyrazolo[3,4-d]pyrimidine Derivatives on Four Mammalian Cancer Cell Lines


 ,
,
Abstract
:1. Introduction
2. Results
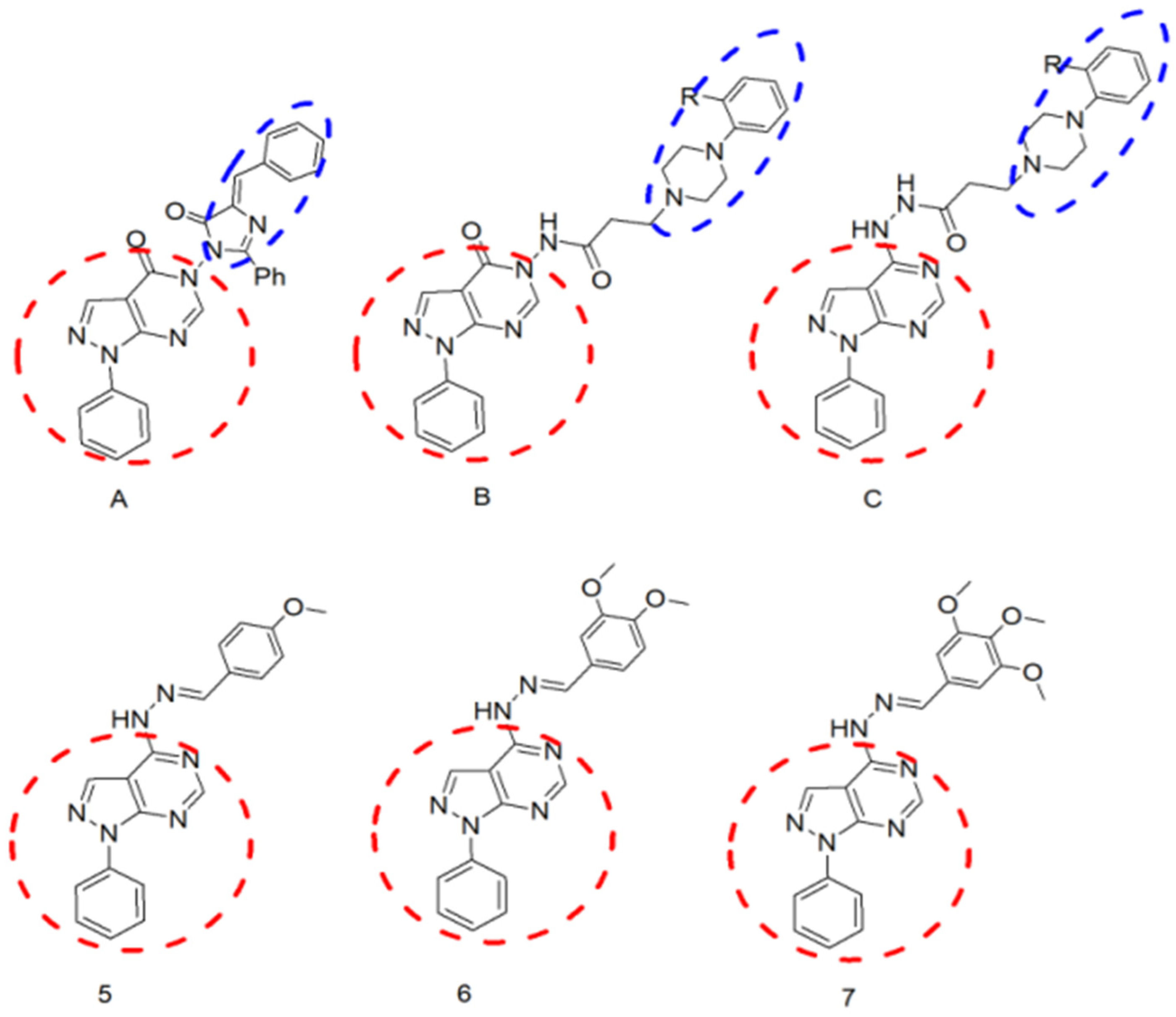
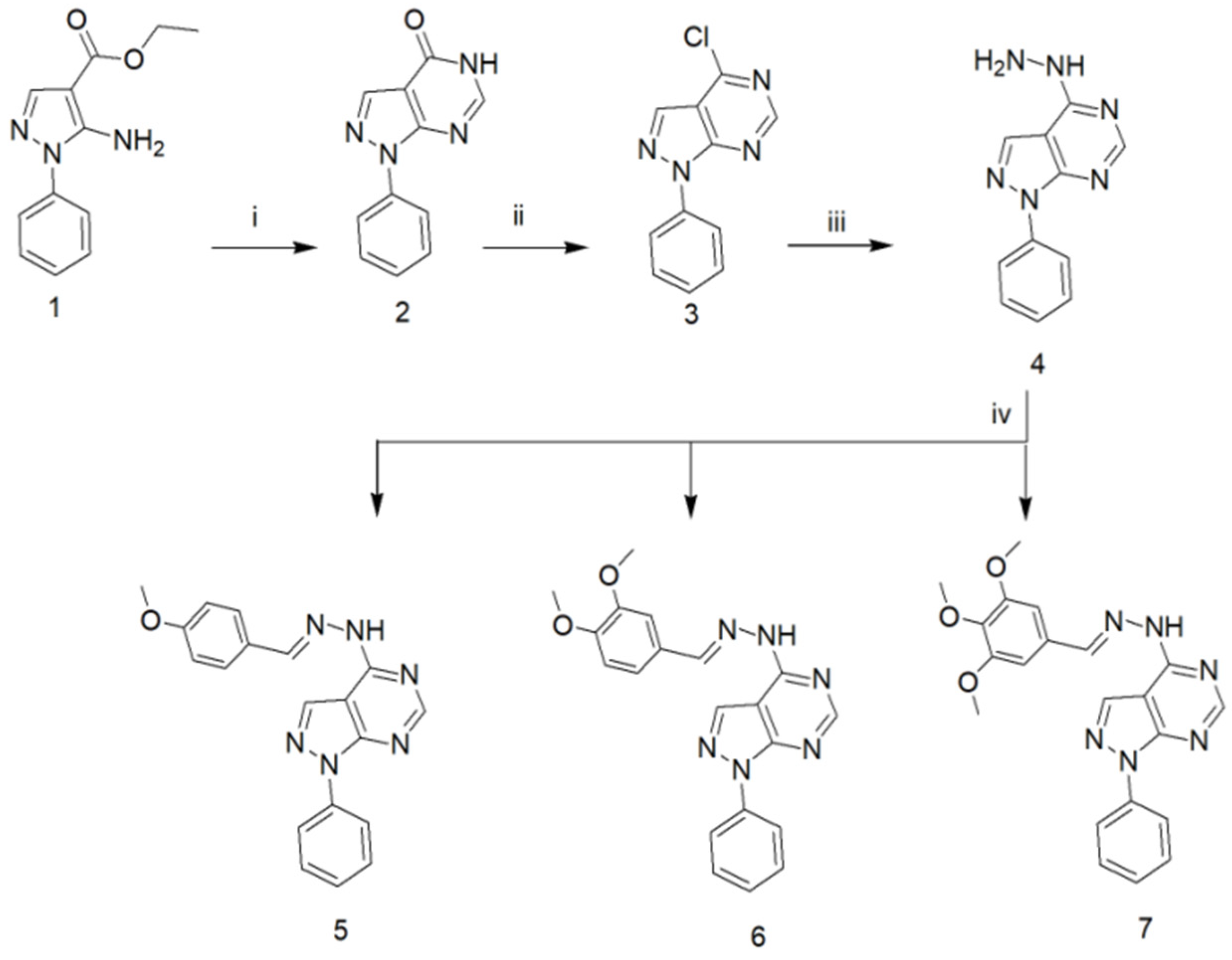
2.1. Chemistry of the Compounds under Investigation in the Current Study
2.2. In Vivo Toxicity Evaluation
2.3. Docking Analysis
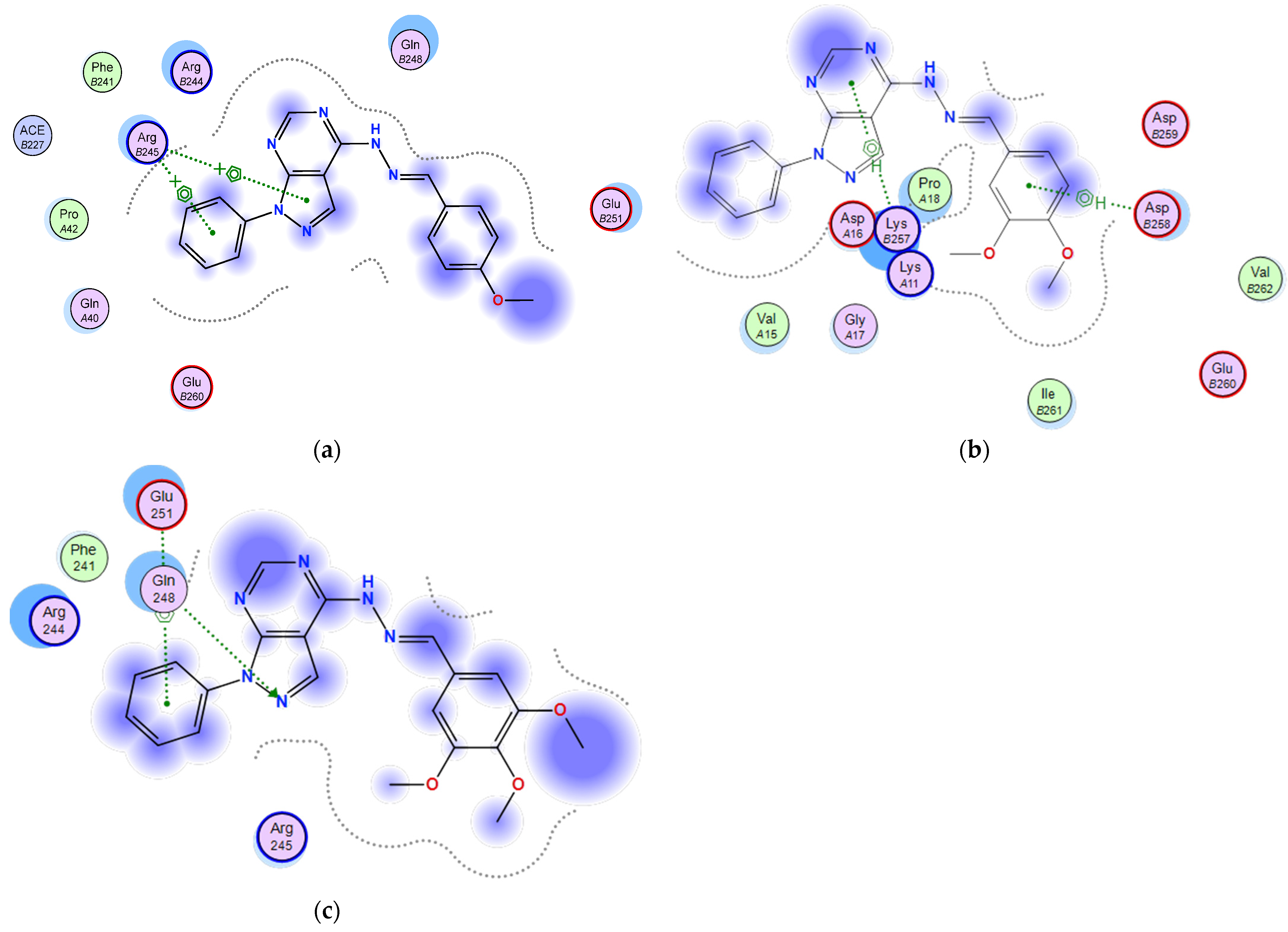
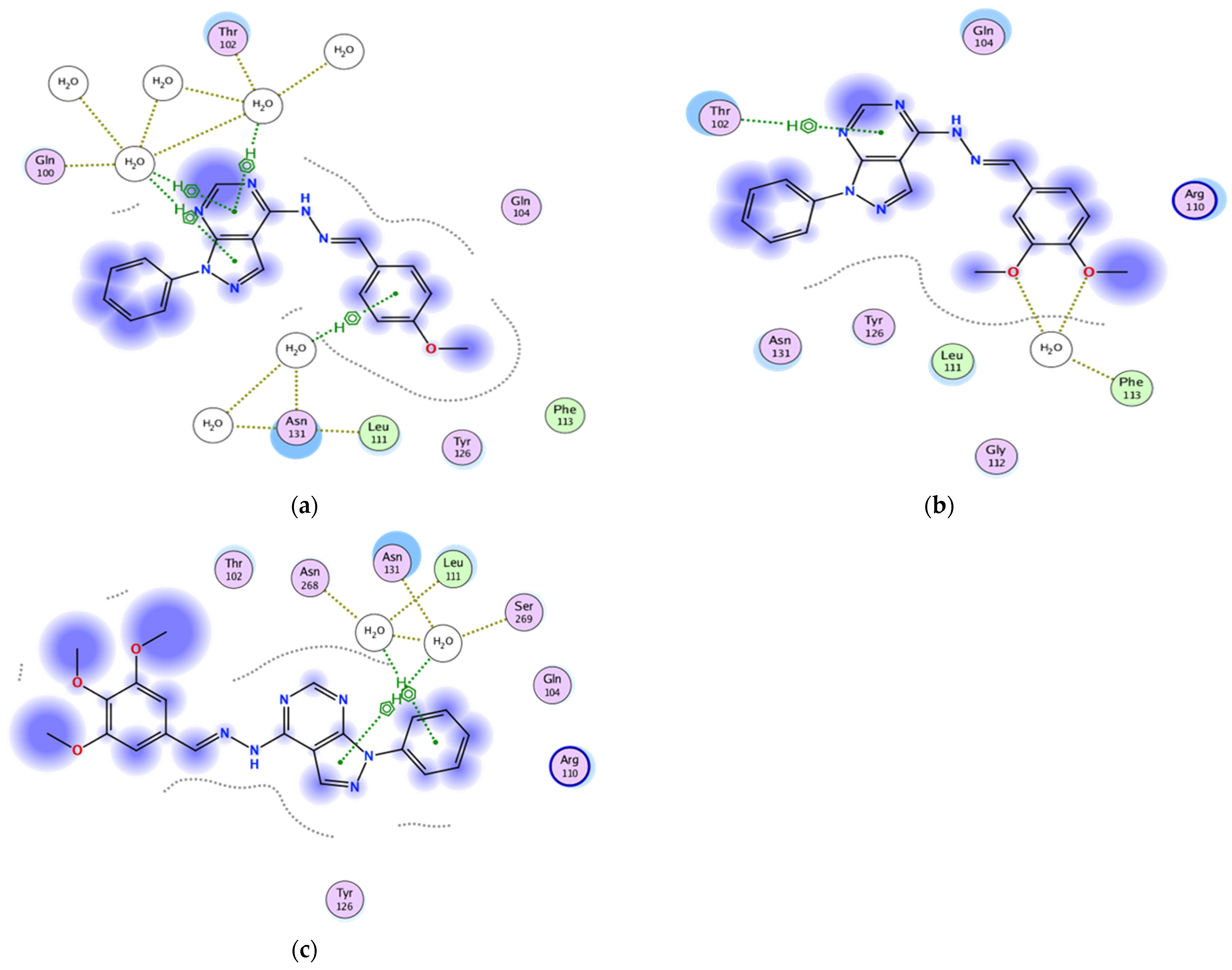
2.3.1. Docking of the Test Compounds (5–7) in the Binding Site of Ki67 Protein
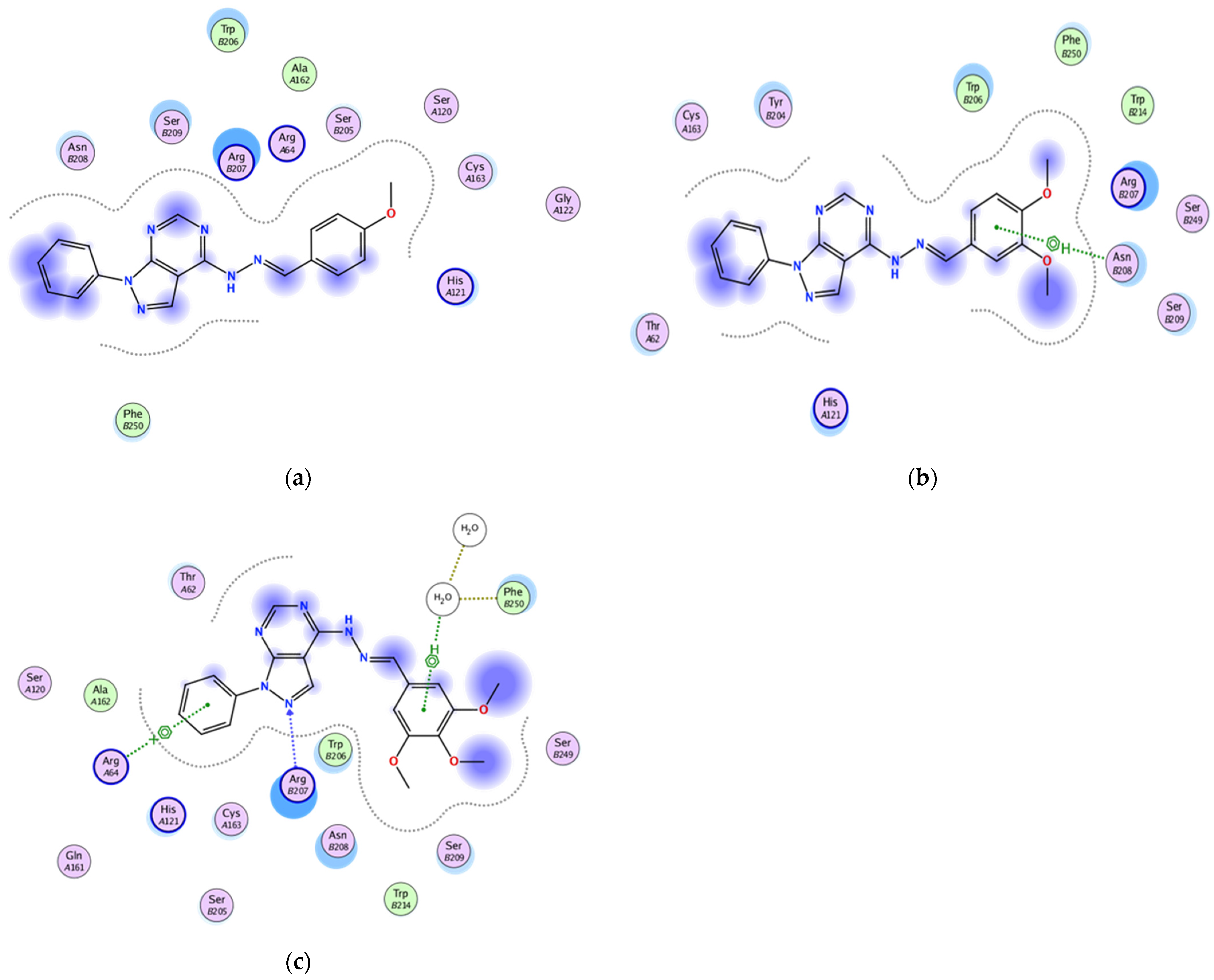
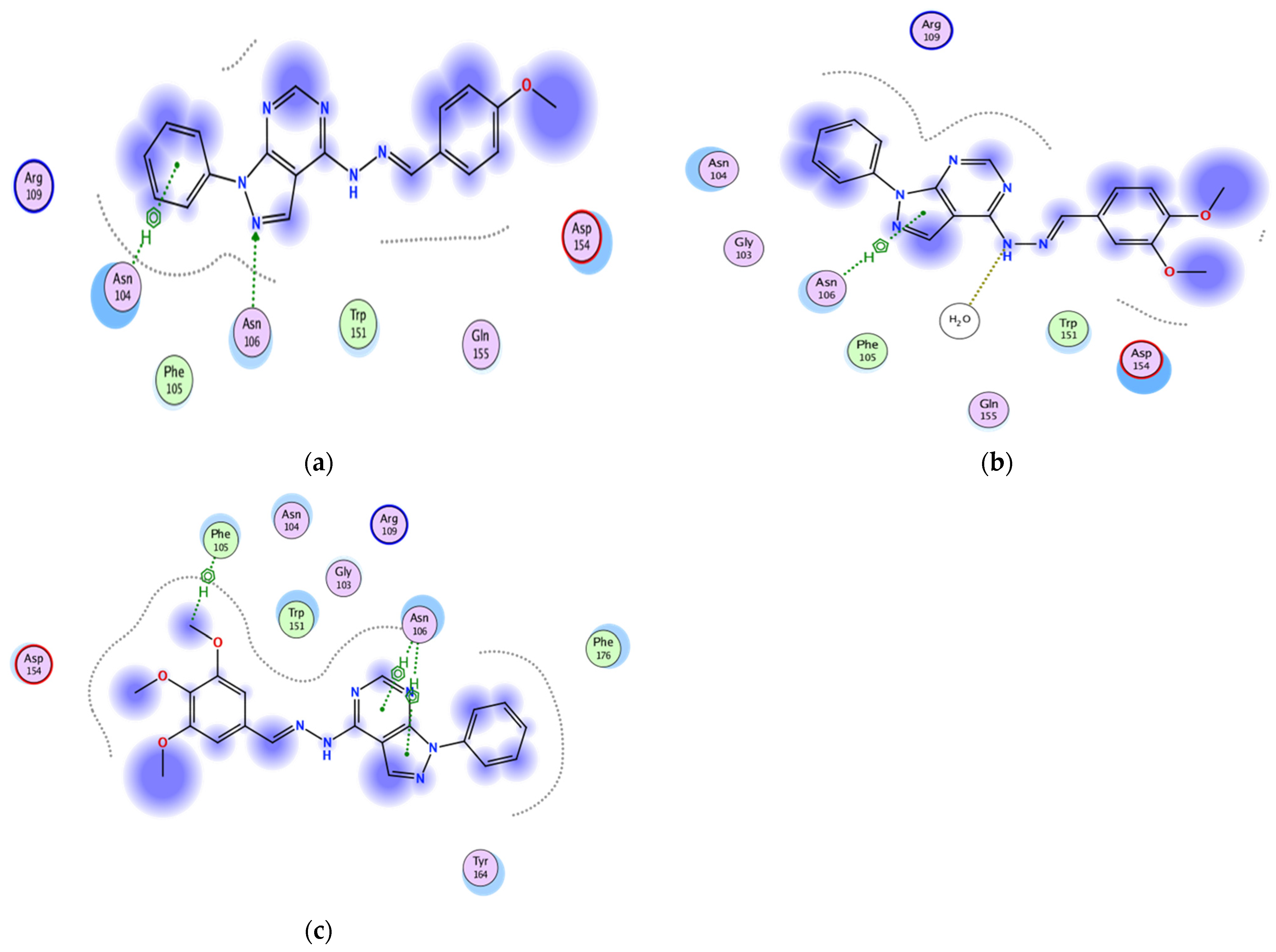
2.3.2. Docking of the Test Compounds (5–7) in the Binding Site of Caspase-3 Protein
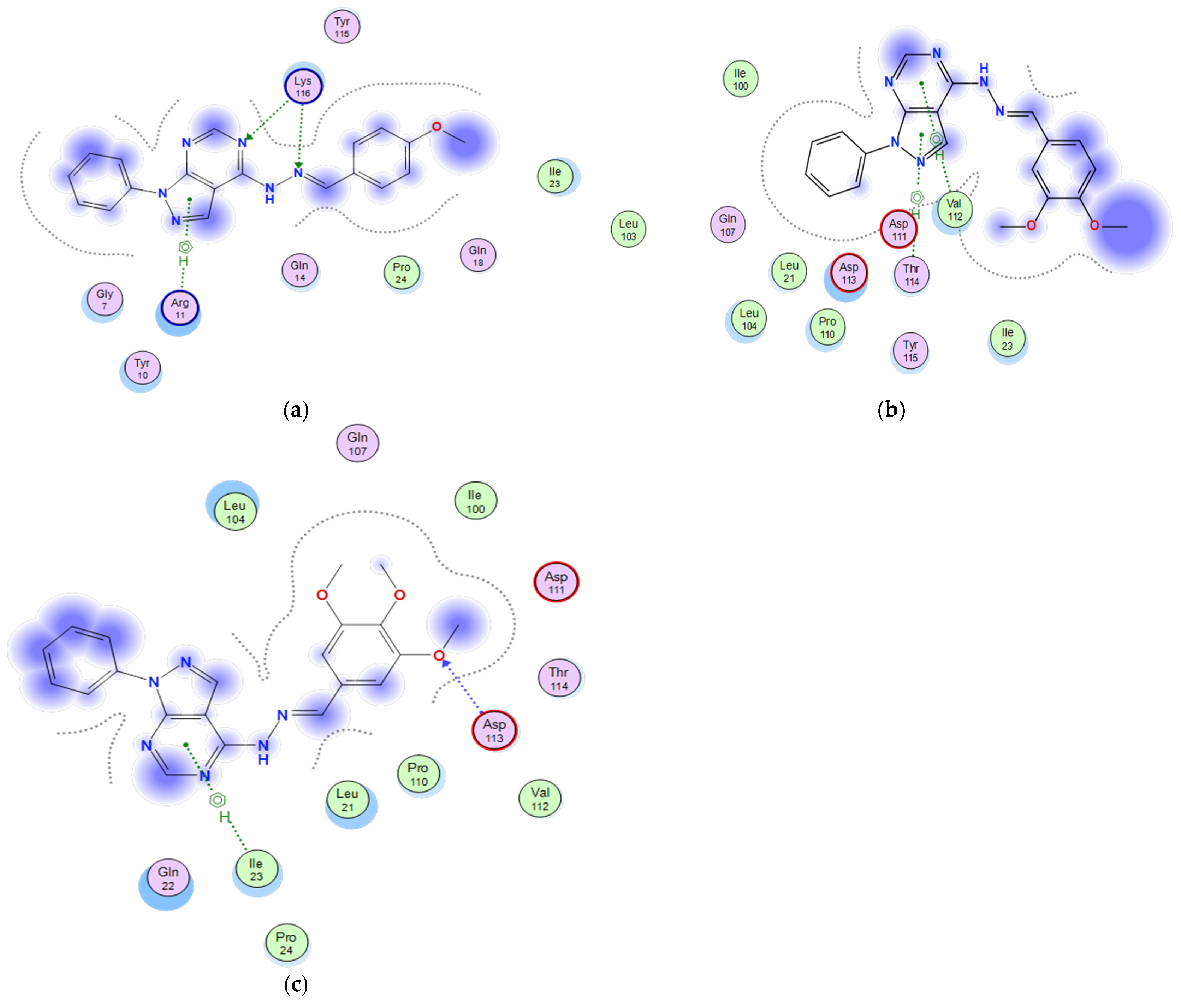
2.3.3. Docking of the Test Compounds (5–7) in the Binding Site of p53 Protein
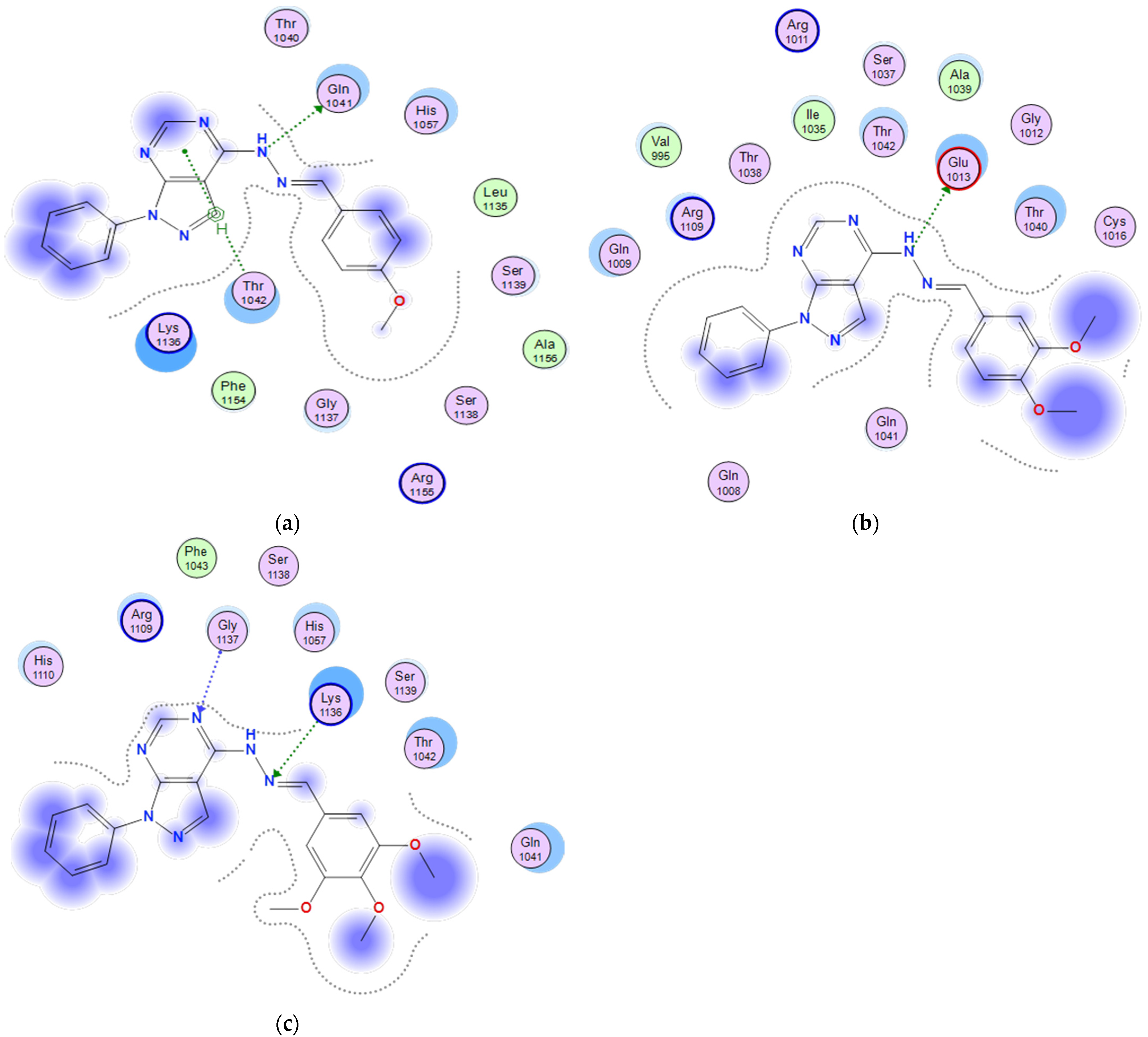
2.3.4. Docking of the Test Compounds (5–7) in the Binding Site of the Bax Protein
2.3.5. Docking of the Test Compounds (5–7) in the Binding Site of Bcl-2 Protein
2.3.6. Docking of the Test Compounds (5–7) in the Binding Site of P21 Protein
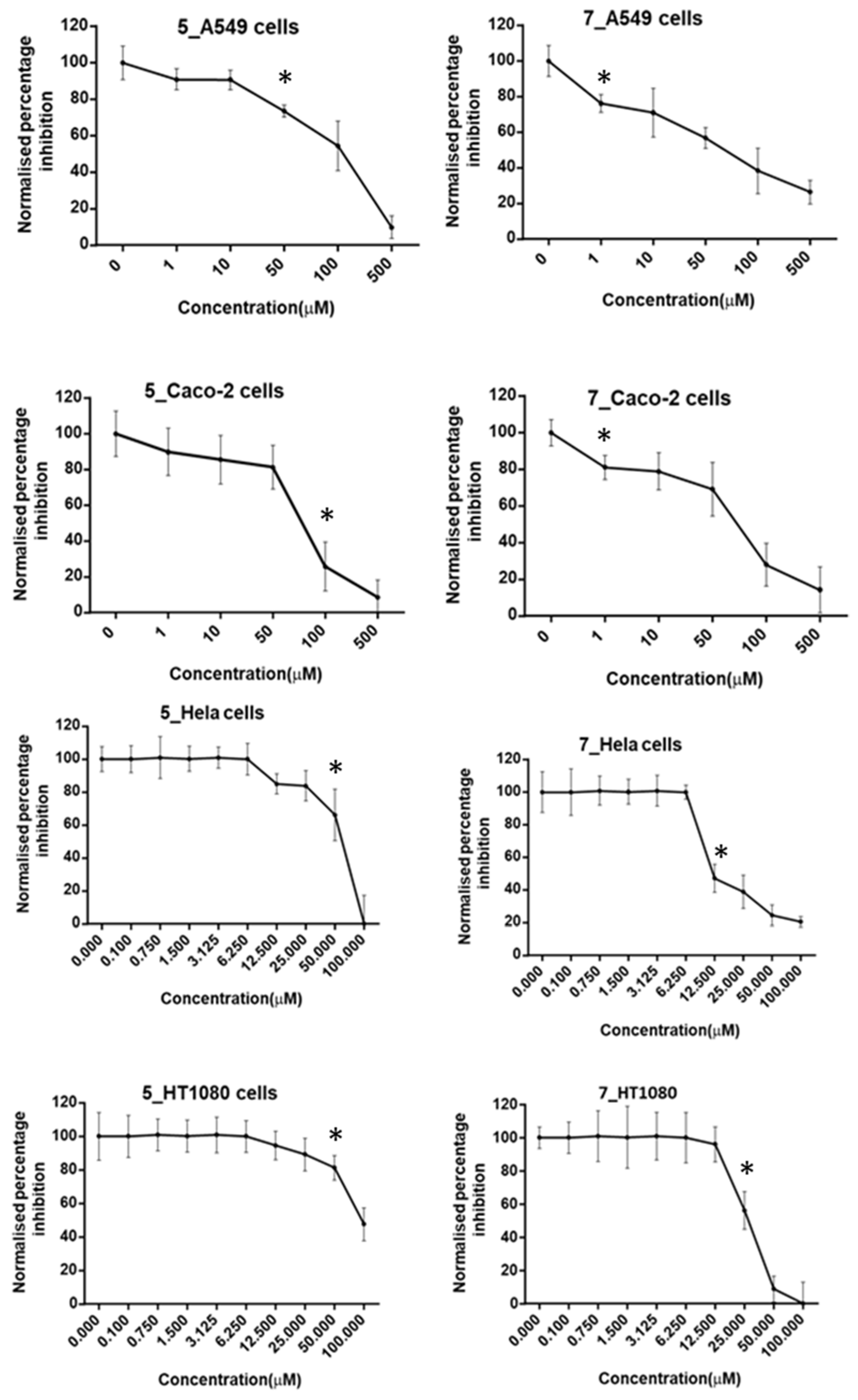
2.4. Cell Viability with MTT
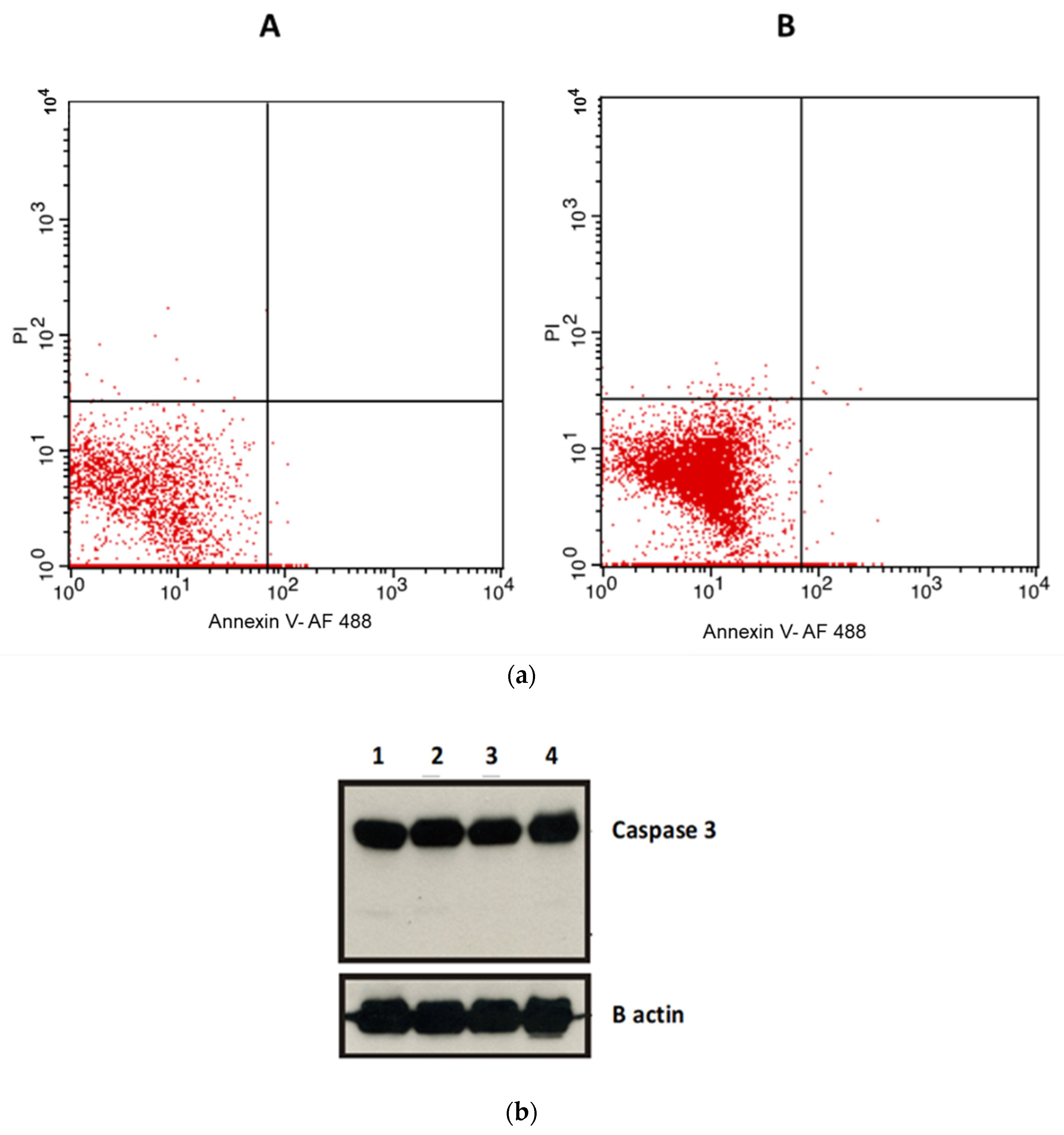
2.5. Apoptosis Assays
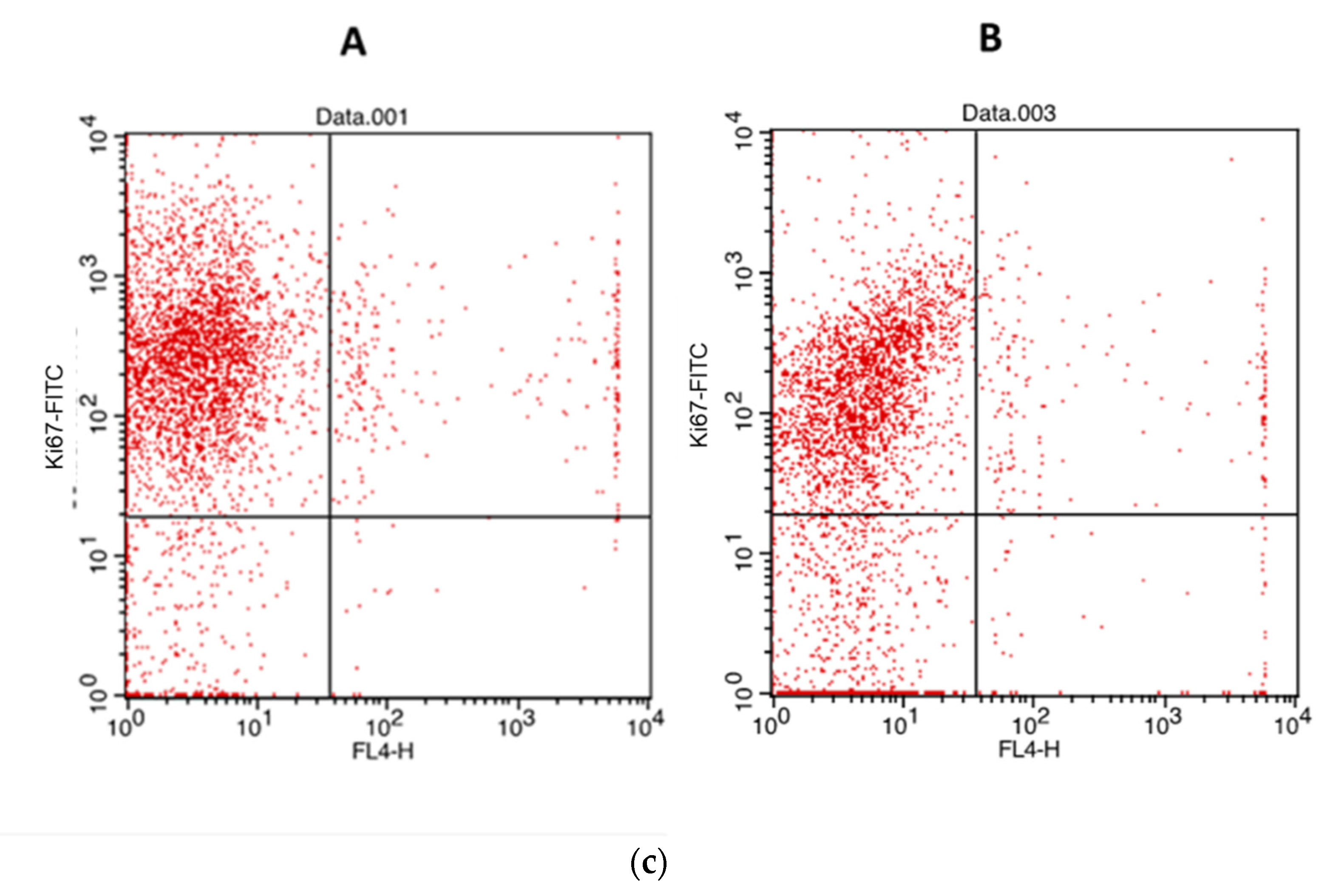
2.6. Proliferation Assay
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. In Vivo Toxicity Evaluation
Assessment of Toxicity Parameters
4.3. Docking Study
Docking Protocol
4.4. Cell Culture
4.5. Experimental Design
4.6. Cell Viability Assays
4.7. Apoptosis Assays
4.8. Cell Proliferation Assay
4.9. Investigation of Caspase-3 Protein Expression Levels Involved in the Anticancer Effect
Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Stefan, D.C.; Elzawawy, A.M.; Khaled, H.M.; Ntaganda, F.; Asiimwe, A.; Addai, B.W.; Wiafe, S.; Adewole, I.F. Developing cancer control plans in Africa: Examples from five countries. Lancet Oncol. 2013, 14, e189–e195. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Khaled, H.M.; Mikhail, N.N.; Baraka, H.; Kamel, H. Cancer incidence in Egypt: Results of the national population-based cancer registry program. J. Cancer Epidemiol. 2014, 2014, 437971. [Google Scholar] [CrossRef] [PubMed]
- El-Zawahry, H.M.; Zeeneldin, A.A.; Samra, M.A.; Mattar, M.M.; El-Gammal, M.M.; Abd El-Samee, A.; Darwish, T. Cost and outcome of treatment of adults with acute myeloid leukemia at the National Cancer Institute-Egypt. J. Egypt Natl. Canc. Inst. 2007, 19, 106–113. [Google Scholar] [PubMed]
- Brown, M.; Cheung, M.; Dickerson, S.; Drewry, D.; Lackey, K.; Peat, A.; Thomson, S.; Veal, J.; Wilson, J. Pyrazolopyrimidines as Kinase Inhibitors. U.S. Patent Application 10/521,910[P], 1 December 2005. [Google Scholar]
- Ghorab, M.; Ragab, F.; Alqasoumi, S.I.; Alafeefy, A.M.; Aboulmagd, S.A. Synthesis of some new pyrazolo[3,4-d]pyrimidine derivatives of expected anticancer and radioprotective activity. Eur. J. Med. Chem. 2010, 45, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinleye, A.; Chen, Y.; Mukhi, N.; Song, Y.; Liu, D. Ibrutinib and novel BTK inhibitors in clinical development. J. Hematol. Oncol. 