
Mitochondrial K+ Transport: Modulation and Functional Consequences


Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo 05508-000, SP, Brazil
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(10), 2935; https://doi.org/10.3390/molecules26102935
Submission received: 21 April 2021
/
Revised: 7 May 2021
/
Accepted: 11 May 2021
/
Published: 14 May 2021
(This article belongs to the Special Issue Compounds Modulating Mitochondrial Ion Channels)
Abstract
:The existence of a K+ cycle in mitochondria has been predicted since the development of the chemiosmotic theory and has been shown to be crucial for several cellular phenomena, including regulation of mitochondrial volume and redox state. One of the pathways known to participate in K+ cycling is the ATP-sensitive K+ channel, MitoKATP. This channel was vastly studied for promoting protection against ischemia reperfusion when pharmacologically activated, although its molecular identity remained unknown for decades. The recent molecular characterization of MitoKATP has opened new possibilities for modulation of this channel as a mechanism to control cellular processes. Here, we discuss different strategies to control MitoKATP activity and consider how these could be used as tools to regulate metabolism and cellular events.
1. Introduction
Mitochondria are widely, and rightfully, recognized as the hubs of energy metabolism, organelles in which key metabolic pathways (including oxidative phosphorylation, the citric acid cycle, β-oxidation, and part of amino acid metabolism) occur. In mitochondria, nutrient breakdown and oxidation is associated with highly efficient ATP generation through oxidative phosphorylation, which involves the reduction of oxygen to water in the electron transport chain, generation of an inner mitochondrial membrane electrochemical potential due to proton pumping, and ATP production coupled with proton re-entry into mitochondria through the ATP synthase [1].
In the 1960s and 1970s, two further mitochondrial functions were uncovered: the generation of superoxide radical anions and other oxidants derived from it [2,3,4,5], and the uptake and release of Ca2+ ions [6,7,8]. Interestingly, the relationship between these two phenomena—redox state and mitochondrial Ca2+ transport—was recognized early on [9,10]: oxidized pyridine nucleotides [11] and membrane protein thiols [12] favor Ca2+ release from mitochondria.
In the 1990s, mitochondrial function gained attention as a regulator of key cellular survival processes. Mitochondrial intermembrane space proteins were found to be involved in apoptosis when released to the cytosol [13,14]. Furthermore, opening of mitochondrial K+ channels was shown to be an effective mechanism to protect against cardiac ischemic damage [15]. These findings and others boosted publications in mitochondrial research, which went from roughly 2000 papers per year to close to 8000 a year today, and still rising [16].
2. The K+ Cycle
Because intracellular K+ concentrations are high, in the range of 140 mM, and the mitochondrial inner membrane has an electrochemical potential in the range of 100 to 200 mV with a negative inside, K+ ions are expected to leak through the membrane at biologically relevant rates, despite the high impermeability of this membrane. When K+ enters mitochondria in the presence of permeable anions (including phosphate, which is present at millimolar concentrations intracellularly), K+ uptake is accompanied by osmotically obligated water and results in organellar swelling [17]. As a result, mechanisms to control K+ concentrations in the matrix were predicted to exist when the chemiosmotic theory was developed [18].
Indeed, mitochondria from all tissues tested possess K+/H+ exchange activity, performed by a protein exchanger that can be pharmacologically modulated [19]. Unfortunately, despite its seminal role in mitochondrial homeostasis, the molecular identity of this exchanger remains debated. Since many key mitochondrial transport pathways were only molecularly identified in the last few years, we hope the molecular identity of this seminal mitochondrial transporter will soon be uncovered.
Surprisingly, in 1991 Inoue et al. discovered, through patch-clamping experiments, that mitochondria had a regulated K+ entry pathway in addition to the K+ leak [20]. This pathway was inhibited by ATP in a Mg2+-dependent manner [21,22]. The newly described mitochondrial ATP-sensitive K+ channel, MitoKATP (Figure 1), presented many characteristics similar to plasma membrane KATP channels (CellKATP’s), including inhibition by sulphonylureas [22,23]. Regulation, however, is not identical, as MitoKATP channels (but not CellKATP’s) are activated by both GTP and GDP, and inhibited by ADP and long-chain acyl-CoA esters [24]. This means MitoKATP’s, unlike CellKATP’s, are not modulated directly by ADP/ATP ratios. Given that the affinity of the channel for ATP is much higher than cellular concentrations and that channel inhibition is also promoted by ADP, MitoKATP regulation in vivo has been proposed to be controlled by cellular levels of guanine nucleotides and long-chain acyl-CoA [24], as well as by oxidants and antioxidants [25].
Functional properties of MitoKATP are still being uncovered, but certainly involve volume homeostasis. Because K+ leak is dependent on inner membrane potentials and these are variable with cell energy demands, a regulated pathway to ensure K+ entry into mitochondria may be necessary to maintain organellar architecture. Volume homeostasis of the mitochondrial matrix through regulated K+ uptake is specifically important to maintain the relationship between the inner and outer mitochondrial membranes, since matrix swelling and contraction can affect the interface between these membranes. Matrix volume regulation may also be important for the maintenance of cristae architecture and function [26].
Structurally, early reconstitution studies demonstrated the existence of two subunits necessary to form the holo-MitoKATP. The first is a pore-forming K+ channel that was named MitoKIR, and the other is an ATP-binding subunit also known to be a receptor for many pharmacological modulators such as sulphonylureas, therefore called MitoSUR [22,26,27,28]. Excitingly, in late 2019 the molecular identity of these channels was identified [29]. The results are compatible with previous structural studies and led to the identification of the genes coding for both subunits, which were termed MITOK and MITOSUR, respectively. This seminal finding allows us new and more specific genetic approaches toward studying the biology and consequences of these channels, which we hope will begin a new era of mechanistic understanding of mitochondrial K+ homeostasis.
