
Microwaves as “Co-Catalysts” or as Substitute for Catalysts in Organophosphorus Chemistry


Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary
Molecules 2021, 26(4), 1196; https://doi.org/10.3390/molecules26041196
Submission received: 10 February 2021
/
Revised: 18 February 2021
/
Accepted: 19 February 2021
/
Published: 23 February 2021
(This article belongs to the Special Issue Catalysts for a Green Chemistry: A Themed Issue Honoring Professor Roger Sheldon for His Contribution in the Field of the Green Chemistry with the Introduction of the E Factor)
Abstract
:The purpose of this review is to summarize the importance of microwave (MW) irradiation as a kind of catalyst in organophosphorus chemistry. Slow or reluctant reactions, such as the Diels-Alder cycloaddition or an inverse-Wittig type reaction, may be performed efficiently under MW irradiation. The direct esterification of phosphinic and phosphonic acids, which is practically impossible on conventional heating, may be realized under MW conditions. Ionic liquid additives may promote further esterifications. The opposite reaction, the hydrolysis of P-esters, has also relevance among the MW-assisted transformations. A typical case is when the catalysts are substituted by MWs, which is exemplified by the reduction of phosphine oxides, and by the Kabachnik–Fields condensation affording α-aminophosphonic derivatives. Finally, the Hirao P–C coupling reaction may serve as an example, when the catalyst may be simplified under MW conditions. All of the examples discussed fulfill the expectations of green chemistry.
The spread of the microwave (MW) technique opened a new chapter in organic chemistry in whole [1,2,3], and also in its specialized fields, such as heterocyclic [3,4,5] and organophosphorus chemistry [2,6]. MW irradiation makes possible more efficient syntheses in terms of reaction time, selectivity, and purity [1,2]. The role of MWs in organic syntheses provoked sharp disputes, however, today there is an agreement that thermal effects are responsible for the beneficial effect of MWs [7,8]. A widely accepted concept is that the efficiency of the MW irradiation is the consequence of the statistically occurring local overheating in the bulk of the mixture [9,10,11].
During our work, we laid the stress on green chemical aspects within organophosphorus chemistry [12]. This will be summarized in this review article. Besides developing efficient syntheses, we could enhance reluctant or slow reactions by applying the MW irradiation (1). It was another option to promote further certain MW-assisted reactions by performing them in the presence of an ionic liquid additive/catalyst (2). It is another possibility to substitute certain catalysts by MW irradiation (3). Last but not least, MWs may simplify catalytic systems/catalysts (4). In this paper, options (1)–(4) will be discussed in detail, placing the particular reactions into literature context.
1. Microwave Irradiation Allowing the Inverse-Wittig Type Reaction That Is Reluctant on Conventional Heating
The Diels–Alder reaction of 1-phenyl-1,2-dihydrophosphinine oxide 1 with dienophiles, like N-phenylmaleimide and dimethyl acetylenedicarboxylate (DMAD), resulted in the formation of the respective phosphabicyclo[2.2.2]octene derivatives (2) and (3). The microwave (MW) technique was useful in shortening the reaction times and making the syntheses efficient. The MW-promoted reactions were 25 times faster than the thermal variations (Scheme 1) [13].
However, the 1-(2,4,6-triisopropylphenyl-1,2-dihydrophosphinine) oxide (4) [14] underwent an inverse-Wittig type transformation in reaction with DMAD to afford the corresponding β-oxophosphorane 5 (Scheme 2) [15,16].
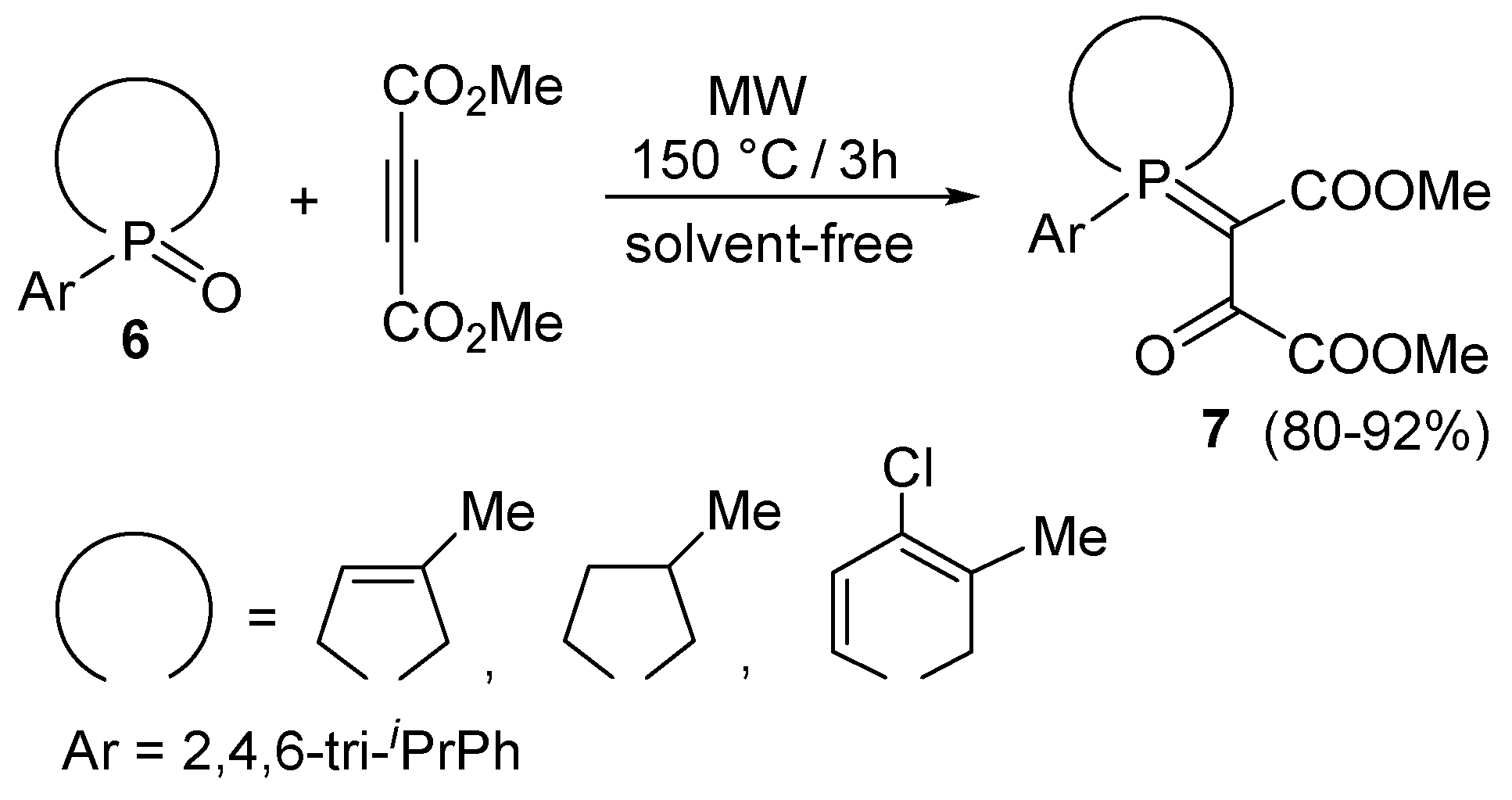
Then, the new protocol seemed to be of a more general value [17]. The use of the MW technique was advantageous not only in the inverse Wittig-type reaction of 2,4,6-triisopropylphenyl-3-phospholene oxide but also in the reaction of 2,4,6-triisopropylphenylphospholane oxide and 2,4,6-triisopropylphenyl-1,2-dihydrophosphinine oxide (all represented by formula 6) to afford β-oxophosphoranes 7 (Scheme 3) [18,19].
In the novel inverse Wittig-type reactions, MW irradiation made possible reactions at 150 °C for 3 h that were rather reluctant on conventional heating. Hence, MW acted as a kind of catalyst.
In the above case, the nature of the 1,2-dihydrophosphinine oxide determined its reactivity towards DMAD. Moreover, both the Diels–Alder cycloaddition and the inverse-Wittig type reaction took place efficiently on MW irradiation without the application of any catalyst.
2. Microwave-Assisted, Ionic Liquid-Catalyzed Direct Esterification of P-Oxoacids That Is Otherwise Impossible under Thermal Conditions
Phosphinic and phosphonic acids cannot be involved in direct esterification under common conditions. Only a few examples are known for the direct esterification of P-acids. These reactions required forcing conditions and were not efficient [22,23,24,25,26]. However, it was found by us that the P-acids can be esterified on MW irradiation.
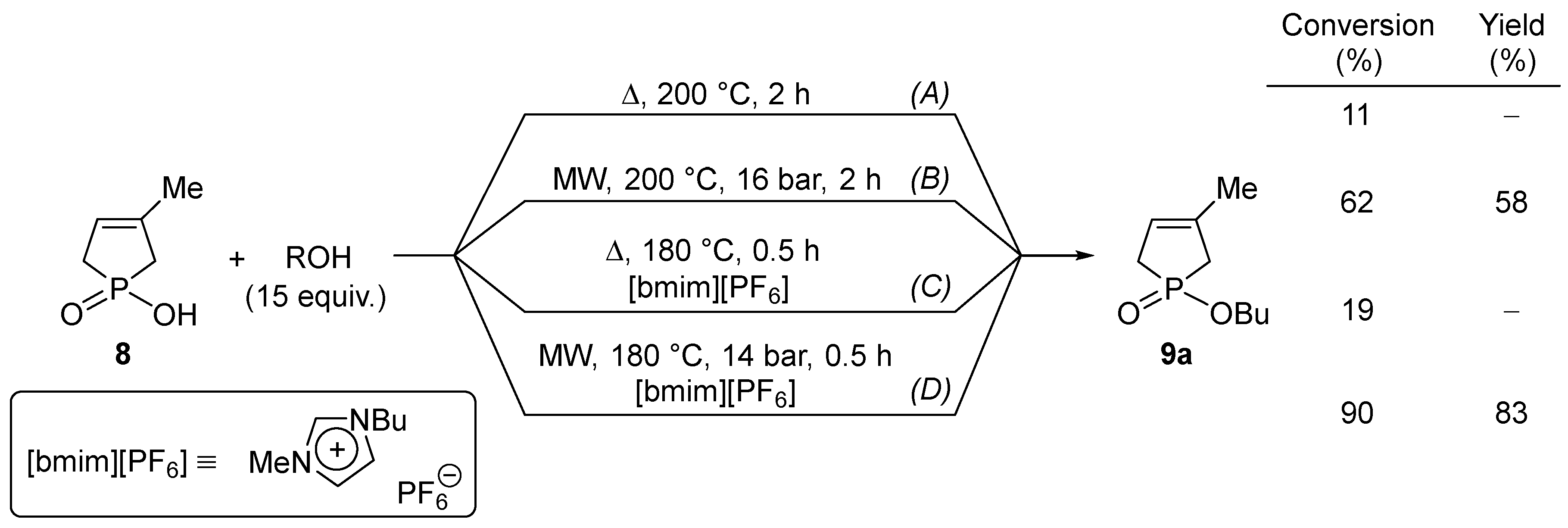
The esterification of 1-hydroxy-3-phospholene oxide (8) with a series of alcohols at 180–235 °C afforded the alkyl phosphinates (9) in yields of 71–95% (Scheme 4/B vs. A, Table 1/Entries 1, 3, 5, 7, 9 and 11) [27,28,29]. The esterifications were less efficient with volatile and sterically hindered alcohols. The relatively high temperature required means a limitation that may be overcome by applying ionic liquids (ILs) as catalysts. It was found that in the presence of 10% of [bmim][PF6] as an additive, the esterifications took place at a lower temperature, and became faster and more efficient in shorter reaction times (Scheme 4/D, Table 1/Entries 2, 4, 6, 8, 10 and 12) [30]. The thermal direct esterifications were also somewhat promoted by the ionic liquid additive (Scheme 4/C).
Then, the MW-promoted IL-catalyzed direct esterifications were extended to other ring phosphinic acids, like 1-hydroxy-3,4-dimethyl-3-phospholene oxide (10), 1-hydroxy-phospholane oxides (12 and 14), as well as, a 1-hydroxy-1,2,3,4,5,6-hexahydrophosphinine oxide (16) (Table 2) [29,30,31].
Using IL-catalysis, even phenols could be the reactants in the MW-assisted esterification of cyclic phosphinic acids [32].