2013, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Antonello, A.; Tarozzi, A.; Morroni, F.; Cavalli, A.; Rosini, M.; Hrelia, P.; Bolognesi, M.L.; Melchiorre, C. Multitarget-directed drug design strategy: A novel molecule designed to block epidermal growth factor receptor (EGFR) and to exert proapoptotic effects. J. Med. Chem. 2006, 49, 6642–6645. [Google Scholar] [CrossRef]
- Abd El Razik, H.A.; Mroueh, M.; Faour, W.H.; Shebaby, W.N.; Daher, C.F.; Ashour, H.M.; Ragab, H.M. Synthesis of new pyrazolo [3,4-d] pyrimidine derivatives and evaluation of their anti-inflammatory and anticancer activities. Chem. Biol. Drug Des. 2017, 90, 83–96. [Google Scholar] [CrossRef]
- Miao, W.; Guo, L.; Wang, Y. Imatinib-Induced Changes in Protein Expression and ATP-Binding Affinities of Kinases in Chronic Myelocytic Leukemia Cells. Anal. Chem. 2019, 91, 3209–3214. [Google Scholar] [CrossRef] [Green Version]
- Li, J.F.; Zhu, Y.Q.; Wang, X.; Yang, H.Z. Synthesis and herbicidal activities of a series of di(aminopyrazoly) ketone derivatives. J. Heterocycl. Chem. 2007, 44, 749–755. [Google Scholar] [CrossRef]
- El-Dean, A.M.K.; Geies, A.A. Synthesis of Some New Pyrazolotriazines, Pyrazolothiazines and Pyrazolopyrimidines. J. Chem. Res. Synop. 1997, 352–353. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc. Available online: www.chemcomp.com (accessed on 15 May 2015).
- Byeon, I.-J.L.; Li, H.; Song, H.; Gronenborn, A.M.; Tsai, M.-D. Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat. Struct. Mol. Biol. 2005, 12, 987–993. [Google Scholar] [CrossRef]
- Thomas III, M.E.; Grinshpon, R.; Swartz, P.; Clark, A.C. Modifications to a common phosphorylation network provide individualized control in caspases. J. Biol. Chem. 2018, 293, 5447–5461. [Google Scholar] [CrossRef] [Green Version]