Despite the lack of knowledge regarding its molecular identity until recently, MitoKATP had been extensively explored for its protective properties against ischemia reperfusion in the heart and other tissues using pharmacological tools. Considering the central role that this protein and the K+/H+ exchanger have in mitochondrial and cellular homeostasis, we believe that, as a result of its recent molecular characterization, this protein could be even further explored as a target for biological modulation and therapeutic interventions. Here, we review the main pharmacological tools employed to modulate the activity of MitoKATP, to understand its function, signaling responses and therapeutic potential.
3. Activating MitoKATP
3.1. Pharmacological MitoKATP Activators
After ATP-sensitive K+ transport in mitochondria was described, pharmacological properties of the MitoKATP were determined through in situ and reconstitution experiments. At that time, cell membrane ATP-dependent K+ channels (CellKATP) were also being explored as receptors for modulating molecules, and the properties of both channels were assessed and compared. This led to the finding that CellKATP’s and MitoKATP presented vastly different affinities for channel activators such as diazoxide, which is not an effective CellKATP agonist, but opens MitoKATP at micromolar concentrations [15,27] (see Table 1). Importantly, in order to see pharmacological activation, experiments required the presence of physiological MitoKATP inhibitors ATP and Mg2+. This highlights a logical but often overlooked property of the MitoKATP: it is already active in the absence of physiological ATP or ADP concentrations, so adding agonists under these conditions will not uncover expected intracellular effects of channel agonists.
In the presence of physiological inhibitors, both diazoxide and cromakalim were found to be potent MitoKATP agonists. Diazoxide is 2000 times more selective toward the mitochondrial channel when compared to plasma membrane counterparts [25]. This selectivity was decisive to identify mitochondrial channels as mediators of cardiac protection against ischemia reperfusion [15]. When used at concentrations that activate MitoKATP, but not CellKATP, diazoxide protects hearts against damage promoted by ischemia followed by reperfusion, a finding that attracted wide attention to the role of mitochondrial K+ homeostasis in tissue protection under myriad of stressful conditions [15,30,31,32].
Diazoxide and cromakalim belong to a large group of K+ channel opener (KCO) molecules that also includes other compounds such as nicorandil and pinacidil. Although the later activators show some selectivity for MitoKATP (Table 1), they have mixed effects and can still act on CellKATP’s. Furthermore, KCO effects on CellKATP vary with cell types. For example, diazoxide can activate CellKATP in β cells, while pinacidil cannot [33,34]. Moreover, nicorandil also has unspecific vasodilatation effects, since it can be a nitric oxide donor [33]. Diazoxide also displays undesired vasodilatation effects by acting on smooth muscle CellKATP’s [35,36]. As a result, the need to search for even more selective drugs was recognized, and BMS191095 was synthesized based on cromakalim structure as a more specific drug that did not promote vasodilatation and disturb action potentials but still protected murine and canine hearts against ischemic insults [37,38].
Cromakalim and diazoxide act as agonists by interacting with the MitoSUR portion of the channel. This is demonstrated by the fact that isolation of MitoKIR and reconstitution into liposomes generates passive K+ transport inhibited by ATP (although much higher concentrations of ATP are needed than with the complete MitoKATP), but none of these KCOs recover ionic flux under these conditions. The results of isolated subunit reconstitution studies thus demonstrate the interaction of the channel agonists with MitoSUR. This same set of experiments also identified p-diethylaminoetylbenzoate as an activator of isolated MitoKIR, also efficient in isolated mitochondria [39].
3.2. Physiological MitoKATP Modulation
Although not the central focus of this review, we should stress that in addition to having many well-studied pharmacological activators, MitoKATP also is physiologically regulated, as these regulators may inspire new pharmacological intervention approaches. Indeed, MitoKATP channels are controlled by kinases, and are activated in response to PKC agonists [42]. MitoKATP channels are also modulated by respiratory complex II activity, and may respond to endogenous complex II inhibitors such as malonate [43].
MitoKATP channel activity is strongly sensitive to redox state. While oxidants such as superoxide radicals, H2O2 and S-nitrosothiols activate the channels, thiol reductants such as N-acetylcysteine, 2-mercaptopropionylglycine and dithiothreitol [25] inhibit the channel directly, suggesting it has redox-sensitive thiols. NADPH is also a MitoKATP inhibitor [44,45]. This redox-regulation of the channel is in keeping with its role in modulating mitochondrial oxidant production [46,47]. We hope that the now uncovered molecular identity of the channel will help future studies in determining the mechanisms of MitoKATP modulation by these compounds, including the identification of its redox sensors. These studies may also open new windows of opportunities for the design of novel KCOs.
3.3. Physiological Consequences of MitoKATP Opening
As discussed previously, activating MitoKATP allows K+ entry into the mitochondrial matrix which, accompanied by anion transport, promotes mitochondrial swelling though uptake of osmotically obligated water. This is important for the maintenance of mitochondrial structure and regulation of the intermembrane space architecture [48]. Thus, activating the MitoKATP can prevent structural damage to mitochondria caused by excessive contraction under pathological situations and preserve transport properties through the mitochondrial membrane, including the transport of ADP and ATP (Figure 2) [44,45].
K+ cycling in mitochondria is expected to result in a decrease in inner membrane potentials through the combined action of K+ entering through MitoKATP and exiting in exchange for protons, uncoupling electron transport from ATP synthesis. Uncoupling promoted by this cycling is limited due to low K+ transport rates through MitoKATP in most tissues, and it promotes only a 1–2% decrease in mitochondrial inner membrane potentials in the heart [49]. Despite the small changes in ΔΨm, even low levels of uncoupling can significantly modify oxidant production rates (Figure 2) [49,50].