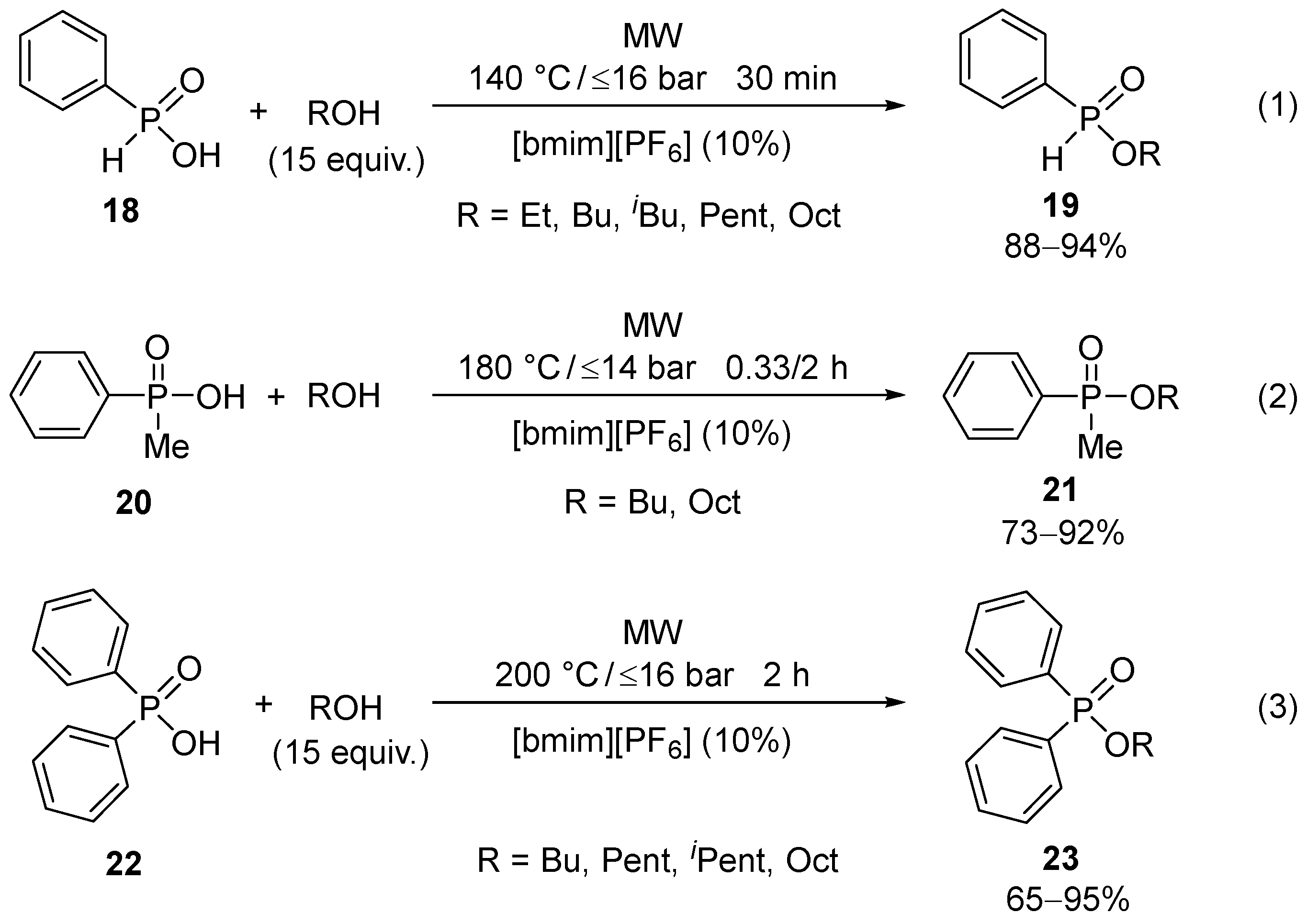
The esterification of the reactive phenyl-H-phosphinic acid (18) took place at a temperature of 160–190 °C to provide the phosphinates (19) in good (73–90%) yields [33]. The presence of [bmim][PF6] had a beneficial effect on the outcome (Scheme 5/(1)) [34]. Methyl-phenylphosphinic acid (20), and what is more, the sterically hindered diphenylphosphinic acid (22), could also be efficiently esterified in the presence of an IL (Scheme 5/(2) and (3)) [34,35].
It is noteworthy that thiobutanol could also be used under MW-assistance to afford the corresponding thiophosphinates [36]; however, the direct amidations of phosphinic acids were reluctant even on MW irradiation [37].
Our next targets were the phosphonic acids. In the first approach, the alkyl phenyl-H-phosphinates (19) were oxidized, then the ester-acid were esterified. However, we could not be satisfied with the efficiency (Scheme 6) [38].
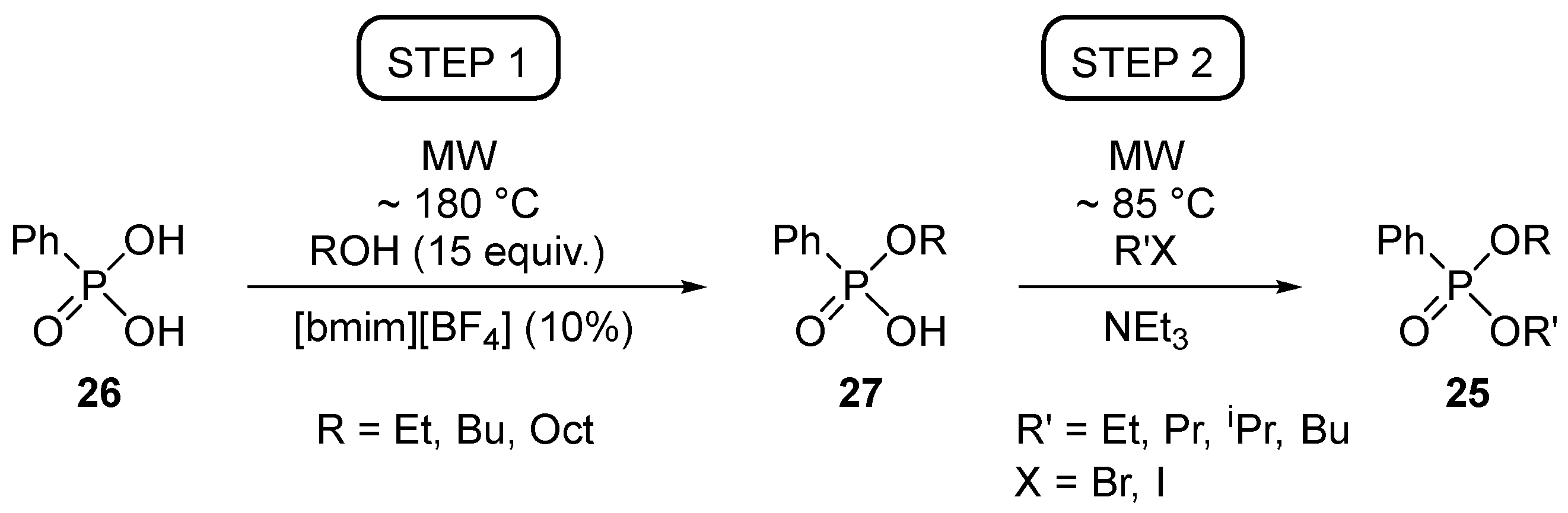
For this, the direct esterification of phenylphosphonic acid was studied in detail. We learned that the MW-assisted and IL-catalyzed protocol furnished the monoesters (27) in good selectivities and in acceptable yields (Table 3). At the same time, the diesterification to species 25 was not efficient (Table 4). [Bmim][PF6] was somewhat less efficient than [bmim][BF4] [34,39].
As an alternative possibility, phenylphosphonic acid (26) was also subjected to alkylating esterifications using BuBr. These reactions were complete and selective for the diesterification only when BuBr was used in a 5-fold quantity at 120 °C (Table 5) [39].
At the same time, the monoalkyl phenylphosphonates (27) could be esterified further in reaction with alkyl halides under MW irradiation (Table 6) [39].
In this way, phenylphosphonates with (two) different alkyl groups (28) could also be prepared (Table 7). The isolated yields of products 28 were mostly around 70% [39].
It follows from our results that the method of choice for the diesterification of phosphonic acid 26 is when the first HO group of the phosphonic acid (26) is esterified with alcohol under MW conditions [34,39], while the second HO function (as in 27) is converted to alkoxy by alkylation using an alkyl halogenide (Scheme 7) [39].
It is noteworthy that the MW-assisted continuous flow esterification of a H-phosphinic acid was also elaborated [40].
We were successful in modeling the rate-enhancing effect of MWs. The esterification of phenyl-H-phosphinic acid and 1-hydroxy-3-methyl-3-phospholene 1-oxide served as the model reactions [41,42].
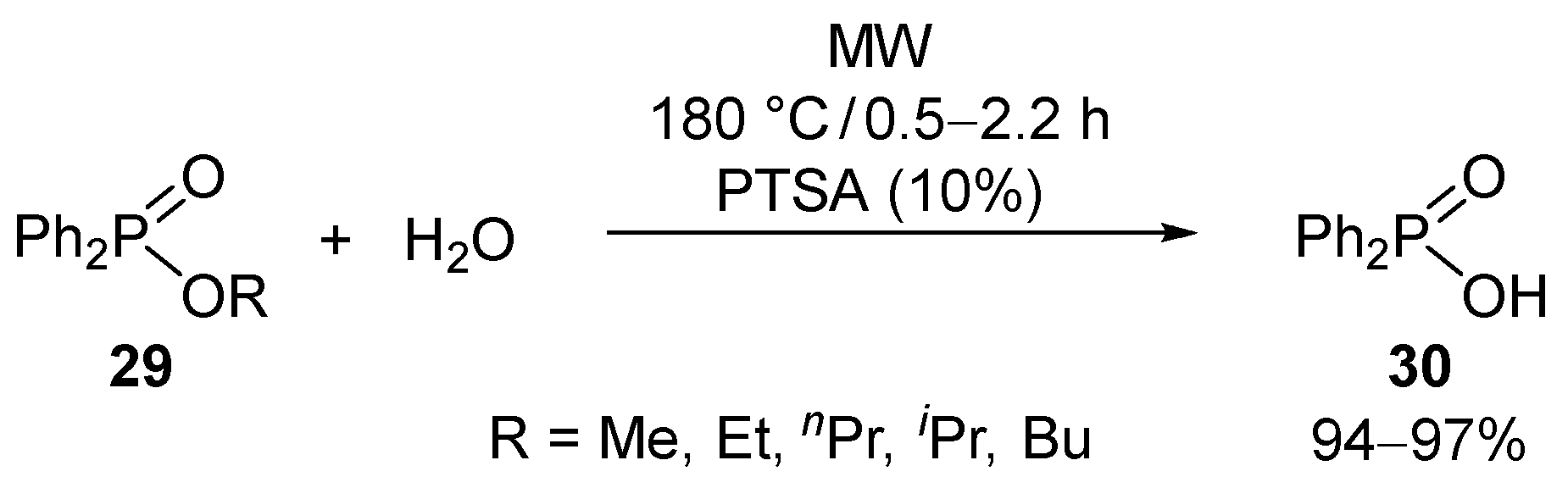
The opposite reaction of esterification is hydrolysis that is of high importance also in the sphere of P-esters [43,44]. The hydrolysis of phosphinates and phosphonates is carried out, in most cases, under acidic conditions [45,46,47,48,49,50,51,52], but, among other possibilities, base-catalyzed cases also occur [53,54,55,56,57]. It was a new approach to perform the hydrolyses under MW irradiation. Alkyl diphenylphosphinates (29) were hydrolyzed at 180 °C in the presence of 10% of p-toluenesulfonic acid (PTSA) as the catalyst (Scheme 8) [58]. For the esters with n-alkyl substituents, completion of the hydrolysis required 1.5–2.2 h; however, with the i-propyl ester, the hydrolysis took place after 0.5 h. This latter experience is due to the realization of the AAl1 mechanism. The acid (30) was isolated in yields of 94–97%.
A comparative thermal experiment afforded the phosphinic acid (30) in a lower conversion of 24%, indicating the beneficial effect of MWs. This is the consequence of the local overheating [27,28] and the better MW absorbing effect of PTSA.
In conclusion, MW irradiation made possible the otherwise impossible direct esterification of phosphinic acids, and the monoesterification of phosphonic acids. MW irradiation proved to be a useful tool in overcoming the enthalpy of activation barriers higher than 130 kJ mol–1 [27,28]. This is due to the beneficial effect of the statistically occurring local overheating [41,42]. On the other hand, the MWs were beneficial also in the acid-catalyzed hydrolysis of phosphinates.
3. Microwave as a Substitute for the Catalysts in the Deoxygenation of Phosphine Oxides
Besides the widely applied trichlorosilane (Cl3SiH) and phenylsilane (PhSiH3) [59,60], the use of the cheaper ethoxysilanes, (EtO)3SiH and (EtO)2MeSiH, as well as 1,1,3,3-tetramethyldisiloxane (TMDS) and polymethylhydrosiloxane, called also as methylpolysiloxane ([PMHS or MPS] represented by formula [–O–SiH(Me)–]n), offers alternative possibilities [61,62]. However, these silanes are of low reactivity. Beller et al. tested different acids as catalysts in the deoxygenation of triphenylphosphine oxide (31) with diethoxymethylsilane as the reductant (Table 8) [61]. No deoxygenation occurred in the lack of a catalyst. Adding 15 mol% of benzoic acid to the mixture, triphenylphosphine (32) was obtained in a yield of 6%. However, in the presence of the diphenyl ester of phosphoric acid as the catalyst, the yield of PPh3 was 75%. Moreover, it was observed that the use of a P-ester-acid catalyst with an electron-withdrawing substituent in the phenyl ring led to the quantitative reduction.
Different tertiary phosphine oxides (33) were reduced to the respective phosphines (34) using (EtO)3SiH in the presence of titanium(IV) isopropoxide as the catalyst (Table 9). The deoxygenations were performed in tetrahydrofuran (THF). On heating at 67 °C, the completion required 1 h, if the silane was applied in a 3-fold excess [62].