- Golovenko, D.; Bräuning, B.; Vyas, P.; Haran, T.E.; Rozenberg, H.; Shakked, Z. New insights into the role of DNA shape on its recognition by p53 proteins. Structure 2018, 26, 1237–1250.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, M.A.; Robin, A.Y.; Gibson, L.; Li, M.X.; Sandow, J.J.; Iyer, S.; Webb, A.I.; Westphal, D.; Dewson, G.; Adams, J.M. BAX activation: Mutations near its proposed non-canonical BH3 binding site reveal allosteric changes controlling mitochondrial association. Cell Rep. 2019, 27, 359–373.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, E.P.; Seo, H.-S.; Guerra, R.M.; Bird, G.H.; Dhe-Paganon, S.; Walensky, L.D. Crystal structures of anti-apoptotic BFL-1 and its complex with a covalent stapled peptide inhibitor. Structure 2018, 26, 153–160.e4. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.G.; Zipfel, S.; Ramey, K.; Vivian, R.; Schrier, A.; Karki, K.K.; Katana, A.; Kato, D.; Kobayashi, T.; Martinez, R. Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®. Bioorg. Med. Chem. Lett. 2019, 29, 2428–2436. [Google Scholar] [CrossRef]
- Chhikara, B.S.; Ashraf, S.; Mozaffari, S.; St Jeans, N.; Mandal, D.; Tiwari, R.K.; Ul-Haq, Z.; Parang, K. Phenylpyrazalopyrimidines as Tyrosine Kinase Inhibitors: Synthesis, Antiproliferative Activity, and Molecular Simulations. Molecules 2020, 25, 2135. [Google Scholar] [CrossRef]
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The novel NF-kappaB inhibitor DHMEQ synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ROS-dependent mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef]