Mitochondria produce oxidants through a few distinct pathways, and a very important source of reactive oxygen species is electron leakage in the electron transport chain, which can generate superoxide radical anions and hydrogen peroxide [51]. High ΔΨm is needed for electron leakage, as it often depends on reverse activity of mitochondrial complexes [5,52,53], which is thermodynamically feasible only at high ΔΨm. Indeed, MitoKATP activation was found to significantly decrease mitochondrial H2O2 release in different tissues [45]. Further studies indicated that, in addition to regulating mitochondrial oxidant production, MitoKATP channels were also redox-sensitive, opened by superoxide radicals, H2O2 and S-nitrosothiols, and closed by thiol reductants and NADPH [25,44]. As a result, they participate in an elegant redox-sensitive pathway regulating mitochondrial oxidant production. This, added to the volume modulating functions of MitoKATP, is probably the most important physiological role for this channel (Figure 2).
While MitoKATP opening effects are sufficient to significantly impact mitochondrial oxidant production, the small changes in ΔΨm are insufficient to change isolated mitochondrial Ca2+ uptake under physiological respiring conditions. However, Ca2+ uptake is decreased by MitoKATP activity in non-respiring mitochondria in which ΔΨm is supported by the reverse activity of the ATP synthase [52,53]. Excessive Ca2+ entry can lead to accumulation of the ion in the mitochondrial matrix and opening of the mitochondrial permeability transition pore (MPTP). This causes a disruption of mitochondrial inner membrane integrity that promotes cell death [54,55]. Since MPTP opening is facilitated by oxidation of protein thiols [12] and enhanced mitochondrial oxidant production [11], it is not surprising that there is evidence to support that MitoKATP activation can prevent MPTP opening [56].
3.4. Beneficial Effects of MitoKATP Activators under Pathological Conditions
MPTP opening leads to loss of oxidative phosphorylation capacity as well as release of pro-death mitochondrial proteins, and is a cause of ischemic tissue damage [57]. MitoKATP’s protective effects against MPTP are certainly part of the reason why KCOs are strongly protective against ischemic damage in the heart and other tissues [15,31].
During ischemia, the interruption of proper blood flow leads to a decrease in oxygen supply that suppresses the activity of the electron transport chain; consequently, proton motive force is quickly dissipated. Under these conditions, ATP synthase can work its reverse activity and hydrolyze ATP, an activity that partially re-establishes ΔΨm [58,59]. As cytosolic Ca2+ concentration rises during ischemia [60], this increase in the driving force (ΔΨm) can promote mitochondrial Ca2+ accumulation and lead to MPTP opening. Upon reperfusion, cardiac cells are known to present an increase in oxidant production induced by ischemia [61], which also favors MPTP opening.
Activation of MitoKATP has been proposed to block ischemic damage by maintaining proper membrane transport properties and preventing ATP entry to the mitochondrial matrix [44]. This blocks ATP hydrolysis by the reverse activity of the ATP synthase, with transient re-establishment of ΔΨm that could prevent ATP loss and excessive mitochondrial Ca2+ accumulation [62]. Moreover, pharmacological activators of MitoKATP can also block excessive production of oxidants in mitochondria, preventing myocadiac ischemic damage. The highly specific MitoKATP activator BMS-191095 was also shown to additionally act to prevent ischemic damage beyond cardiac cell effects by inhibiting platelet aggregation [32].
Ischemic protection by MitoKATP opening occurs not only in the heart: results in murine neurons and brain [63] show that diazoxide can protect these cells against hypoxic damage associated with ischemia by preventing repolarization of mitochondria [31].
MitoKATP-mediated protection is also not limited to ischemia reperfusion. Indeed, diazoxide treatment also protected murine heart against hypertrophy by controlling oxidant generation [64], and prevented neuronal injury in models of the metabolic disease methylmalonic acidemia by preventing MPTP opening [65,66]. Overall, given its effects modulating mitochondrial redox state and preventing MPTP without overt changes in oxidative phosphorylation, MitoKATP can act as an effective protective pathway under many different pathological conditions.
Indeed, protective mechanisms mediated by MitoKATP are also part of endogenous signaling pathways. Preconditioning is a protective intervention where hearts undergo brief and non-damaging periods of ischemia [45], which significantly decrease damage associated with a subsequent larger and typically damaging ischemic event. The protection promoted by preconditioning can be blocked by MitoKATP inhibitors [33,34,67,68], indicating that this protein actively participates in this process. Another key protein in preconditioning signaling is protein kinase C (PKC). PKC activation leads to an increase in MitoKATP activity, and PMA, an activator of PKC, has been shown to indirectly promote opening of MitoKATP in intact cells [42].
4. Inhibiting MitoKATP
Prior to the identification of MitoKATP as their receptor, molecules such as glyburide (also known as glybenclamide) and 5-hydroxydecanoate (5-HD) were known to reverse ischemic protection induced by KCOs or preconditioning [67,68]. Interestingly, after MitoKATP was identified, initial studies in isolated mitochondria failed to see selective inhibition of MitoKATP by these compounds [69]. Later studies showed that, counterintuitively, MitoKATP could not be inhibited pharmacologically when open by the absence of physiological inhibitors. Instead, channel modulation by glyburide and 5-HD required incubation in the presence of physiological inhibitors such as ATP and Mg2+ or long chain acyl-CoA, in addition to the presence of either physiological activators, such as guanine nucleotides, or pharmacological agonists, such as diazoxide [70]. Under these appropriate conditions, both 5-HD and glyburide were shown to effectively close the channel.
Because of its selectivity, 5-HD became the most employed tool as a MitoKATP antagonist. Both molecules are proposed to act through the MitoSUR subunit and cannot inhibit MitoKIR alone [71]. This is expected for glyburide, considering it is a sulphonylurea [71]. 4-aminopyridine, a general K+ inhibitor, requires much higher concentrations than all other inhibitors listed in order to function on MitoKATP [20]. Another well-described modulator known to act directly on the MitoKIR subunit is tetraphenylphosphonium (TPP+) [49], although this is not a selective inhibitor, and has been shown to act in CellKATP’s as well [72]. This is of interest, since TPP+ is often used to monitor mitochondrial inner membrane potentials with electrodes [73]. Information on MitoKATP inhibitors mentioned here are listed in Table 2.