The reduction of aryl-diphenylphosphine oxides with (EtO)3SiH in the presence of Ti(OiPr)4 in benzene at reflux provided the corresponding phosphine in 90% yield after a reaction time of 30 min [63].
The deoxygenation of triphenylphosphine oxide (31) was also elaborated using TMDS in the presence of catalysts. Copper(II) triflate was a suitable promoter in the reduction of the P=O unit at 100 °C in PhMe [64]. Without a catalyst, even measuring in TMDS in a 12 equivalents’ quantity, no reduction occurred. The use of 15 mol% of the (PhO)2P(O)OH in boiling PhMe also promoted deoxygenation [61]. Using 10 mol% of Ti(OiPr)4 at 100 °C, triphenylphosphine (32) was formed in a conversion of 86%. In the presence of 1% of InBr3 as the catalyst, the reduction was quantitative at 100 °C [65] (Table 10).
It is noteworthy that there is no need for any catalyst if the reduction of triphenylphosphine oxide (31) with TMDS is performed in a solvent-free manner under MW irradiation. After treatment at 200 °C for 6.5 h, the reduction was quantitative. On conventional heating at 175 °C, there was a need for a 1-day reaction time. As the reduction takes place in the absence of catalyst, the MW-assisted approach may be regarded as a “green” protocol (Table 11) [66,67]. Practically, MW irradiation substituted the catalyst.
The reduction of ring phosphine oxides, such as 3-methyl-1-phenyl-2-phospholene oxide (35a), was performed using TMDS together with InBr3 as the catalyst in PhMe at 100 °C [65]. The deoxygenation also took place in the absence of catalyst in PhMe at reflux [68]. The solvent- and catalyst-free reduction of 1-phenyl-3-phospholene oxide 35b was complete after a shorter reaction time in both the thermal and MW-promoted variations (Table 12) [66].
The next user-friendly silane, PMHS can be used in the deoxygenation of triphenylphosphine oxide (31). At 290 °C in the absence of any solvent, triphenylphosphine (32) was isolated in an 86% yield [69]. It is a disadvantage that the application of PMHS requires a relatively high temperature. Applying the reducing agent in a 12 equivalents’ quantity at 100 °C in PhMe, no reduction took place after 2 h. At the same time, applying Cu(OTf)2 as the promoter at 100 °C for 15 h, the deoxygenation took place [64]. Applying 15 mol% of (PhO)2P(O)OH as the catalyst at reflux for 1 day, the conversion was incomplete, and the phosphine (32) was obtained in a 35% yield [62]. Applying PMHS at a temperature of 175 °C on conventional heating in the absence of any catalyst and solvent, completion of the reduction required 17 h. In the MW-promoted version, 8 h was enough for an almost quantitative outcome (Table 13).
The first deoxygenation of 1-phenyl-2-phospholene oxide 35a with PMHS was performed in the absence of any solvent at 250 °C [69]. Then, this transformation was carried out in PhMe at reflux 6 h [68]. The deoxygenation of 1-phenyl-3-phospholene oxide 35b was also investigated under thermal and MW-promoted, in most cases, solvent-free conditions. These deoxygenations were complete at 110 °C after 4 and 2 h, respectively (Table 14) [66,67]. The outcome of the deoxygenation of the dimethylphospholene oxide 35c was better in PhMe at reflux.
Optimum conditions for the reduction of 1-alkyl-3-methyl-3-phospholene 1-oxides by PhSiH3, TMDS, and PMHS were also evaluated [70].
TMDS and PMHS are user-friendly and low-cost reducing agents. The lower reactivity can be compensated by a solvent-free and MW-assisted protocol. MWs can be regarded as a kind of promoter substituting efficient catalysts.
4. Microwave as a Substitute for Catalysts in the Kabachnik–Fields Reaction
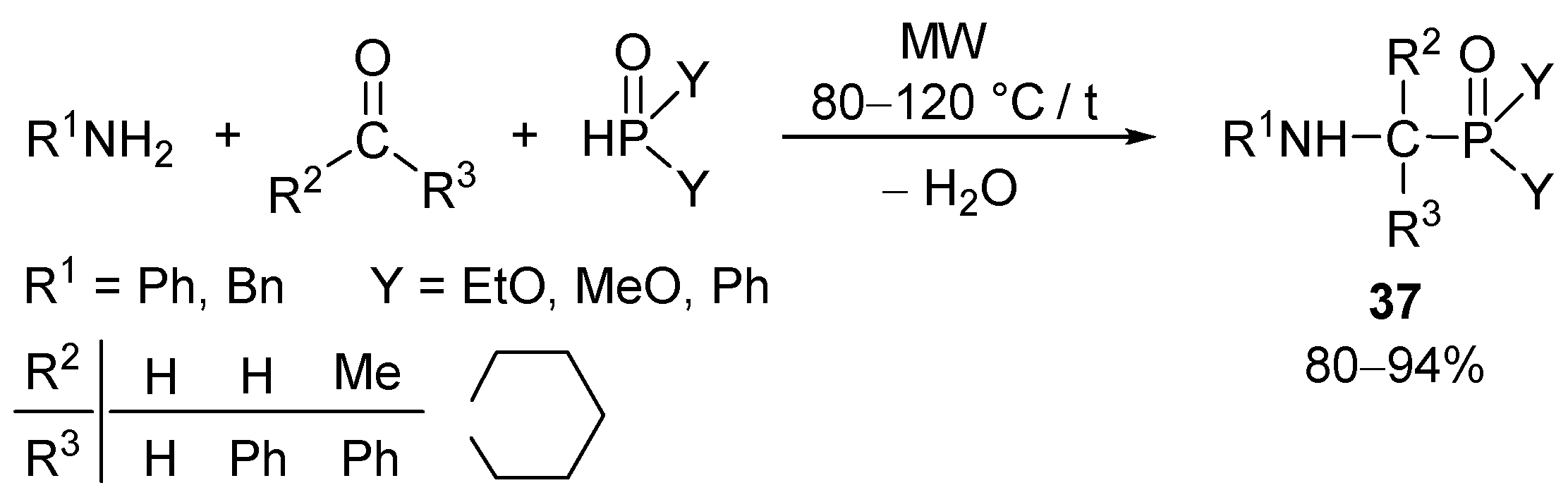
α-Aminophosphonic acids, the P-analogues of α-amino acids, are of importance due to their potential biological activity, which is the consequence of their enzyme inhibitory properties [71]. The major method for the synthesis of α-aminophosphonates is the Kabachnik–Fields condensation of amines, aldehydes or ketones, and dialkyl phosphites [72,73]. α-Aminophosphonates (37, Y = RO) and α-aminophosphine oxides (37, Y = Ph) were synthesized by the solvent- and catalyst-free MW-assisted phospha-Mannich reaction of primary amines, oxo compounds, and dialkyl phosphites or diphenylphosphine oxide. Earlier preparations utilized special catalysts, such as, tetra-tert-butyl-substituted phthalocyanine—AlCl3 complex [74], magnesium perchlorate [75], metal triflates (M(OTf)n, M = Li, Mg, Al, Cu and Ce) [76], indium(III) triflate [77], bismuth(I) nitrate [78], samarium(II) iodide [79], ceric ammonium nitrate (CAN) [80], indium(III) chloride [81], and a variety of lanthanide (Yb, Sm, Sc, La) triflates [82], which mean cost and environmental burden. It was found that under MW conditions, there is no need for any catalyst (Scheme 9) [83].
Starting from heterocyclic amines: pyrrolidine, piperidine, morpholine and piperazine derivatives or heterocyclic >P(O)H species, N-heterocyclic [84] and P-heterocyclic [85] α-aminophosphonates were obtained. 3-Amino-6-methyl-2H-pyran-2-ones were also suitable amino derivatives in Kabachnik–Fields reaction with formaldehyde and dialkyl phosphites or diphenylphosphine oxide [86]. As special cases, α-aminophosphonates with different alkoxy groups [87], α-aminophosphinates [88], and α-aminophoshonates with sterically demanding α-aryl substituents [89] were also synthesized under MW-assisted conditions. Moreover, carboxylic amides could also be used under solvolytic conditions [90]. It is noteworthy that the phospha-Mannich reactions may also be performed in the presence of the T3P® activating agent [91].
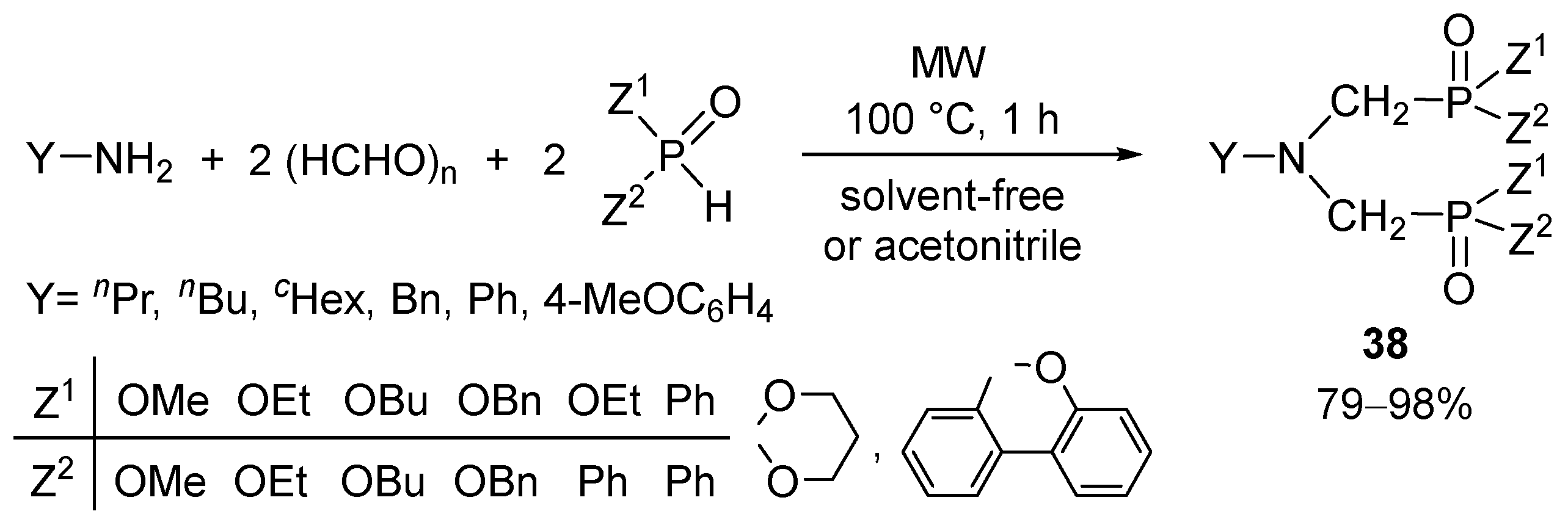
Primary amines are suitable components for bis(Kabachnik–Fields) condensations [92]. In these distances, alkyl or arylamines were reacted with two equivalents of the formaldehyde and the >P(O)H reagents to afford the bis(Z1Z2P(O)CH2)amines (38) (Scheme 10) [93,94,95]. Most of the reactions could be carried out in a solvent-free manner.
The bisphosphinoyl derivatives (38, Z1 = Z2 = Ph) were transformed after double-deoxygenation to bis(phosphines) that were useful in the synthesis of ring platinum complexes [94,95,96] α-, β- and γ-amino acids (or esters) were also utilized in the double Kabachnik–Fields condensation to furnish the bis(phosphono- or phosphinoyl) products [97,98].
The α-aminophosphonates may be formed via imine of α-hydroxyphosphonate intermediates [72,92,99,100]. α-Hydroxyphosphonates may be formed in a reversible manner from the corresponding oxo compound and dialkyl phosphite [101,102]. It was a somewhat surprising experience that the α-hydroxyphosphonates could be converted to the respective α-aminophosphonates by reaction with primary amines under MW conditions. This was promoted by a favorable adjacent group effect [103,104].
In summary, a wide range of mono- and bis Kabachnik–Fields reactions were carried out under MW-assisted and catalyst-free conditions, and mostly in a solvent-free manner.
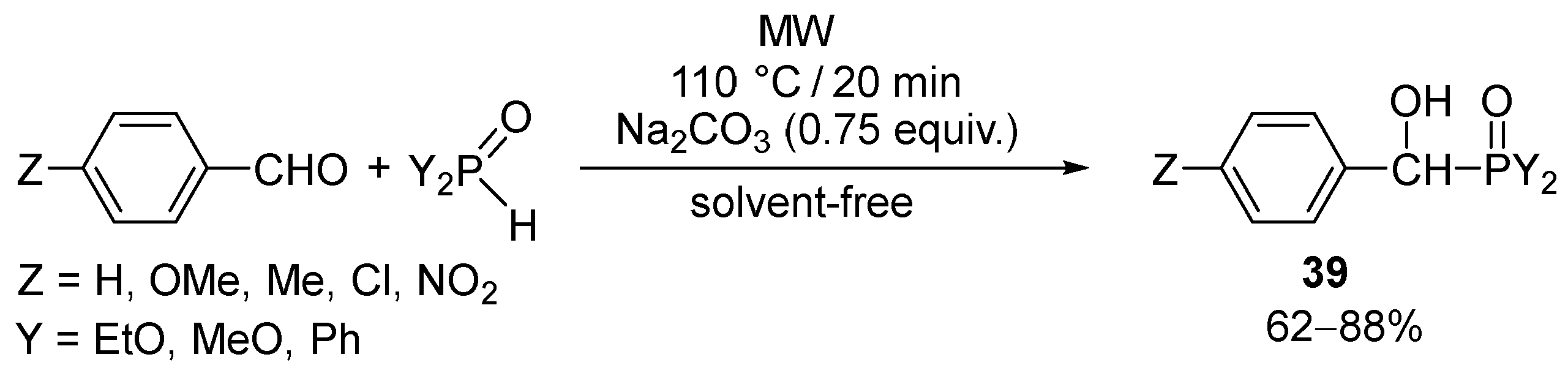
α-Aryl-α-hydroxyphosponates [105] mentioned above as intermediates in the phospha-Mannich condensations, along with α-aryl-α-hydroxyphosphine oxides (39), were synthesized in a catalytic and solvent-free MW-assisted Pudovik reaction comprising the addition of >P(O)H species to aryl aldehydes (Scheme 11) [106].