- Cui, W.; Yu, C.H.; Hu, K.Q. In vitro and in vivo effects and mechanisms of celecoxib-induced growth inhibition of human hepatocellular carcinoma cells. Clin. Cancer Res. 2005, 11, 8213–8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Song, E.; Jiang, M.; Song, Y. Celecoxib and Afatinib synergistic enhance radiotherapy sensitivity on human non-small cell lung cancer A549 cells. Int. J. Radiat. Biol. 2021, 97, 170–178. [Google Scholar] [CrossRef]
- Snyder, J.A.; Ha, Y.; Olsofka, C.; Wahdan, R. Both actin and myosin inhibitors affect spindle architecture in PtK1 cells: Does an actomyosin system contribute to mitotic spindle forces by regulating attachment and movements of chromosomes in mammalian cells? Protoplasma 2010, 240, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Fioravanti, A.; La Motta, C.; Orlandi, P.; Canu, B.; Di Desidero, T.; Mugnaini, L.; Sartini, S.; Cosconati, S.; Frati, R.; et al. Antiproliferative and proapoptotic activity of CLM3, a novel multiple tyrosine kinase inhibitor, alone and in combination with SN-38 on endothelial and cancer cells. Biochem Pharm. 2011, 81, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Hammer, A.; Gustafson, L.W. Implementation of p16/Ki67 dual stain cytology in a Danish routine screening laboratory: Importance of adequate training and experience. Cancer Med. 2020, 9, 8235–8242. [Google Scholar] [CrossRef]