5. Modulation of the Mitochondrial K+/H+ Exchanger
The activity of a mitochondrial K+/H+ exchanger has been predicted since the proposal of the chemiosmotic theory [18]. An exit route is necessary for K+, since it would be predicted to leak across the inner membrane in the presence of the electrochemical gradient. Experimentally, K+/H+ exchange was initially observed as K+ efflux from mitochondria caused by suspending these organelles in hypotonic sucrose [74]. This already indicates the importance of this pathway in maintaining mitochondrial structure and counterbalancing osmotic swelling caused by K+ uptake.
Physiologically, K+/H+ exchange activity is known to be reversibly inhibited by intramitochondrial Mg2+ [75]. When K+ enters the matrix through leakage or MitoKATP, it is accompanied by osmotically obligated water, promoting swelling. This dilutes intramitochondrial Mg2+ and activates K+/H+ exchange, ensuring volume homeostasis [75].
Some tools have been proposed in the literature to control exchanger activity. Quinine and quinacrine are reversible inhibitors at relatively low concentrations [76], while N,N’-dicyclohexylcarbodiimide (DCCD) can irreversibly inactivate the exchanger [77]. DCCD is a non-selective drug that reacts with carboxylic groups in several bioenergetically relevant targets, including other ion transport proteins [78], while quinine and quinacrine are K+ transport modulators, but do not have strict selectivity for mitochondrial K+/H+ exchange. Interestingly, DCCD only reacts with the active mitochondrial K+/H+ exchanger; therefore Mg2+ and quinine protect the exchanger from DCCD inactivation [79].
Mitochondrial K+/H+ exchange inhibitors block excessive mitochondrial contraction in isolated mitochondria [76]. Although this suggests an interesting role of the exchanger in maintaining mitochondrial architecture and preserving inner membrane transport properties, its potential as a target for therapeutical interventions has not yet been explored, mostly due to the lack of specific pharmacological tools to modulate this transport and the lack of knowledge regarding the molecular identity of the exchanger.
6. Conclusions
Considering the central relevance that mitochondria have in nearly every cell in the human body, the mitochondrial K+ cycle, and more specifically MitoKATP as a key component of it, are powerful targets to control key mitochondrial functions, including the preservation of volume, architecture and redox state. Given the already numerous known MitoKATP pharmacological regulators and the potential for new drug discovery and design, as well as the recent molecular identification of the channel protein subunits, we believe the applications for mitochondrial KCOs will expand largely in the near future.
Author Contributions
Both authors participated in manuscript preparation. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by grants #2020/06970-5 and #2013/07937-8 from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Centro de Pesquisa, Inovação e Difusão de Processos Redox em Biomedicina (CEPID Redoxoma), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). O.R.P., Jr. is supported by CAPES and FAPESP fellowships.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Nicholls, D.G.; Ferguson, S.J. Quantitative Bioenergetics. Bioenergetics 2013, 27–51. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.R.; Chausse, B.; Da Cunha, F.M.; Luévano-Martínez, L.A.; Marazzi, T.B.M.; Pessoa, P.S.; Queliconi, B.B.; Kowaltowski, A.J. Mitochondrial compartmentalization of redox processes. Free Radic. Biol. Med. 2012, 52, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium transport and signaling in mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Vercesi, A.; Bababunmi, E.A. Regulation of Ca2+ release from mitochondria by the oxidation reduction state of pyridine nucleotides. Proc. Natl. Acad. Sci. USA 1978, 75, 1690–1694. [Google Scholar] [CrossRef] [Green Version]
- Vercesi, A.E.; Castilho, R.F.; Kowaltowski, A.J.; de Oliveira, H.C.F.; de Souza-Pinto, N.C.; Figueira, T.R.; Busanello, E.N.B. Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic. Biol. Med. 2018, 129, 1–24. [Google Scholar] [CrossRef]
- Gogvadze, V.; Schweizer, M.; Richter, C. Control of the pyridine nucleotide-linked Ca2+ release from mitochondria by respiratory substrates. Cell Calcium 1996, 19, 521–526. [Google Scholar] [CrossRef]