Dialkyl phosphites could also be reacted with α-ketophosphonates to result in the formation of dronate analogue α-hydroxybisphosphonates in the presence of diethylamine and in the absence of any solvent [107,108].
It was found that the Pudovik reaction may also be realized at room temperature in a solvent-free manner. However, these methods required special catalysts, such as piperazine [109], magnesium chloride/3 equivalents of triehylamine [110], barium hydroxide [111], sodium carbonate [112], potassium phosphate [113], sodium-modified fluoroapatite [114], and silica-supported tungstic acid [115].
Moreover, the work-up needed a considerable quantity of solvents. Hence, these methods cannot be regarded as green. The author of this review together with co-workers developed an indeed environmentally-friendly method by crystallizing the products from the mixtures [116].
5. Microwave Irradiation Allowing the Simplification of the Catalysts in the Hirao Reaction
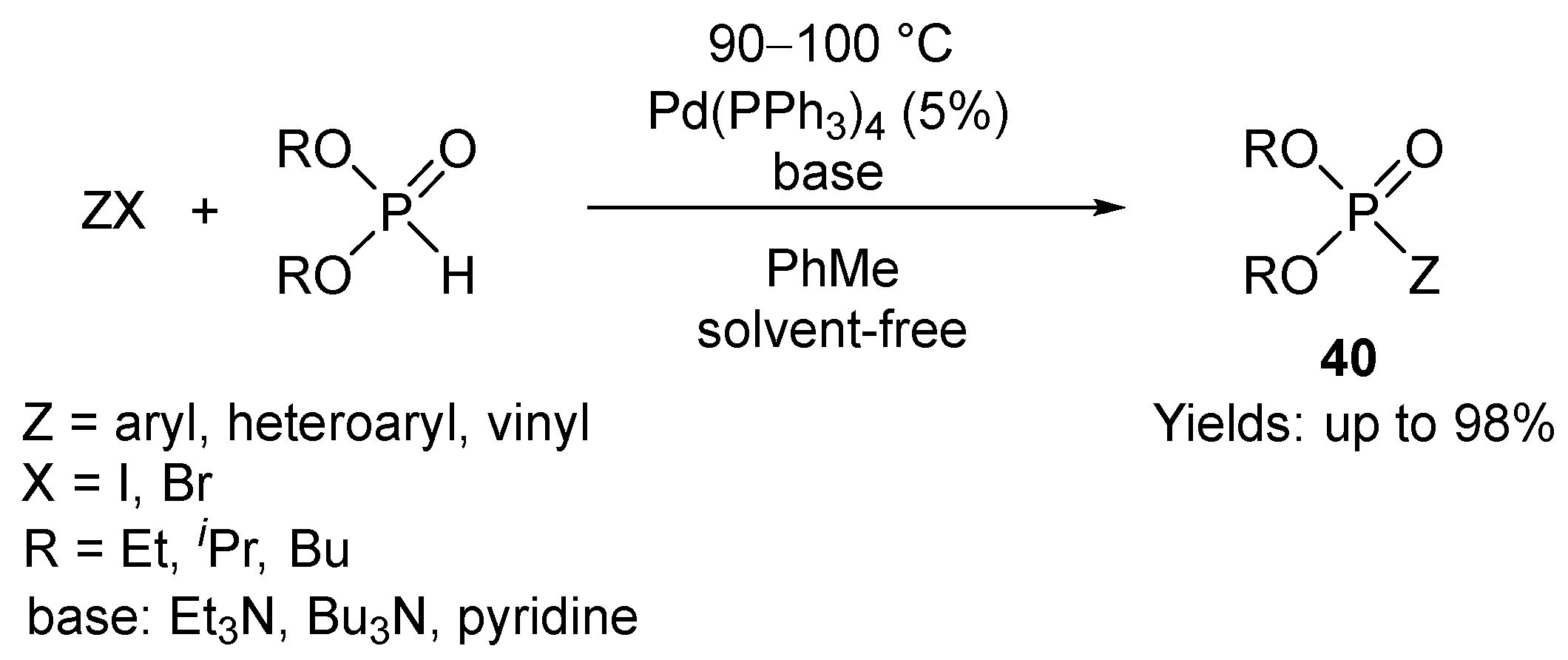
The Hirao reaction of aryl, heteroaryl, and vinyl halogenides (or other derivatives) with dialkyl phosphites, alkyl H-phosphinates, and secondary phosphine oxides is an important method for the synthesis of phosphonates, phosphinates, and phosphine oxides, respectively [117,118,119]. The original Hirao P–C coupling aimed at the synthesis of arylphosphonates reacting aryl- and vinyl halogenides with dialkyl phosphites in the presence of tetrakis(triphenylphosphine)palladium (Scheme 12) [120,121,122]. The P–C coupling reaction was then extended to other substrates [123,124,125,126,127,128,129,130,131,132,133,134,135] that was followed by further variations involving different >P(O)H reagents, Pd(II)-, or other metal (Ni(II) and Cu(II)) salts as catalyst precursors together with mono- and bidentate P-ligands, bases (mostly amines) and solvents.
Instead of Pd(PPh3)4, the application of Pd salts together with P-ligands was spread. In such cases, the Pd(0) catalyst is formed in situ from the components. From among the Pd salts, Pd(OAc)2 is the most suitable. The Pd(OAc)2/P-ligand combination was often utilized in the preparation of arylphosphonates, and this approach was more suitable than the classical Pd(PPh3)4 catalyst [136,137,138,139,140,141,142,143,144,145,146,147,148]. PPh3, dppp, dppb, dppe, dppf, and BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl were the typical P-ligands applied.
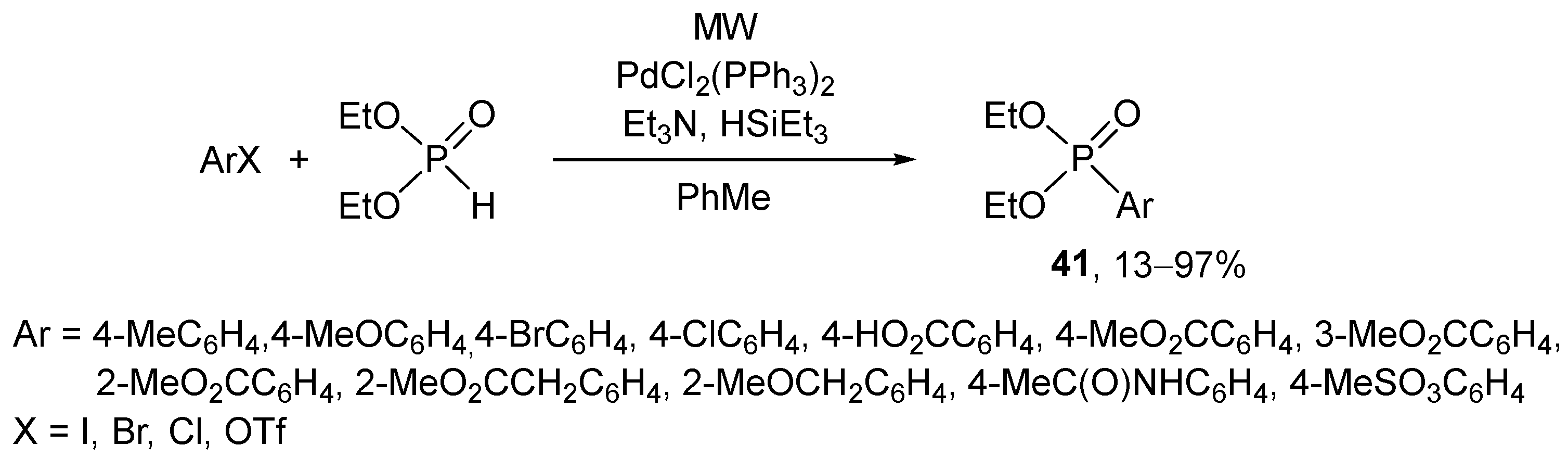
“Greener” and more efficient protocols were developed for the Hirao reaction in the last twenty years. The MW technique also proved to be useful in the Hirao reactions. The first MW-assisted P–C coupling took place between aryl halides/triflates and diethyl phosphite in the presence of bis(triphenylphosphine)palladium dichloride, triethylamine, and triethylsilane as the reductant (Scheme 13) [149]. The Hirao reaction was performed in a domestic microwave device.
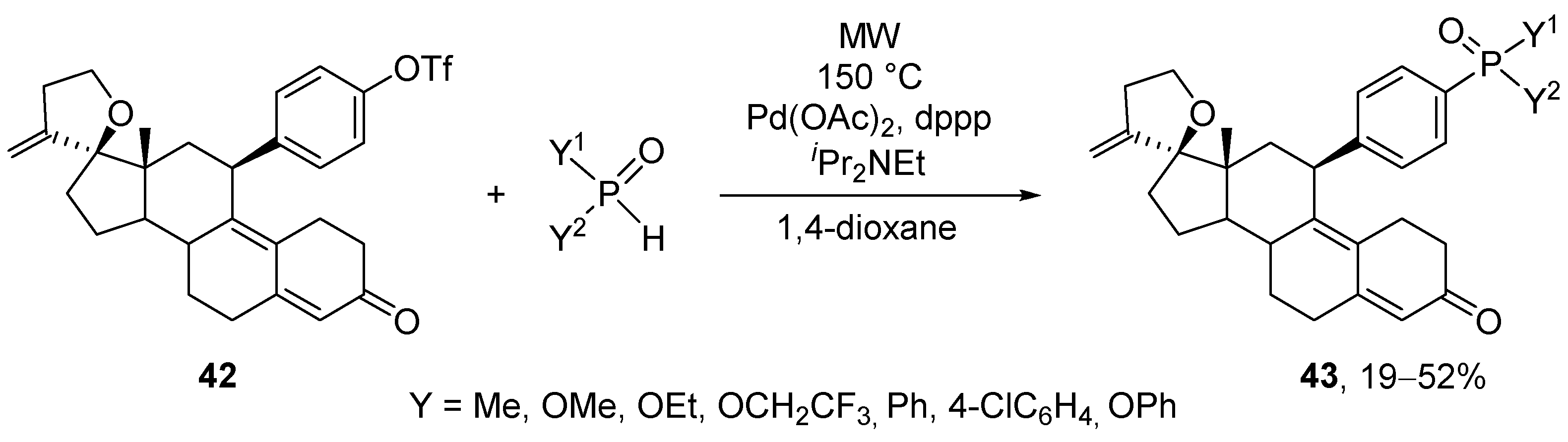
The MW-assisted Hirao reaction of dialkyl phosphites with aryl and vinyl halides/triflates was also studied in the presence of Pd(PPh3)4 [150]. The best results (72–96%) were obtained using Cs2CO3 in THF. The MW protocol was also applied in the synthesis of P-functionalized 11β-aryl-substituted steroids (43) that are progesterone receptor antagonists (Scheme 14) [151].