- La Rosa, S.; Bonzini, M.; Sciarra, A.; Asioli, S.; Maragliano, R.; Arrigo, M.; Foschini, M.P.; Righi, A.; Maletta, F.; Motolese, A.; et al. Exploring the Prognostic Role of Ki67 Proliferative Index in Merkel Cell Carcinoma of the Skin: Clinico-Pathologic Analysis of 84 Cases and Review of the Literature. Endocr. Pathol. 2020. [Google Scholar] [CrossRef]
- Brönimann, S.; Pradere, B.; Karakiewicz, P.; Abufaraj, M. An overview of current and emerging diagnostic, staging and prognostic markers for prostate cancer. Expert Rev. Mol. Diagn. 2020, 20, 841–850. [Google Scholar] [CrossRef]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Basolo, F.; Castellone, M.D.; Melillo, R.M. Efficient inhibition of RET/papillary thyroid carcinoma oncogenic kinases by 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2). J. Clin. Endocrinol. Metab. 2003, 88, 1897–1902. [Google Scholar] [CrossRef] [Green Version]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Belfield, A.; Goldberg, D.M. Normal ranges and diagnostic value of serum 5′nucleotidase and alkaline phosphatase activities in infancy. Arch. Dis. Child. 1971, 46, 842–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doumas, B.T.; Watson, W.A.; Biggs, H.G. Albumin standards and the measurement of serum albumin with bromcresol green. Clin. Chim. Acta 1971, 31, 87–96. [Google Scholar] [CrossRef]
- Watson, D.; Rogers, J.A. A study of six representative methods of plasma bilirubin analysis. J. Clin. Pathol. 1961, 14, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fossati, P.; Prencipe, L. Serum triglycerides determined colorimetrically with an enzyme that produces hydrogen peroxide. Clin. Chem. 1982, 28, 2077–2080. [Google Scholar] [CrossRef]