- Singh, B.K.; Tripathi, M.; Pandey, P.K.; Kakkar, P. Alteration in mitochondrial thiol enhances calcium ion dependent membrane permeability transition and dysfunction in vitro: A cross-talk between mtThiol, Ca2+, and ROS. Mol. Cell. Biochem. 2011, 357, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996, 184, 1331–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Oliveira, M.F. Mitochondria: New developments in pathophysiology. Mol. Aspects Med. 2020, 71. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K+ cycle. Biochim. Biophys. Acta Bioenerg. 2003, 1606, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- DiResta, D.J.; Kutschke, K.P.; Hottois, M.D.; Garlid, K.D. K+, H+ exchange and volume homeostasis in brown adipose tissue mitochondria. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1986, 251. [Google Scholar] [CrossRef]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Dołowy, K.; Szewczyk, A. Matrix Mg2+ regulates mitochondrial ATP-dependent potassium channel from heart. FEBS Lett. 2005, 579, 1625–1632. [Google Scholar] [CrossRef] [Green Version]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar] [CrossRef]
- Szewczyk, A.; Mikołajek, B.; Pikuła, S.; Nałecz, M.J. Potassium channel openers induce mitochondrial matrix volume changes via activation of ATP-sensitive K+ channel. Pol. J. Pharmacol. 1993, 45, 437–443. [Google Scholar] [PubMed]
- Paucek, P.; Yarov-Yarovoy, V.; Sun, X.; Garlid, K.D. Inhibition of the mitochondrial K(ATP) channel by long-chain acyl-CoA esters and activation by guanine nucleotides. J. Biol. Chem. 1996, 271, 32084–32088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facundo, H.T.F.; de Paula, J.G.; Kowaltowski, A.J. Mitochondrial ATP-sensitive K+ channels are redox-sensitive pathways that control reactive oxygen species production. Free Radic. Biol. Med. 2007, 42, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Bajgar, R.; Seetharaman, S.; Kowaltowski, A.J.; Garlid, K.D.; Paucek, P. Identification and Properties of a Novel Intracellular (Mitochondrial) ATP-sensitive Potassium Channel in Brain. J. Biol. Chem. 2001, 276, 33369–33374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Sun, X.; Schindler, P.A. The mitochondrial KATP channel as a receptor for potassium channel openers. J. Biol. Chem. 1996, 271, 8796–8799. [Google Scholar] [CrossRef] [Green Version]
- Mironova, G.D.; Skarga, Y.Y.; Grigoriev, S.M.; Negoda, A.E.; Kolomytkin, O.V.; Marinov, B.S. Reconstitution of the mitochondrial ATP-dependent potassium channel into bilayer lipid membrane. J. Bioenerg. Biomembr. 1999, 31, 159–163. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Alberici, L.C.; Paim, B.A.; Zecchin, K.G.; Mirandola, S.R.; Pestana, C.R.; Castilho, R.F.; Vercesi, A.E.; Oliveira, H.C.F. Activation of the mitochondrial ATP-sensitive K+ channel reduces apoptosis of spleen mononuclear cells induced by hyperlipidemia. Lipids Health Dis. 2013, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Lu, C.; Wan, R.; Auyeung, W.W.; Mattson, M.P. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 2002, 22, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.R.; Park, J.W.; Jung, I.S.; Yi, K.Y.; Yoo, S.E.; Chung, H.J.; Yun, Y.P.; Kwon, S.H.; Shin, H.S. BMS-191095, a cardioselective mitochondrial KATP opener, inhibits human platelet aggregation by opening mitochondrial KATP channels. Arch. Pharm. Res. 2005, 28, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R. KATP channel openers: Structure-activity relationships and therapeutic potential. Med. Res. Rev. 2004, 24, 213–266. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Rapposelli, S.; Calderone, V. Cardiac ATP-Sensitive Potassium Channels: A Potential Target for an Anti-Ischaemic Pharmacological Strategy. Cardiovasc. Hematol. Agents Med. Chem. 2012, 5, 79–90. [Google Scholar] [CrossRef]
- Nichols, C.G.; Lederer, W.J. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am. J. Physiol. Hear. Circ. Physiol. 1991, 261, 30–36. [Google Scholar] [CrossRef]
- Ashcroft, S.J.H.; Ashcroft, F.M. Properties and functions of ATP-sensitive K-channels. Cell. Signal. 1990, 2, 197–214. [Google Scholar] [CrossRef]
- Grover, G.J.; D’Alonzo, A.J.; Garlid, K.D.; Bajgar, R.; Lodge, N.J.; Sleph, P.G.; Darbenzio, R.B.; Hess, T.A.; Smith, M.A.; Paucek, P.; et al. Pharmacologic characterization of BMS-191095, a mitochondrial KATP opener with no peripheral vasodilator or cardiac action potential shortening activity. J. Pharmacol. Exp. Ther. 2001, 297, 1184–1192. [Google Scholar]
- Grover, G.J.; D’Alonzo, A.J.; Darbenzio, R.B.; Parham, C.S.; Hess, T.A.; Bathala, M.S. In vivo characterization of the mitochondrial selective kATP opener (3R)-trans-4-((4-chlorophenyl)-N-(1H-imidazol-2-ylmethyl) dimethyl-2H-1-benzopyran-6-carbonitril monohydrochloride (BMS-191095): Cardioprotective, hemodynamic, and electrophysiological ef. J. Pharmacol. Exp. Ther. 2002, 303, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Mironova, G.D.; Negoda, A.E.; Marinov, B.S.; Paucek, P.; Costa, A.D.T.; Grigoriev, S.M.; Skarga, Y.Y.; Garlid, K.D. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J. Biol. Chem. 2004, 279, 32562–32568. [Google Scholar] [CrossRef] [Green Version]