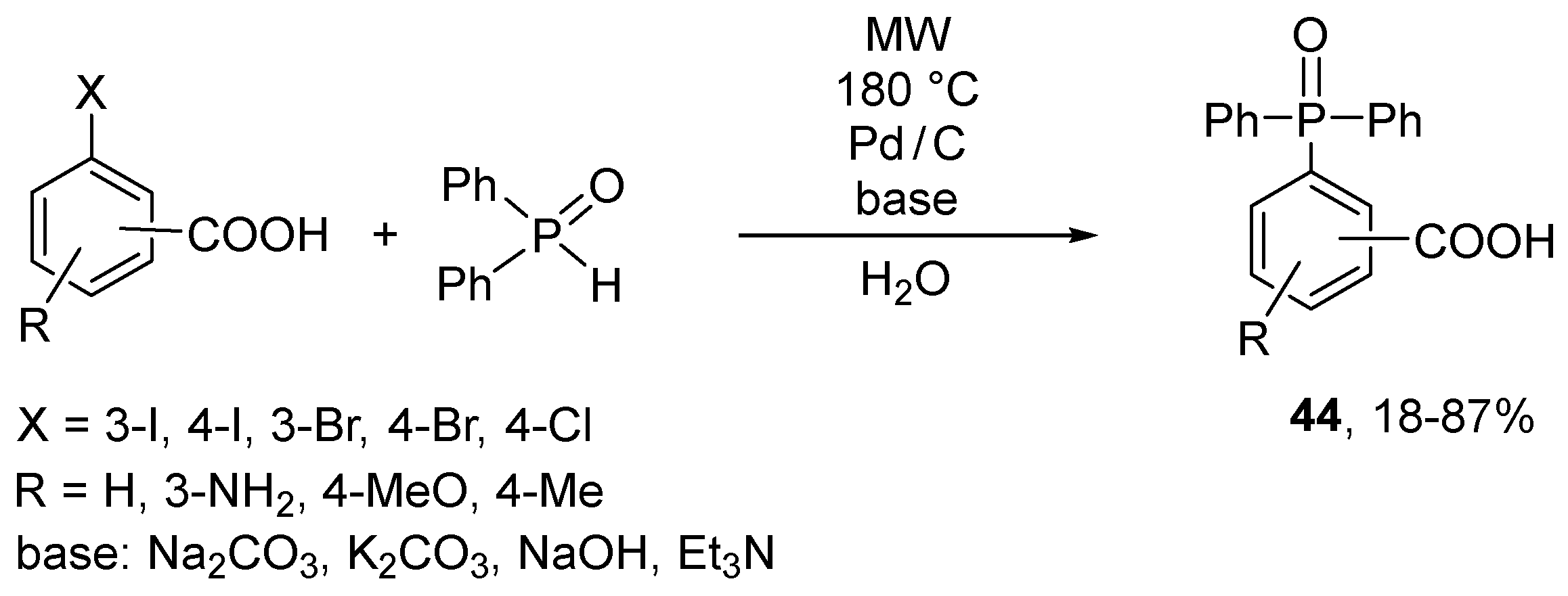
A few arylboronic acids and arylfluoroborates were coupled with dialkyl phosphites using the Pd(OAc)2 or Pd(O2CCF3)2/dmphen catalyst combination, and p-benzoquinone in the absence of a base [152]. The authors assumed the role of the reoxidant in the catalytic cycle. The author of this review believes that the application of an oxidant in the P–C coupling is mistaken. Instead, a reductive agent may be useful. A new cyclodiphosphazane-containing Pd catalyst was tested in the preparation of triarylphosphine oxides from aril bromides and diphenylphosphine oxide [153]. The use of this exotic promoter and Cs2CO3 as a base in acetonitrile under MW irradiation gave Ph3P=O in yields of 46–95%. It is noteworthy that the coupling of iodo- and bromobenzoic acids with diphenylphosphine oxide could be performed in water using Pd/C catalyst under MW conditions (Scheme 15) [154]. In this case, the tetrabutylammonium bromide additive had no influence on the outcome.
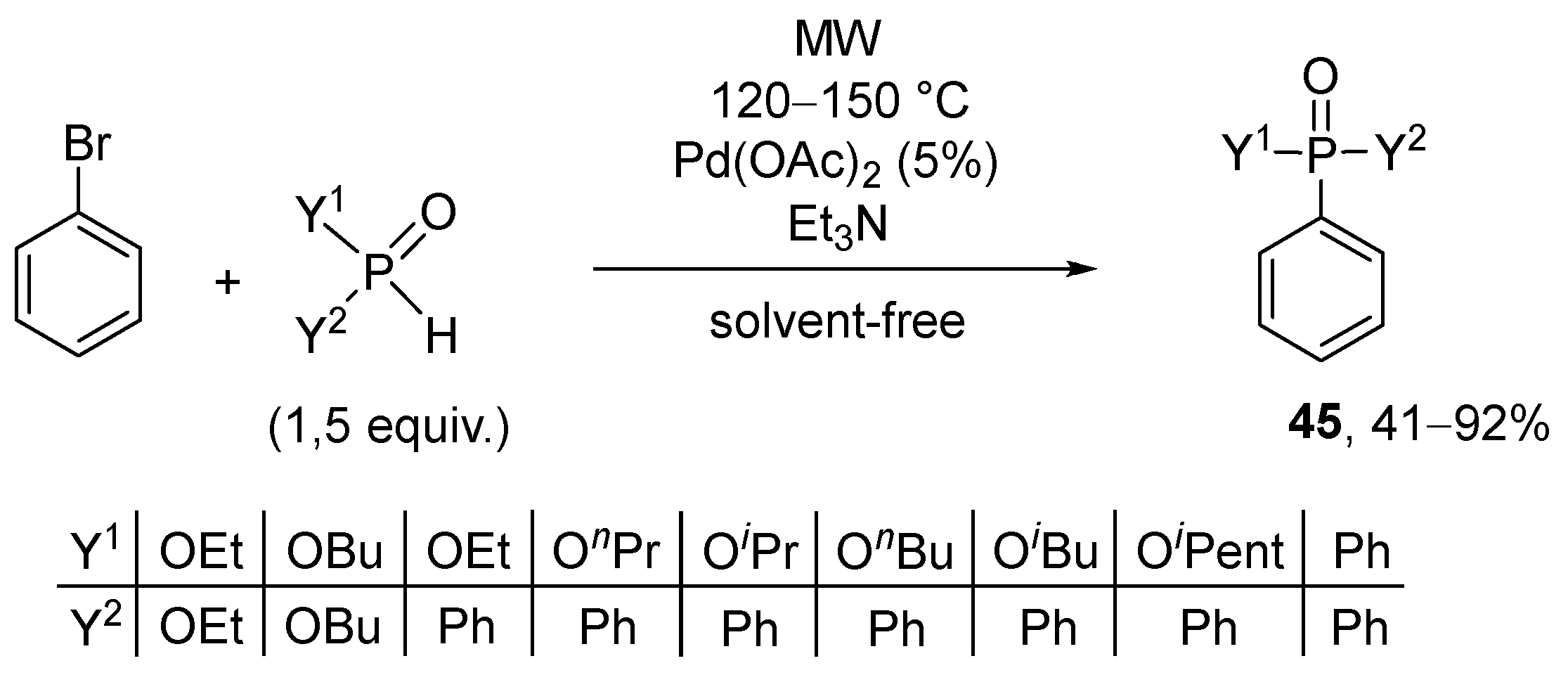
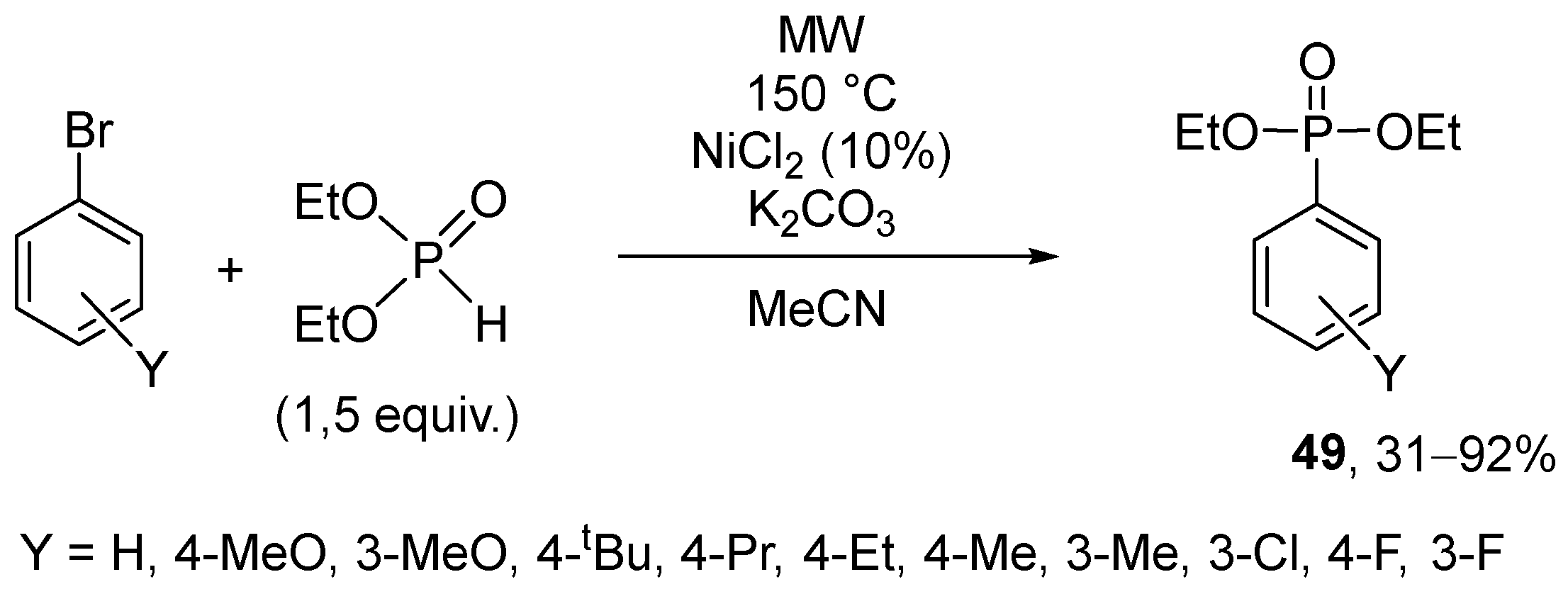
It was recognized by the Keglevich group that the Hirao cross-couplings may take place in the presence of Pd(OAc)2 without the addition of the usual P-ligands under solvent-free and MW-assisted conditions (Scheme 16) [155]. The reaction of bromobenzene and 1.5 equivalents of the dialkyl phosphites, phenyl-H-phosphinates, and diphenylphosphine oxide was carried out in the presence of 5% of Pd(OAc)2 and 1.1 equivalents of triethylamine. The relevance of the MW technique was pointed out by comparative thermal experiments. The conversion was almost complete at 120 °C, but the best results were obtained at 150 °C.
In the next stage, the Pd(OAc)2-promoted “P-ligand-free” Hirao reactions were extended to different bromoarenes (Table 15) [156]. The experience was that both electron-releasing and electron-withdrawing substituents decreased the reactivity, as in these instances, higher temperatures (175–200 °C) were necessary to obtain the aryl phosphonates in acceptable yields (69–92%) (Table 15). The reactions of the methoxy- and alkyl-substituted bromoarenes with a decreased reactivity required, in most cases, a temperature of 200 °C, and the application of 10% of the Pd(OAc)2 catalyst.
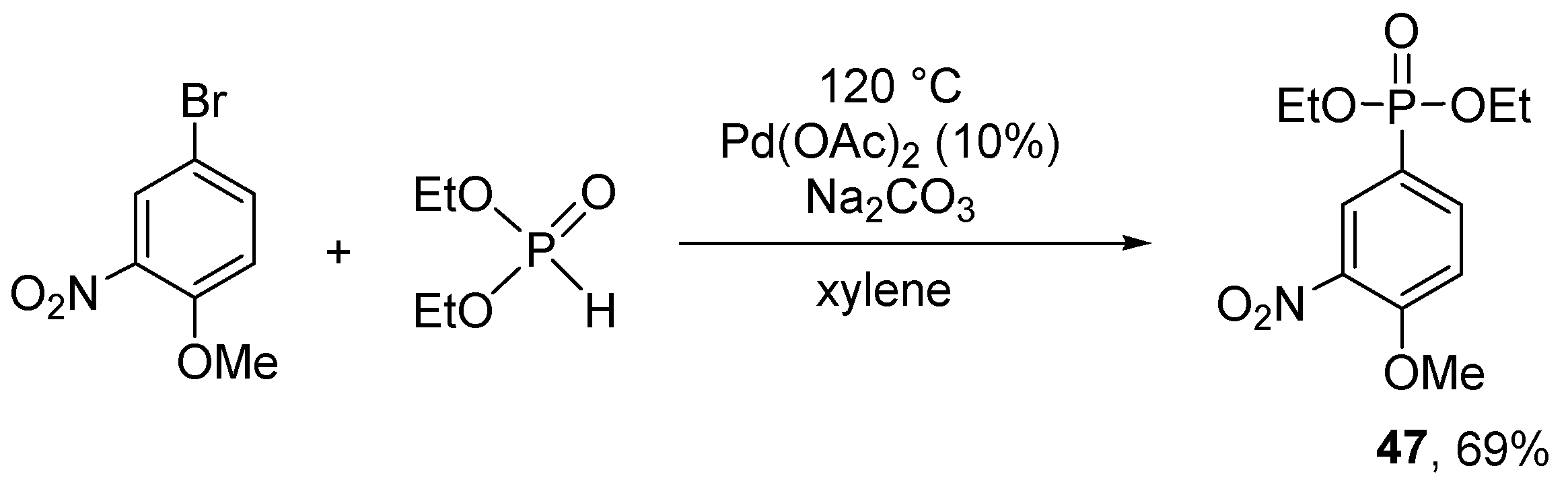
Recently, Hirao et al. have publised another “P-ligand-free” reaction (Scheme 17) [157]. According to this, diethyl phosphite was coupled with 2-nitro-5-bromoanisole applying Pd(OAc)2 as the catalyst and Na2CO3 in xylene at 120 °C to furnish the respective aryl phosphonate (47) in a yield of 69% after 24 h.
Xiao and his co-workers described the Pd-catalyzed cross-coupling of an arylsulfinate salt with dialkyl phosphites applying PdCl2 without the usual P-ligands in DMF–DMSO [158]. The arylphosphonates were prepared in good yields using silver carbonate as the oxidant under MW irradiation. Here, it is noted again that there is no need for an oxidant during the P–C coupling. The Pd(OAc)2-promoted “P-ligand-free” protocol was extended to the Hirao reaction of heteroaryl bromides [159], and the reactivity of the substrates was studied in detail [160].