- Fenech, G.; Tommasini, A. Method of colorimetric determination of urea. Boll Chim. Farm 1952, 91, 391–395. [Google Scholar]
- Lustgarten, J.A.; Wenk, R.E. Simple, rapid, kinetic method for serum creatinine measurement. Clin. Chem. 1972, 18, 1419–1422. [Google Scholar] [CrossRef]
- Abel, L.L.; Levy, B.B. Brodie, and F.E. Kendall. A simplified method for the estimation of total cholesterol in serum and demonstration of its specificity. J. Biol. Chem. 1952, 195, 357–366. [Google Scholar] [CrossRef]
- Ye, T.H.; Yang, F.F.; Zhu, Y.X.; Li, Y.L.; Lei, Q.; Song, X.J.; Xia, Y.; Xiong, Y.; Zhang, L.D.; Wang, N.Y.; et al. Inhibition of Stat3 signaling pathway by nifuroxazide improves antitumor immunity and impairs colorectal carcinoma metastasis. Cell Death Dis. 2017, 8, e2534. [Google Scholar] [CrossRef]
- Zhu, Y.; Ye, T.; Yu, X.; Lei, Q.; Yang, F.; Song, Y.; Liu, L.; Deng, H.; Gao, T.; Peng, C. Nifuroxazide exerts potent anti-tumor and anti-metastasis activity in melanoma. Sci. Rep. 2016, 6, 20253. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Hu, M.; Lei, Q.; Xia, Y.; Zhu, Y.; Song, X.; Li, Y.; Jie, H.; Liu, C.; Xiong, Y.; et al. Nifuroxazide induces apoptosis and impairs pulmonary metastasis in breast cancer model. Cell Death Dis. 2015, 6, e1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Zeng, A.; Fang, A.; Song, L.; Fan, C.; Zeng, C.; Ye, T.; Chen, H.; Tu, C.; Xie, Y. Nifuroxazide induces apoptosis, inhibits cell migration and invasion in osteosarcoma. Investig. New Drugs 2019, 37, 1006–1013. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | 5 | 6 | 7 | |