- Crestanello, J.A.; Doliba, N.M.; Babsky, A.M.; Doliba, N.M.; Niibori, K.; Osbakken, M.D.; Whitman, G.J.R. Opening of potassium channels protects mitochondrial function from calcium overload. J. Surg. Res. 2000, 94, 116–123. [Google Scholar] [CrossRef]
- Teshima, Y.; Akao, M.; Baumgartner, W.A.; Marbán, E. Nicorandil prevents oxidative stress-induced apoptosis in neurons by activating mitochondrial ATP-sensitive potassium channels. Brain Res. 2003, 990, 45–50. [Google Scholar] [CrossRef]
- Sato, T.; O’Rourke, B.; Marbán, E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ. Res. 1998, 83, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtovich, A.P.; Brookes, P.S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, P.; Kowaltowski, A.J.; Laclau, M.N.; Seetharaman, S.; Paucek, P.; Boudina, S.; Thambo, J.B.; Tariosse, L.; Garlid, K.D. Mechanisms by which opening the mitochondrial ATP-sensitive K+ channel protects the ischemic heart. Am. J. Physiol. Hear. Circ. Physiol. 2002, 283, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, R.S.; Facundo, H.T.F.; Kowaltowski, A.J. Mitochondrial K+ transport and cardiac protection during ischemia/reperfusion. Braz. J. Med. Biol. Res. 2005, 38, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queliconi, B.B.; Wojtovich, A.P.; Nadtochiy, S.M.; Kowaltowski, A.J.; Brookes, P.S. Redox regulation of the mitochondrial KATP channel in cardioprotection. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1309–1315. [Google Scholar] [CrossRef]
- Ferranti, R.; Da Silva, M.M.; Kowaltowski, A.J. Mitochondrial ATP-sensitive K+ channel opening decreases reactive oxygen species generation. FEBS Lett. 2003, 536, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Beavis, A.D.; Brannan, R.D.; Garlid, K.D. Swelling and contraction of the mitochondrial matrix. I. A structural interpretation of the relationship between light scattering and matrix volume. J. Biol. Chem. 1985, 260, 13424–13433. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Seetharaman, S.; Paucek, P.; Garlid, K.D. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am. J. Physiol. Hear Circ. Physiol. 2001, 280, 649–657. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Tahara, E.B.; Navarete, F.D.T.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Wong, R.; Steenbergen, C.; Murphy, E. Mitochondrial permeability transition pore and calcium handling. Methods Mol. Biol. 2012, 810, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes-Filho, S.L.; Amigo, I.; Prado, F.M.; Ferreira, N.C.; Koike, M.K.; Pinto, I.F.D.; Miyamoto, S.; Montero, E.F.S.; Medeiros, M.H.G.; Kowaltowski, A.J. Caloric restriction protects livers from ischemia/reperfusion damage by preventing Ca2+-induced mitochondrial permeability transition. Free Radic. Biol. Med. 2017, 110, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.T.; Jakob, R.; Costa, C.L.; Andrukhiv, K.; West, I.C.; Garlid, K.D. The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J. Biol. Chem. 2006, 281, 20801–20808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Rego, A.C.; Vesce, S.; Nicholls, D.G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1-7 neural cells. Cell Death Differ. 2001, 8, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Classen, J.B.; Mergner, W.J.; Costa, M. ATP hydrolysis by ischemic Mitochondria. J. Cell. Physiol. 1989, 141, 53–59. [Google Scholar] [CrossRef]
- Kihara, Y.; Grossman, W.; Morgan, J.P. Direct measurement of changes in intracellular calcium transients during hypoxia, ischemia, and reperfusion of the intact mammalian heart. Circ. Res. 1989, 65, 1029–1044. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, M.M.; Sartori, A.; Belisle, E.; Kowaltowski, A.J. Ischemic preconditioning inhibits mitochondrial respiration, increases H2O2 release, and enhances K+ transport. Am. J. Physiol. Hear Circ. Physiol. 2003, 285, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korge, P.; Honda, H.M.; Weiss, J.N. Protection of cardiac mitochondria by diazoxide and protein kinase C: Implications for ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2002, 99, 3312–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Lacza, Z.; Rajapakse, N.; Horiguchi, T.; Snipes, J.; Busija, D.W. MitoKATP opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am. J. Physiol. Hear Circ. Physiol. 2002, 283, 1005–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, A.M.B.; de Lacerda Alexandre, J.V.; Araújo, M.T.S.; David, C.E.B.; Ponte Viana, Y.I.; Coelho, B.N.; Caldas, F.R.L.; Varela, A.L.N.; Kowaltowski, A.J.; Facundo, H.T. Diazoxide Modulates Cardiac Hypertrophy by Targeting H2O2 Generation and Mitochondrial Superoxide Dismutase Activity. Curr. Mol. Pharmacol. 2020, 13, 76–83. [Google Scholar] [CrossRef]