Moreover, the mechanism of the Pd-catalyzed Hirao reactions carried out using the P-reactant in excess was investigated experimentally and by quantum chemical calculations [161]. It was found that if Pd(OAc)2 is applied in a quantity of 10%, the >P(O)H reactant should be used in a quantity of 1.3 equivalents. 10% of the P-species reduces Pd(II) to Pd(0), while 20% covers the P-ligand that is the trivalent tautomeric form (>P–OH) of the >P(O)H reagent. The whole catalytic cycle involving oxidative addition, ligand exchange, and reductive elimination was adapted to our model, and the elemental steps were refined [161]. The formation of the “PdP2” catalyst and its activity were investigated under a separate cover [162]. It turned out that the Ar2POH ligands with 2-MePh or 3,5-diMePh substituents are more advantageous than the Ph one, as the steric hindrance prevents the tricoordination of the Pd [162].
It was also found that NiCl2 may also be a suitable catalyst in the P–C coupling of bromobenzene and a series of >P(O)H reagents (Scheme 18) [163]. The experiments were carried out at 150 °C on MW irradiation. Using 1.5 equivalents of NEt3 in a solvent-free manner, completion of the reaction of diethyl phosphite and bromobenzene required 2 h, and the diethyl phenylphosphonate was isolated in a 67% yield. The use of K2CO3 in acetonitrile was more advantageous: in the presence of 5% NiCl2, after a reaction time of 45 min the yield of the corresponding product was 92%. Applying phenyl-H-phosphinates, the diphenylphosphinates were isolated in yields of 84–89%. Diphenylphosphine oxide and other aryl-substituted secondary phosphine oxides served as additional reagents in the “P-ligand-free” P–C couplings under discussion.
The NiCl2-catalyzed phosphonylation of a series of bromoarenes gave similar results as those in the presence of Pd(OAc)2. However, the scope of the aryl bromides was more limited (Scheme 19) [163].
The nature of the Ni-catalyst, its formation, and the mechanism of the NiCl2-catalyzed P–C coupling reactions was also studied experimentally and by theoretical calculations [163]. It was found that in these cases, a Ni(II)(PY2OH)2 type catalyst is formed, and Ni(II) is converted to Ni(IV) in the oxidative addition step. This surprising finding was also proved to be true for earlier Ni-catalyzed instances [164] carried out originally in the presence of Zn or Mg reductants [165,166,167]. Hence, the Ni(II) → Ni(IV) conversion may be of more general value instead of the earlier assumed Ni(0) → Ni(II) protocol [168].
Both the Pd- and the Ni-catalyzed protocols elaborated by the Keglevich group are suitable for the coupling of bromoarenes and different >P(O)H reagents. Considering the conditions, costs, and toxicity, one can conclude that the application of Pd(OAc)2 is more attractive, but the use of NiCl2 may be a good alternative as well. The recent developments and extensions of the P–C coupling reactions open a new horizon since there is no need to add sensitive and expensive P-ligands.
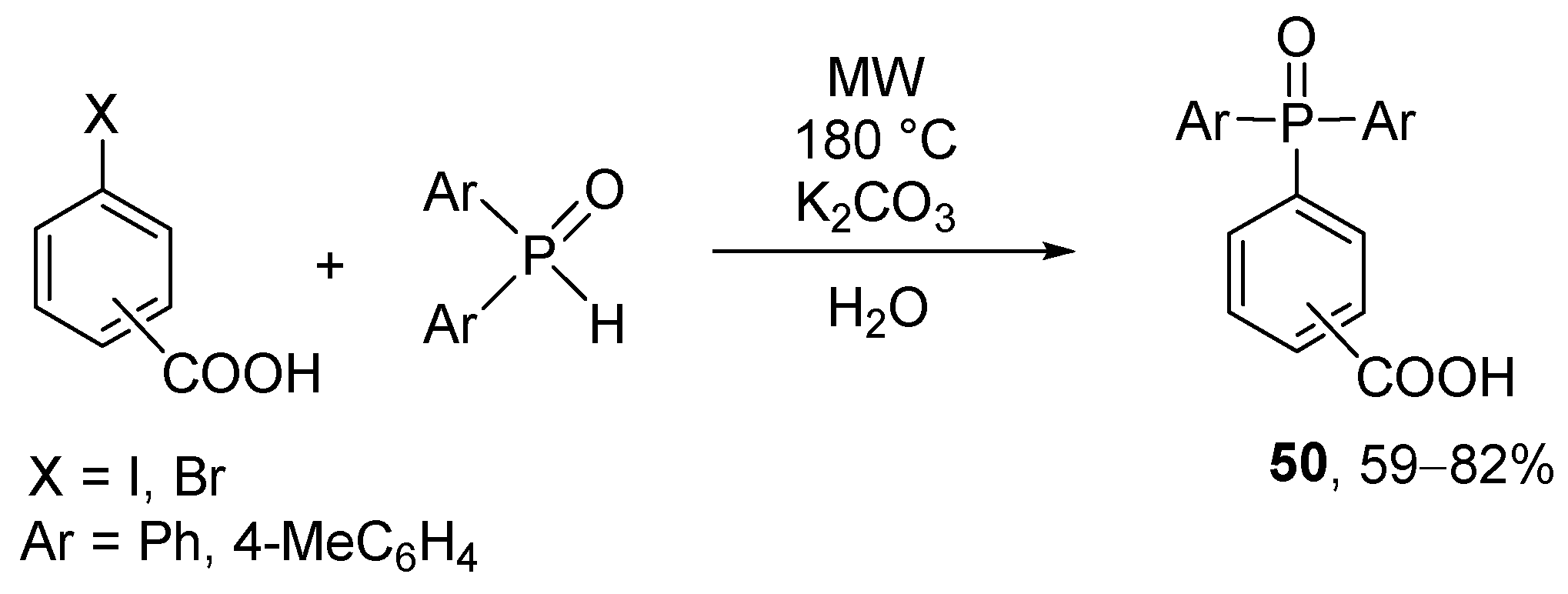
A catalyst-free method was developed for the P–C coupling of halobezoic acids and secondary phosphine oxides in water as the medium (Scheme 20) [169]. 4-Iodo-, 3-bromo-, and 4-bromobenzoic acids were coupled with diaryl phosphine oxides under MW conditions at 180 °C for 1–6 h in the presence of K2CO3. The only limitation of this “green” P–C coupling reaction is the low water-solubility of the P-reagents.
In summary, the Hirao reaction utilizing a series of suitably substituted aryl derivatives and different >P(O)H reagents along with a Pd or Ni catalyst provides arylphosphonates, tertiary phosphine oxides, and related compounds that may be useful intermediates in synthetic organic chemistry. The chemistry discussed hides interesting green chemical aspects, such as MW activation, solvent- and catalyst-free protocols, as well as mechanistic delicates.
Funding
This project was supported by the National Research, Development and Innovation Office (K119202 and K134318).
Data Availability Statement
The data presented in this study are available in the references.
Acknowledgments
G.K. is grateful to all co-authors appearing in the cited references.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de la Hoz, A.; Loupy, A. (Eds.) Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Keglevich, G. (Ed.) Milestones in Microwave Chemistry—SpringerBriefs in Molecular Science; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Guenin, E.; Meziane, D. Microwave assisted phosphorus organic chemistry: A review. Curr. Org. Chem. 2011, 15, 3465–3485. [Google Scholar] [CrossRef]
- Cravotto, G.; Tagliapietra, S.; Caporaso, M.; Garella, D.; Borretto, E.; Di Stilo, A. Recent advances in the cyclization of N-heterocycles: The role of enabling techniques (review). Chem. Heterocyclic Compd. 2013, 49, 811–826. [Google Scholar] [CrossRef]
- Maiuolo, L.; De Nino, A.; Algieri, V.; Nardi, M. Microwave-assisted 1,3-dipolar cyclo-addition: Recent advances in synthesis of isoxazolidines. Mini-Rev. Org. Chem. 2017, 14, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Maiuolo, L.; Algieri, V.; Russo, B.; Tallarida, M.A.; Nardi, M.; Di Gioia, M.L.; Merchant, Z.; Merino, P.; Delso, I.; De Nino, A. Synthesis, biological and in silico evaluation of pure nucleobase-containing spiro (indane-isoxazolidine) derivatives as potential inhibitors of MDM2-p53 interaction. Molecules 2019, 24, 2909. [Google Scholar] [CrossRef] [Green Version]
- Kappe, C.O.; Pieber, B.; Dallinger, D. Microwave effects in organic synthesis: Myth or reality? Angew. Chem. Int. Ed. 2013, 52, 1088–1094. [Google Scholar] [CrossRef]
- De la Hoz, A.; Díaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Microwave-assisted organic synthesis: General considerations and transformations of heterocyclic compounds. Curr. Org. Chem. 2010, 14, 1050–1074. [Google Scholar] [CrossRef]
- Kuhnert, N. Microwave-assisted reactions in organic synthesis—Are there any nonthermal microwave effects? Angew. Chem. Int. Ed. 2002, 41, 1863–1866. [Google Scholar] [CrossRef]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Matthews, NC, USA, 2002; p. 23. [Google Scholar]
- Keglevich, G.; Greiner, I. The meeting of two disciplines: Organophosphorus and green chemistry. Curr. Green Chem. 2014, 1, 2–16. [Google Scholar] [CrossRef]
- Keglevich, G.; Dudás, E. Microwave promoted efficient synthesis of 2-phosphabicyclo[2.2.2]octadiene- and octene 2-oxides under solventless conditions in Diels-Alder reaction. Synth. Commun. 2007, 37, 3191–3199. [Google Scholar] [CrossRef]
- Keglevich, G.; Forintos, H.; Sipos, M.; Dobó, A.; Ludányi, K.; Vékey, K.; Tungler, A.; Tőke, L. The synthesis of six-membered P-heterocycles with sterically demanding substituent on the phosphorus atom. Heteroatom Chem. 2001, 12, 528–533. [Google Scholar] [CrossRef]
- Keglevich, G.; Forintos, H.; Körtvélyesi, T.; Tőke, L. Inverse Wittig reaction of oxaphosphetes formed by the [2+2] cycloaddition of arylphosphine oxides and dimethyl acetylenedicarboxilate (DMAD). J. Chem. Soc. Perkin Trans. 2002, 1, 26–27. [Google Scholar] [CrossRef]
- Keglevich, G. 4+2] Versus [2+2] Cycloadditions in the sphere of P-heterocycles as useful synthetic tools. Curr. Org. Chem. 2002, 6, 891–912. [Google Scholar] [CrossRef]
- Keglevich, G.; Körtvélyesi, T.; Forintos, H.; Vaskó, Á.G.; Izvekov, V.; Tőke, L. New evidences on the structure of the product from the reaction of cyclic 2,4,6-trialkylphenylphosphine oxides with dimethyl acetylenedicarboxylate (DMAD); ring-opening by an inverse Wittig reaction type protocol. Tetrahedron 2002, 58, 3721–3727. [Google Scholar] [CrossRef]
- Keglevich, G.; Dudás, E.; Sipos, M.; Lengyel, D.; Ludányi, K. Efficient synthesis of cyclic β-oxophosphoranes by the microwave-assisted reaction of cyclic phosphine oxides and dialkyl acetylenedicarboxylate. Synthesis 2006, 8, 1365–1369. [Google Scholar] [CrossRef]
- Keglevich, G.; Forintos, H.; Körtvélyesi, T. Synthesis and reactions of β-oxophosphoranes/ylides containing a cyclic or acyclic P-moiety. Curr. Org. Chem. 2004, 8, 1245–1261. [Google Scholar] [CrossRef]
- Keglevich, G.; Körtvélyesi, T.; Forintos, H.; Lovas, S. Structure and stability of oxaphosphetes formed as intermediates in the reaction of tertiary phosphine oxides and acetylenic derivatives. J. Chem. Soc. Perkin Trans. 2002, 10, 1645–1646. [Google Scholar] [CrossRef]
- Keglevich, G.; Körtvélyesi, T.; Ujvári, A.; Dudás, E. A quantum chemical study on the reaction of 1-aryl-1,2-dihydrophosphinine oxides with dimethyl acetylenedicarboxylate. J. Organomet. Chem. 2005, 690, 2497–2503. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Ranasinghe, M.G.; Jenkins, I.D. Mono- and dialkylation of isopropyl phosphinate—A simple preparation of alkylphosphinate esters. Phosphorus Sulfur Silicon 1996, 115, 255–259. [Google Scholar] [CrossRef]
- McBride, J.J., Jr.; Mais, A. Preparing Esters of Phosphinic Acids. U.S. Patent 3,092,650, 4 June 1963. [Google Scholar]
- Cherbuliez, E.; Weber, G.; Rabinowitz, J. Recherches sur la formation etla transformation des esters XLVII. Sur les monoesters de divers acides du phosphore et sur leur vitesse de scission à différents pH. Helv. Chim. Acta 1963, 46, 2464–2470. [Google Scholar] [CrossRef]
- Nifant’ev, E.E.; Levitan, L.P. Convenient synthesis of alkyl hypophosphites. Zh. Obshch. Khim. 1965, 35, 758. [Google Scholar]
- Denham, H.; Stiles, A.R. Allyl-Type Phosphinates and Polymers of the Same; Patent and Trademark Office: Washington, DC, USA, 1953.