|---|---|---|---|---|
| Cholesterol (mg/dL) | 70 ± 3.1 | 71 ± 4.8 | 69 ± 7.8 | 71 ± 5.8 |
| Triglyceride (mg/dL) | 74 ± 5.1 | 72 ± 8.6 | 75 ± 6.5 | 77 ± 7.5 |
| Bilirubin (mg/dL) | 0.12 ± 0.13 | 0.11 ± 0.12 | 0.12 ± 0.12 | 0.11± 0.32 |
| Albumin (mg/dL) | 4.5 ± 0.12 | 4.4 ± 0.37 | 4.6 ± 1.8 | 4.6 ±1.1 |
| ALT (U/L) | 44 ± 4.1 | 43 ± 5.2 | 44 ± 5.7 | 42 ± 7.2 |
| AST (U/L) | 103 ± 4.6 | 102 ± 8.7 | 100 ± 9.8 | 103 ± 8.8 |
| ALP (U/L) | 1.92 ± 0.47 | 2.1 ± 0.52 | 2 ± 0.12 | 1.8 ± 0.88 |
| Urea (U/L) | 5.8 ± 0.23 | 5.7 ± 0.98 | 5.8 ± 0.80 | 5.88 ± 0.90 |
| Creatinine (mg/dL) | 28 ± 2.5 | 27 ± 4.2 | 29 ± 3.86 | 28 ± 3.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othman, E.M.; Bekhit, A.A.; Anany, M.A.; Dandekar, T.; Ragab, H.M.; Wahid, A. Design, Synthesis, and Anticancer Screening for Repurposed Pyrazolo[3,4-d]pyrimidine Derivatives on Four Mammalian Cancer Cell Lines. Molecules 2021, 26, 2961. https://doi.org/10.3390/molecules26102961
Othman EM, Bekhit AA, Anany MA, Dandekar T, Ragab HM, Wahid A. Design, Synthesis, and Anticancer Screening for Repurposed Pyrazolo[3,4-d]pyrimidine Derivatives on Four Mammalian Cancer Cell Lines. Molecules. 2021; 26(10):2961. https://doi.org/10.3390/molecules26102961
Chicago/Turabian StyleOthman, Eman M., Amany A. Bekhit, Mohamed A. Anany, Thomas Dandekar, Hanan M. Ragab, and Ahmed Wahid. 2021. "Design, Synthesis, and Anticancer Screening for Repurposed Pyrazolo[3,4-d]pyrimidine Derivatives on Four Mammalian Cancer Cell Lines" Molecules 26, no. 10: 2961. https://doi.org/10.3390/molecules26102961