- Maciel, E.N.; Kowaltowski, A.J.; Schwalm, F.D.; Rodrigues, J.M.; Souza, D.O.; Vercesi, A.E.; Wajner, M.; Castilho, R.F. Mitochondrial permeability transition in neuronal damage promoted by Ca2+ and respiratory chain complex II inhibition. J. Neurochem. 2004, 90, 1025–1035. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Maciel, E.N.; Fornazari, M.; Castilho, R.F. Diazoxide protects against methylmalonate-induced neuronal toxicity. Exp. Neurol. 2006, 201, 165–171. [Google Scholar] [CrossRef]
- Toombs, C.F.; Moore, T.L.; Shebuski, R.J. Limitation of infarct size in the rabbit by ischaemic preconditioning is reversible with glibenclamide. Cardiovasc. Res. 1993, 27, 617–622. [Google Scholar] [CrossRef]
- Auchampach, J.A.; Grover, G.J.; Gross, G.J. Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovasc. Res. 1992, 26, 1054–1062. [Google Scholar] [CrossRef]
- Beavis, A.D.; Lu, Y.; Garlid, K.D. On the regulation of K+ uniport in intact mitochondria by adenine nucleotides and nucleotide analogs. J. Biol. Chem. 1993, 268, 997–1004. [Google Scholar] [CrossRef]
- Jabůrek, M.; Yarov-Yarovoy, V.; Paucek, P.; Garlid, K.D. State-dependent inhibition of the mitochondrial K(ATP) channel by glyburide and 5-hydroxydecanoate. J. Biol. Chem. 1998, 273, 13578–13582. [Google Scholar] [CrossRef]
- Szewczyk, A.; Wójcik, G.; Lobanov, N.A.; Nalecz, M.J. The mitochondrial sulfonylurea receptor: Identification and characterization. Biochem. Biophys. Res. Commun. 1997, 230, 611–615. [Google Scholar] [CrossRef]
- Zhang, H.; Bolton, T.B.; Piekarska, A.E.; McPherson, G.A. The electrophysiological effects of tetraphenylphosphonium on vascular smooth muscle. Eur. J. Pharmacol. 1998, 347, 119–123. [Google Scholar] [CrossRef]
- Kamo, N.; Muratsugu, M.; Hongoh, R.; Kobatake, Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 1979, 49, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D. Unmasking the mitochondrial K H exchanger: Swelling-induced K+-loss. Biochem. Biophys. Res. Commun. 1978, 83, 1450–1455. [Google Scholar] [CrossRef]
- Garlid, K.D. On the mechanism of regulation of the mitochondrial K+/H+ exchanger. J. Biol. Chem. 1980, 255, 11273–11279. [Google Scholar] [CrossRef]
- Jung, D.W.; Farooqui, T.; Utz, E.; Brierley, G.P. Effects of quinine on K+ transport in heart mitochondria. J. Bioenerg. Biomembr. 1984, 16, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; DiResta, D.J.; Beavis, A.D.; Martin, W.H. On the mechanism by which dicyclohexylcarbodiimide and quinine inhibit K+ transport in rat liver mitochondria. J. Biol. Chem. 1986, 261, 1529–1534. [Google Scholar] [CrossRef]
- Azzi, A.; Casey, R.P.; Nałęcz, M.J. The effect of N,N′-dicyclohexylcarbodiimide on enzymes of bioenergetic relevance. BBA Rev. Bioenerg. 1984, 768, 209–226. [Google Scholar] [CrossRef]
- Martin, W.H.; DiResta, D.J.; Garlid, K.D. Kinetics of inhibition and binding of dicyclohexylcarbodiimide to the 82,000-dalton-mitochondrial K+/H+ antiporter. J. Biol. Chem. 1986, 261, 12300–12305. [Google Scholar] [CrossRef]
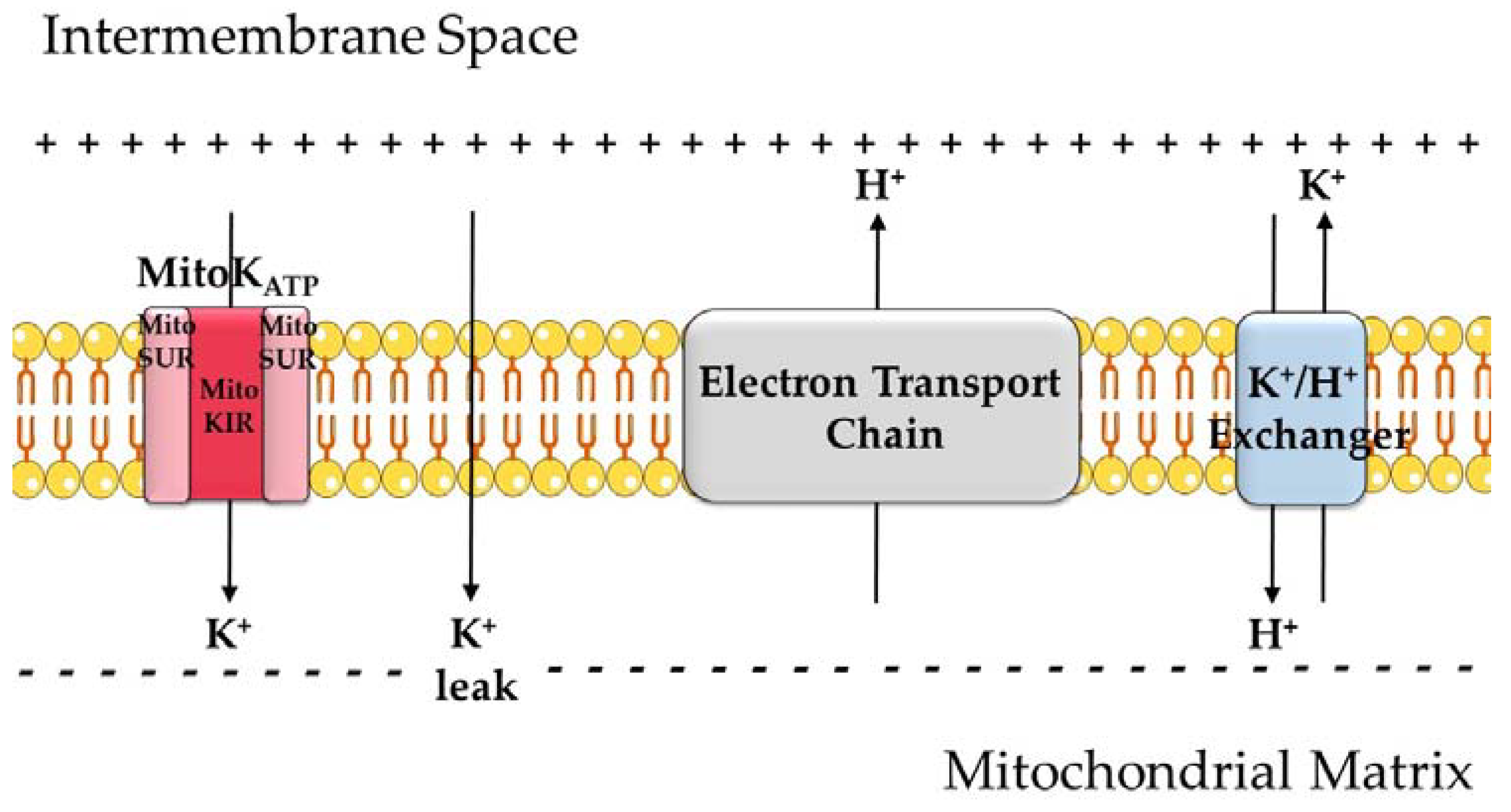
Figure 1.
An overview of mitochondrial K+ transport. Because of proton pumping by the electron transport chain (ETC) coupled to the reduction of oxygen and oxidation of coenzymes, mitochondria have an electrochemical gradient, with a negatively charged matrix. K+ leak across the inner membrane is quantitatively relevant due to the high concentrations of this ion in the cytosol and this electrochemical gradient. K+ ions can also enter the matrix through the ATP-sensitive K+ channel (KATP or MitoKATP). The channel is formed by two subunits: MitoKIR (or MitoK) and MitoSUR. K+ is removed from the matrix in exchange for H+.
Figure 1.
An overview of mitochondrial K+ transport. Because of proton pumping by the electron transport chain (ETC) coupled to the reduction of oxygen and oxidation of coenzymes, mitochondria have an electrochemical gradient, with a negatively charged matrix. K+ leak across the inner membrane is quantitatively relevant due to the high concentrations of this ion in the cytosol and this electrochemical gradient. K+ ions can also enter the matrix through the ATP-sensitive K+ channel (KATP or MitoKATP). The channel is formed by two subunits: MitoKIR (or MitoK) and MitoSUR. K+ is removed from the matrix in exchange for H+.