- Keglevich, G.; Kiss, N.Z.; Mucsi, Z.; Körtvélyesi, T. Insights into a surprising reaction: The microwave-assisted direct esterification of phosphinic acids. Org. Biomol. Chem. 2012, 10, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Mucsi, Z.; Kiss, N.Z.; Keglevich, G. A quantum chemical study on the mechanism and energetics of the direct esterification, thioesterification and amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide. RSC Adv. 2014, 4, 11948–11954. [Google Scholar] [CrossRef] [Green Version]
- Kiss, N.Z.; Böttger, É.; Drahos, L.; Keglevich, G. Microwave-assisted direct esterification of cyclic phosphinic acids. Heteroatom Chem. 2013, 24, 283–288. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Keglevich, G. Microwave-assisted direct esterification of cyclic phosphinic acids in the presence of ionic liquids. Tetrahedron Lett. 2016, 57, 971–974. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E.; Kiss, N.Z.; Jablonkai, E.; Hegedűs, L.; Grün, A.; Greiner, I. Microwave-assisted esterification of phosphinic acids. Curr. Org. Chem. 2011, 15, 1802–1810. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Rádai, Z.; Tihanyi, I.; Szabó, T.; Keglevich, G. Microwave-assisted direct esterification of a cyclic phosphinic acid with phenols. Mendeleev Commun. 2018, 28, 31–32. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Novel synthesis of phosphinates by the microwave-assisted esterification of phosphinic acids. Synth. Commun. 2009, 39, 2392–2404. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Keglevich, G. Direct esterification of phosphinic and phosphonic acids enhanced by ionic liquid additives. Pure Appl. Chem. 2019, 91, 59–65. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Henyecz, R.; Jablonkai, E.; Keglevich, G. Synthesis of n-butyl ester and n-butylamide of methyl-phenylphosphinic acid—Two case studies. Synth. Commun. 2016, 46, 766–774. [Google Scholar] [CrossRef]
- Keglevich, G.; Kiss, N.Z.; Drahos, L.; Körtvélyesi, T. Direct esterification of phosphinic acids under microwave conditions; Extension to the synthesis of thiophosphinates and new mechanistic insights. Tetrahedron Lett. 2013, 54, 466–469. [Google Scholar] [CrossRef]
- Keglevich, G.; Kiss, N.Z.; Körtvélyesi, T. Microwave-assisted functionalization of phosphinic acids; Amidations versus esterifications. Heteroatom Chem. 2013, 24, 91–99. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Mucsi, Z.; Böttger, É.; Drahos, L.; Keglevich, G. A three-step conversion of phenyl-1H-phosphinic acid to dialkyl phenylphosphonates including two microwave-assisted direct esterification steps. Curr. Org. Synth. 2014, 11, 767–772. [Google Scholar] [CrossRef]
- Henyecz, R.; Kiss, A.; Mórocz, V.; Kiss, N.Z.; Keglevich, G. Synthesis of phosphonates from phenylphosphonic acid and its monoesters. Synth. Commun. 2019, 49, 2642–2650. [Google Scholar] [CrossRef] [Green Version]
- Kiss, N.Z.; Henyecz, R.; Keglevich, G. Continuous flow esterification of a H-phosphinic acid, and transesterification of H-phosphinates and H-phosphonates under microwave conditions. Molecules 2020, 25, 719. [Google Scholar] [CrossRef] [Green Version]
- Keglevich, G.; Kiss, N.Z.; Mucsi, Z. A comparative study on the thermal and microwave-assisted direct esterification of phenyl-H-phosphinic acid—Modeling the rate enhancing effect of MWs. Curr. Phys. Chem. 2016, 6, 307–311. [Google Scholar] [CrossRef]
- Keglevich, G.; Greiner, I.; Mucsi, Z. An interpretation of the rate enhancing effect of microwaves—Modelling the distribution and effect of local overheating—A case study. Curr. Org. Chem. 2015, 19, 1436–1440. [Google Scholar] [CrossRef]
- Frank, A.W. Phosphonous acids (thio-, seleno analogs) and deivatives. In Organic Phosphorus Compounds; Kosolapoff, G.M., Maier, L., Eds.; J. Wiley & Sons, Inc.: New York, NY, USA, 1973; Volume 4, pp. 264–265. [Google Scholar]
- Houben, J.; Weyl, T.; Büchel, K.H.; Padeken, H.G. Methoden der Organischen Chemie (Houben-Weyl), Phosphor-Verbindungen II; Weyl, H., Regitz, M., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1982; ISBN 3132171042. [Google Scholar]
- Desai, J.; Wang, Y.; Wang, K.; Malwal, S.R.; Oldfield, E. Isoprenoid biosynthesis inhibitors targeting bacterial cell growth. Chem. Med. Chem. 2016, 11, 2205–2215. [Google Scholar] [CrossRef]
- Tcarkova, K.V.; Artyushin, O.I.; Bondarenko, N.A. Synthetic routes to bis(3-aminophenyl) phosphinic acid. Phosphorus Sulfur Silicon 2016, 191, 1520–1522. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Bölcskei, A.; Drahos, L.; Kraszni, M.; Balogh, G.T. Synthesis and proton dissociation properties of arylphosphonates; A microwave-assisted catalytic Arbuzov reaction with aryl bromides. Heteroatom Chem. 2012, 23, 574–582. [Google Scholar] [CrossRef]
- Gavande, N.; Yamamoto, I.; Salam, N.K.; Ai, T.-H.; Burden, P.M.; Johnston, G.A.R.; Hanrahan, J.R.; Chebib, M. Novel Cyclic Phosphinic Acids as GABAC ρ Receptor Antagonists: Design, Synthesis, and Pharmacology. ACS Med. Chem. Lett. 2011, 2, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Haake, P.; Hurst, G. Reactions of phosphinates. The acid-catalyzed and acid-inhibited hydrolysis of p-nitrophenyl diphenylphosphinate. J. Am. Chem. Soc. 1966, 88, 2544–2550. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z.; Harsági, N.; Szigetvári, Á.; Kiss, N.Z. A study on the acidic hydrolysis of cyclic phosphinates: 1-Alkoxy-3-phospholene 1-oxides, 1-ethoxy-3-methylphospholane 1-oxide, and 1-ethoxy-3-methyl-1,2,3,4,5,6-hexahydrophosphinine 1-oxide. Heteroatom Chem. 2017, 28, e21394. [Google Scholar] [CrossRef]
- Harsági, N.; Rádai, Z.; Kiss, N.Z.; Szigetvári, Á.; Keglevich, G. Two step acidic hydrolysis of dialkyl arylphosphonates. Mendeleev Commun. 2020, 30, 38–39. [Google Scholar] [CrossRef]
- Harsági, N.; Rádai, Z.; Szigetvári, Á.; Kóti, J.; Keglevich, G. Optimization and a kinetic study on the acidic hydrolysis of dialkyl α-hydroxybenzylphosphonates. Molecules 2020, 25, 3793. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Nayler, R.A. Hydrolysis of phosphinic esters: General-base catalysis by imidazole. J. Chem. Soc. B 1971, 1967–1972. [Google Scholar] [CrossRef]
- Rahil, J.; Haake, P. Rates and mechanism of the alkaline hydrolysis of a sterically hindered phosphinate ester. Partial reaction by nucleophilic attack at carbon. J. Org. Chem. 1981, 46, 3048–3052. [Google Scholar] [CrossRef]
- Cook, R.D.; Farah, S.; Ghawi, L.; Itani, A.; Rahil, J. The influence of the changing of P=O to P=S and POR to PSR on the reactivity of phosphinate esters under alkaline hydrolysis conditions. Can. J. Chem. 1986, 64, 1630–1637. [Google Scholar] [CrossRef]
- Wróblewski, A.E.; Verkade, J.G. 1-oxo-2-oxa-1-phosphabicyclo[2.2.2]octane: A new mechanistic probe for the basic hydrolysis of phosphate esters. J. Am. Chem. Soc. 1996, 118, 10168–10174. [Google Scholar] [CrossRef]
- Cevasco, G.; Thea, S. The quest for carbanion-promoted dissociative pathways in the hydrolysis of aryl phosphinates. J. Chem. Soc. Perkin. Trans 1993, 2, 1103–1106. [Google Scholar] [CrossRef]
- Harsági, N.; Szőllősi, B.; Kiss, N.Z.; Keglevich, G. MW irradiation and ionic liquids as green tools in hydrolyses and alcoholyses. Green Proc. Synth. 2021, 10. in press. [Google Scholar] [CrossRef]
- Kovács, T.; Keglevich, G. The reduction of tertiary phosphine oxides by silanes. Curr. Org. Chem. 2017, 21, 569–585. [Google Scholar] [CrossRef] [Green Version]
- Kovács, T.; Keglevich, G. The deoxygenation of phosphine oxides under green chemical conditions. Phosphorus Sulfur Silicon 2016, 191, 359–366. [Google Scholar] [CrossRef]
- Li, Y.H.; Lu, L.Q.; Das, S.; Pisiewicz, S.; Junge, K.; Beller, M. Highly chemoselective metal-free reduction of phosphine oxides to phosphines. J. Am. Chem. Soc. 2012, 134, 18325–18329. [Google Scholar] [CrossRef] [PubMed]
- Coumbe, T.; Lawrence, N.J.; Muhammad, F. Titanium (IV) catalysis in the reduction of phosphine oxides. Tetrahedron Lett. 1994, 35, 625–628. [Google Scholar] [CrossRef]
- Onodera, G.; Matsumoto, H.; Milton, M.D.; Nishibayashi, Y.; Uemura, S. Ruthenium-catalyzed formation of aryl(diphenyl)phosphine reactions of propargylic diphenylphosphine oxide. Org. Lett. 2005, 7, 4029–4032. [Google Scholar] [CrossRef]
- Li, Y.H.; Das, S.; Zhou, S.L.; Junge, K.; Beller, M. General and selective copper-catalyzed reduction of tertiary and secondary phosphine oxides: Convenient synthesis of phosphines. J. Am. Chem. Soc. 2012, 134, 9727–9732. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, L.; Metay, E.; Delbrayelle, D.; Mignani, G.; Lemaire, M. Reduction of phosphine oxides to phosphines with the InBr3/TMDS system. Tetrahedron 2012, 68, 3151–3155. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The deoxygenation of phosphine oxides under green chemical conditions. Heteroatom Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Binder, J.; Falk, A.; Uhlig, F.; Keglevich, G. A study on the deoxygenation of phosphine oxides by different silane derivatives. Curr. Org. Synth. 2016, 13, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Keglevich, G.; Kovács, T. Silanes as reagents for the deoxygenation of tertiary phosphine oxides—A case study for the deoxygenation of 5-membered cyclic phosphine oxides. Curr. Green Chem. 2014, 1, 182–188. [Google Scholar] [CrossRef]
- Fritzsche, H.; Hasserodt, U.; Korte, F. Reduktion organischer Verbindungen des fünfwertigen Phosphors zu Phosphinen, I. Reduktion tertiärer Phosphinoxyde zu tertiären Phosphinen mit Silanen. Chem. Ber. 1964, 97, 1988–1993. [Google Scholar] [CrossRef]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Keglevich, G. A study on the deoxygenation of trialkyl-, dialkyl-phenyl- and alkyl-diphenyl phosphine oxides by hydrosilanes. Heteroatom Chem. 2017, 28, e21376. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields reaction: Mechanism and synthetic use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef] [Green Version]
- Kafarski, P.; Gorniak, M.G.V.; Andrasiak, I. Kabachnik–Fields reaction under green conditions—A critical overview. Curr. Green Chem. 2015, 2, 218–222. [Google Scholar] [CrossRef]
- Matveeva, E.D.; Podrugina, T.A.; Tishkovskaya, E.V.; Tomilova, L.G.; Zefirov, N.S. A novel catalytic three-component synthesis (Kabachnick-Fields reaction) of α-aminophosphonates from ketones. Synlett 2003, 2321–2324. [Google Scholar] [CrossRef]
- Wu, J.; Sun, W.; Xia, H.-G.; Sun, X. A facile and highly efficient route to α-amino phosphonates via three-component reactions catalyzed by Mg(ClO4)2 or molecular iodine. Org. Biomol. Chem. 2006, 4, 1663–1666. [Google Scholar] [CrossRef] [PubMed]
- Firouzabadi, H.; Iranpoor, N.; Sobhani, S. Metal triflate-catalyzed one-pot synthesis of α-aminophosphonates from carbonyl compounds in the absence of solvent. Synthesis 2004, 16, 2692–2696. [Google Scholar] [CrossRef]
- Ghosh, R.; Maiti, S.; Chakraborty, A.; Maiti, D.K. In(OTf)3 catalysed simple one-pot synthesis of α-amino phosphonates. J. Mol. Catal. A 2004, 210, 53–57. [Google Scholar] [CrossRef]
- Bhattacharya, A.K.; Kaur, T. An efficient one-pot synthesis of α-amino phosphonates catalyzed by bismuth nitrate pentahydrate. Synlett 2007, 745–748. [Google Scholar] [CrossRef]
- Xu, F.; Luo, Y.; Deng, M.; Shen, Q. One-pot synthesis of α-amino phosphonates using samarium diiodide as a catalyst precursor. Eur. J. Org. Chem. 2003, 4728–4730. [Google Scholar] [CrossRef]