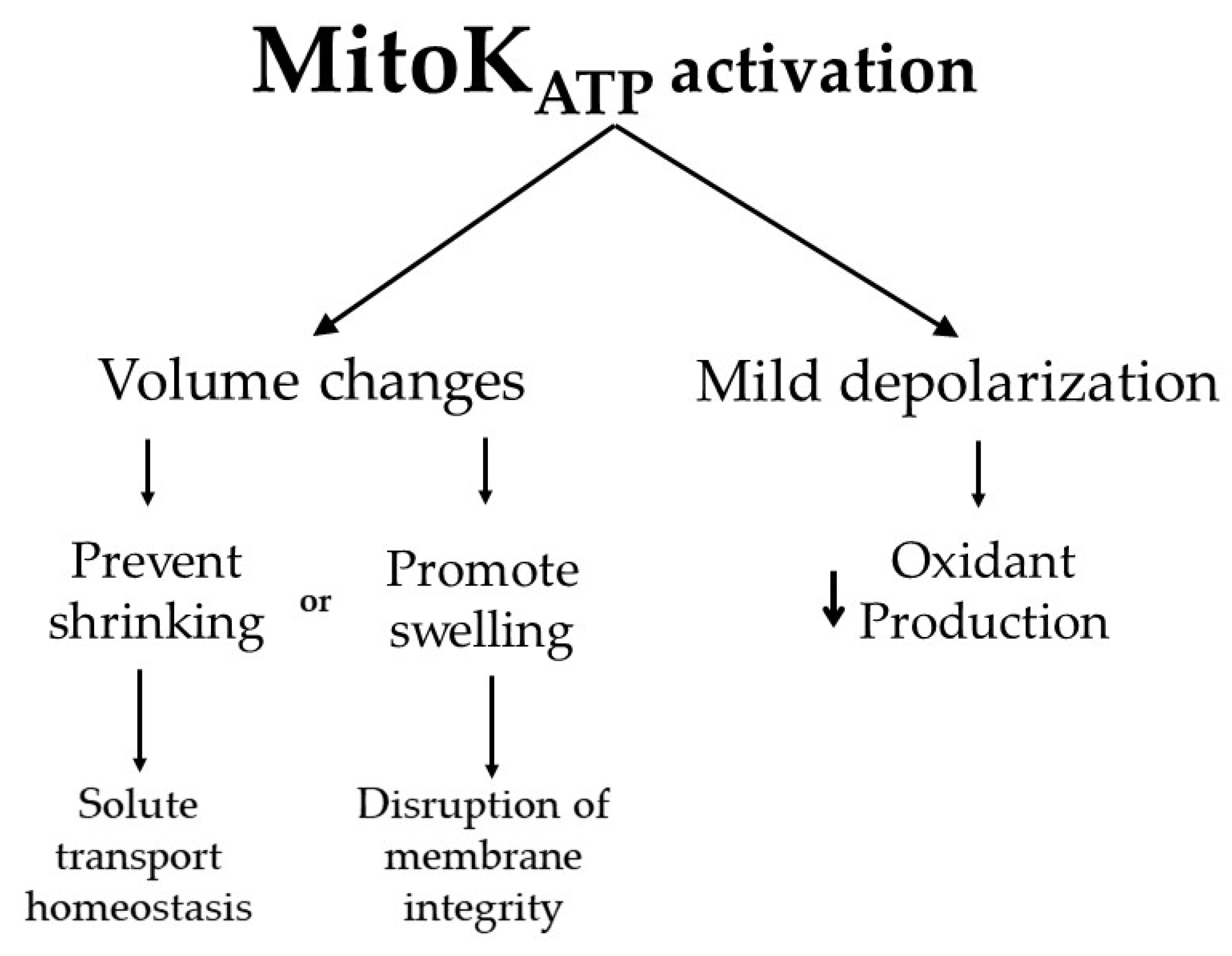
Figure 2.
Functional consequences of MitoKATP activation. K+ entry in the mitochondrial matrix through MitoKATP leads to the uptake of water, which changes mitochondrial volume. This can be important to maintain proper membrane transport properties, while in extreme conditions, excessive swelling (often not specifically a consequence of MitoKATP activity) can lead to membrane integrity disruption. Dilution of matrix components by water uptake also leads to the activation of K+ exit in exchange for protons, which leads to proton exit as the net product, mildly uncoupling mitochondria. This uncoupling prevents oxidant production by the electron transport chain.
Figure 2.
Functional consequences of MitoKATP activation. K+ entry in the mitochondrial matrix through MitoKATP leads to the uptake of water, which changes mitochondrial volume. This can be important to maintain proper membrane transport properties, while in extreme conditions, excessive swelling (often not specifically a consequence of MitoKATP activity) can lead to membrane integrity disruption. Dilution of matrix components by water uptake also leads to the activation of K+ exit in exchange for protons, which leads to proton exit as the net product, mildly uncoupling mitochondria. This uncoupling prevents oxidant production by the electron transport chain.


{kind=link}
{kind=link}
Table 1.
MitoKATP activators.
| Compound Name | Effective Concentrations (μM) | References |
|---|---|---|
| Diazoxide | 30 | Garlid et al., 1997 [15] |
| Pinacidil | 100 | Crestanello et al., 2000 [40] |
| Nicorandil | 100 | Teshima et al., 2003 [41] |
| Cromakalim | 30 | Garlid et al., 1997 [15] |
| BMS191095 | 10 | Grover et al., 2001 [38] |
| p-diethylaminoetylbenzoate | 100 | Mironova et al., 2004 [39] |
| phorbol 12-myristate 13-acetate (PMA) | 0.2 | Sato et al., 1998 [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pereira, O., Jr.; Kowaltowski, A.J. Mitochondrial K+ Transport: Modulation and Functional Consequences. Molecules 2021, 26, 2935. https://doi.org/10.3390/molecules26102935
AMA Style
Pereira O Jr., Kowaltowski AJ. Mitochondrial K+ Transport: Modulation and Functional Consequences. Molecules. 2021; 26(10):2935. https://doi.org/10.3390/molecules26102935
Chicago/Turabian StylePereira, Osvaldo, Jr., and Alicia J. Kowaltowski. 2021. "Mitochondrial K+ Transport: Modulation and Functional Consequences" Molecules 26, no. 10: 2935. https://doi.org/10.3390/molecules26102935