- Ravinder, K.; Vijender Reddy, A.; Krishnaiah, P.; Venkataramana, G.; Niranjan Reddy, V.L.; Venkateswarlu, Y. CAN catalyzed one-pot synthesis of α-amino phosphonates from carbonyl compounds. Synth. Commun. 2004, 34, 1677–1683. [Google Scholar] [CrossRef]
- Ranu, B.C.; Hajra, A.; Jana, U. General procedure for the synthesis of α-amino phosphonates from aldehydes and ketones using indium(III) chloride as a catalyst. Org. Lett. 1999, 1, 1141–1143. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.H.; Kang, J.; Lee, J.K. Lanthanide triflate-catalyzed three component synthesis of α-amino phosphonates in ionic liquids. A catalyst reactivity and reusability study. Chem. Commun. 2001, 1698–1699. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A. Eco-friendly accomplishment of the extended Kabachnik–Fields reaction; a solvent- and catalyst-free microwave-assisted synthesis of α-aminophosphonates and α-aminophosphine oxides. Lett. Org. Chem. 2008, 5, 616–622. [Google Scholar] [CrossRef]
- Prauda, I.; Greiner, I.; Ludányi, K.; Keglevich, G. Efficient synthesis of phosphono- and phosphinoxidomethylated N-heterocycles under solvent-free microwave conditions. Synth. Commun. 2007, 37, 317–322. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A.; Sipos, M.; Ludányi, K.; Greiner, I. Synthesis of cyclic aminomethylphosphonates and aminomethyl-arylphosphinic acids by an efficient microwave-mediated phospha-Mannich approach. Heteroatom Chem. 2008, 19, 207–210. [Google Scholar] [CrossRef]
- Bálint, E.; Takács, J.; Drahos, L.; Juranovič, A.; Kočevar, M.; Keglevich, G. α-Aminophosphonates and α-aminophosphine oxides by the microwave-assisted Kabachnik–Fields reactions of 3-amino-6-methyl-2H-pyran-2-ones. Heteroatom Chem. 2013, 24, 221–225. [Google Scholar] [CrossRef]
- Tajti, Á.; Bálint, E.; Keglevich, G. Synthesis of ethyl octyl α-aminophosphonate derivatives. Curr. Org. Synth. 2016, 13, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Bálint, E.; Tóth, R.E.; Keglevich, G. Synthesis of alkyl α-aminomethyl-phenylphosphinates and N,N-bis(alkoxyphenylphosphinylmethyl)amines by the microwave-assisted Kabachnik–Fields reaction. Heteroatom Chem. 2016, 27, 323–335. [Google Scholar] [CrossRef]
- Hudson, H.R.; Tajti, Á.; Bálint, E.; Czugler, M.; Karaghiosoff, K.; Keglevich, G. Microwave-assisted synthesis of α-aminophosphonates with sterically demanding α-aryl substituents. Synth. Commun. 2020, 50, 1446–1455. [Google Scholar] [CrossRef]
- Tripolszky, A.; Zoboki, L.; Bálint, E.; Kóti, J.; Keglevich, G. Microwave-assisted synthesis of α-aminophosphine oxides by the Kabachnik-Fields reaction applying amides as the starting materials. Synth. Commun. 2019, 49, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Milen, M.; Ábrányi-Balogh, P.; Dancsó, A.; Frigyes, D.; Pongó, L.; Keglevich, G. T3P®-promoted Kabachnik–Fields reaction: An efficient synthesis of α-aminophosphonates. Tetrahedron Lett. 2013, 54, 5430–5433. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Garifzyanov, A.R.; Talan, A.S.; Davletshin, R.R.; Kurnosova, N.V. Synthesis of new liophilic functionalized aminomethylphosphine oxides and their acid-base and membrane-transport properties toward acidic substrates. Russ. J. Gen. Chem. 2009, 79, 1835–1849. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A.; Szöllősy, Á.; Drahos, L. Synthesis of bis(phosphonatomethyl)-, bis(phosphinatomethyl)-, and bis(phosphinoxidomethyl)amines, as well as related ring bis(phosphine) platinum complexes. Synth. Commun. 2011, 41, 2265–2272. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Pintér, G.; Szőllősy, A.; Holczbauer, T.; Czugler, M.; Drahos, L.; Körtvélyesi, T.; Keglevich, G. Synthesis and utilization of the bis(>P(O)CH2)amine derivatives obtained by the double Kabachnik–Fields reaction with cyclohexylamine; Quantum chemical and X-ray study of the related bidentate chelate platinum complexes. Curr. Org. Chem. 2012, 16, 547–554. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Pongrácz, P.; Kollár, L.; Drahos, L.; Holczbauer, T.; Czugler, M.; Keglevich, G. N-benzyl and N-aryl bis(phospha-Mannich adducts): Synthesis and catalytic activity of the related bidentate chelate platinum complexes in hydroformylation. J. Organomet. Chem. 2012, 717, 75–82. [Google Scholar] [CrossRef]
- Bálint, E.; Tripolszky, A.; Jablonkai, E.; Karaghiosoff, K.; Czugler, M.; Mucsi, Z.; Kollár, L.; Pongrácz, P.; Keglevich, G. Synthesis and use of α-aminophosphine oxides and N,N-bis(phosphinoylmethyl)amines—A study on the related ring platinum complexes. J. Organomet. Chem. 2015, 801, 111–121. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Drahos, L.; Keglevich, G. The synthesis of N,N-bis(dialkoxyphosphinoylmethyl)- and N,N-bis(diphenylphosphinoylmethyl) glycine esters by the microwave-assisted double Kabachnik–Fields reaction. Heteroatom Chem. 2013, 24, 510–515. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Kóti, J.; Keglevich, G. Synthesis of N,N-bis(dialkoxyphosphinoylmethyl)- and N,N-bis(diphenylphosphinoylmethyl)-β- and γ-amino acid derivatives by the microwave-assisted double Kabachnik–Fields reaction. Heteroatom Chem. 2015, 26, 106–115. [Google Scholar] [CrossRef]
- Keglevich, G.; Fehérvári, A.; Csontos, I. A study on the Kabachnik–Fields reaction of benzaldehyde, propylamine and diethyl phosphite by in situ Fourier transform (FT) IR spectroscopy. Heteroatom Chem. 2011, 22, 599–604. [Google Scholar] [CrossRef]
- Keglevich, G.; Kiss, N.Z.; Menyhárd, D.; Fehérvári, A.; Csontos, I. A study on the Kabachnik–Fields reaction of benzaldehyde, cyclohexylamine and dialkyl phosphites. Heteroatom Chem. 2012, 23, 171–178. [Google Scholar] [CrossRef]
- Gancarz, R.; Gancarz, I.; Walkowiak, U. On the reversibility of hydroxyphosphonate formation in the Kabachnik–Fields reaction. Phosphorus Sulfur Silicon 1995, 104, 45–52. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z. α-Hydroxyphosphonates as intermediates in the Kabachnik–Fields reaction: New proof of their reversible formation. Tetrahedron Lett. 2020, 61, 151961. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Kaszás, A.; Drahos, L.; Mucsi, Z.; Keglevich, G. A neighbouring group effect leading to enhanced nucleophilic substitution of amines at the hindered α-carbon atom of an α-hydroxyphosphonate. Tetrahedron Lett. 2012, 53, 207–209. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Rádai, Z.; Mucsi, Z.; Keglevich, G. Synthesis of α-aminophosphonates from α-hydroxyphosphonates; A theoretical study. Heteroatom Chem. 2016, 27, 260–268. [Google Scholar] [CrossRef]
- Rádai, Z.; Keglevich, G. Synthesis and reactions of α-hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keglevich, G.; Tóth, V.R.; Drahos, L. Microwave-assisted synthesis of α-hydroxy-benzylphosphonates and -benzylphosphine oxides. Heteroatom Chem. 2011, 22, 15–17. [Google Scholar] [CrossRef]
- Grün, A.; Molnár, I.G.; Bertók, B.; Greiner, I.; Keglevich, G. Synthesis of α-hydroxy-methylenebisphosphonates by the microwave-assisted reaction of α-oxophosphonates and dialkyl phosphites under solventless conditions. Heteroatom Chem. 2009, 20, 350–354. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Molnár, I.G.; Greiner, I. Phenyl-, benzyl- and unsymmetrical hydroxy-methylenebisphosphonates as dronic acid ester analogues from α-oxophosphonates by microwave-assisted synthesis. Heteroatom Chem. 2011, 22, 640–648. [Google Scholar] [CrossRef]
- Kumar, K.S.; Reddy, C.B.; Reddy, M.V.N.; Rani, C.R.; Reddy, C.S. Green chemical synthesis of α-hydroxyphosphonates. Org. Commun. 2012, 5, 50–57. [Google Scholar]
- Tajbakhsh, M.; Khaksar, S.; Tafazoli, Z.; Bekhradnia, A. MgCl2/Et3N base system as a new catalyst for the synthesis of α-hydroxyphosphonate. Chin. J. Chem. 2012, 30, 827–829. [Google Scholar] [CrossRef]
- Nandre, K.P.; Nandre, J.P.; Patil, V.S.; Bhosale, S.V. Barium hydroxide catalyzed greener protocol for the highly efficient and rapid synthesis of α-hydroxyphosphonates under solvent free conditions. Chem. Biol. Interfaces 2012, 2, 314–321. [Google Scholar]
- Kong, D.-L.; Liu, R.-D.; Li, G.-Z.; Zhang, P.-W.; Wu, M.-S. A rapid, convenient, solventless green approach for the synthesis of α-hydroxyphosphonates by grinding. Asian J. Chem. 2014, 26, 1246–1248. [Google Scholar] [CrossRef]
- Kulkarni, M.A.; Lad, U.P.; Desai, U.V.; Mitragotri, S.D.; Wadgaonkar, P.P. Mechanistic approach for expeditious and solvent-free synthesis of α-hydroxy phosphonates using potassium phosphate as catalyst. Compt. Rend. Chim. 2013, 16, 148–152. [Google Scholar] [CrossRef]
- Ramananarivo, H.R.; Solhy, A.; Sebti, J.; Smahi, A.; Zahouily, M.; Clark, J.; Sebti, S. An eco-friendly paradigm for the synthesis of α-hydroxyphosphonates using sodium-modified fluorapatite under solventless conditions. ACS Sustain. Chem. Eng. 2013, 403–409. [Google Scholar] [CrossRef]
- Santhisudha, S.; Sreelakshmi, P.; Jayaprakash, S.H.; Kumar, B.V.; Reddy, C.S. Silica-supported tungstic acid catalyzed synthesis and antioxidant activity of α-hydroxyphosphonates. Phosphorus Sulfur 2015, 190, 1479–1488. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z.; Kiss, N.Z. To date the greenest method for the preparation of α-hydroxyphosphonates from substituted benzaldehydes and dialkyl phosphites. Green Proc. Synth. 2017, 6, 197–201. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. P–C bond formation by coupling reaction utilizing >P(O)H species as the reagents. Curr. Org. Synth. 2014, 11, 429–453. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. Advances and new variations of the Hirao reaction. Org. Prep. Proced. Int. 2014, 46, 281–316. [Google Scholar] [CrossRef]
- Henyecz, R.; Keglevich, G. New developments on the Hirao reactions, especially from ”green” point of view. Curr. Org. Synth. 2019, 16, 523–545. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Stereoselective synthesis of vinylphosphonate. Tetrahedron Lett. 1980, 21, 3595–3598. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Yamada, N.; Ohshiro, Y.; Agawa, T. Palladium-catalyzed new carbon-phosphorus bond formation. Bull. Chem. Soc. Jpn. 1982, 55, 909–913. [Google Scholar] [CrossRef] [Green Version]
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. A novel synthesis of dialkyl arenephosphonates. Synthesis 1981, 56–57. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, J. Palladium-catalyzed reaction of aryl polyfluoroalkanesulfonates with O,O-dialkyl phosphonates. Synthesis 1987, 726–727. [Google Scholar] [CrossRef]
- Ziessel, R.F.; Charbonnière, L.J.; Mameri, S.; Camerel, F. Bridging of bipyridine units by phenylphosphine links: Linear and cyclic oligomers and some acid derivatives. J. Org. Chem. 2005, 70, 9835–9840. [Google Scholar] [CrossRef]
- Kant, M.; Bischoff, S.; Siefken, R.; Gründemann, E.; Köckritz, A. Synthesis and characterization of 4-and 4,4’-phosphorylated 2,2’-bis(diphenylphosphanyl)-1,1’-binaphthyls. Eur. J. Org. Chem. 2001, 477–481. [Google Scholar] [CrossRef]
- Chauhan, S.S.; Varshney, A.; Verma, B.; Pennington, M.W. Efficient synthesis of protected L-phosphonophenylalanine (Ppa) derivatives suitable for solid phase peptide synthesis. Tetrahedron 2007, 48, 4051–4054. [Google Scholar] [CrossRef]
- Petrakis, K.S.; Nagabhushan, T.L. Palladium-catalyzed substitutions of triflates derived from tyrosine-containing peptides and simpler hydroxyarenes forming 4-(diethoxyphosphinyl)phenylalanines and diethyl arylphosphonates. J. Am. Chem. Soc. 1987, 109, 2831–2833. [Google Scholar] [CrossRef]
- Kobayashi, Y.; William, A.D. Palladium- and nickel-catalyzed coupling reactions of α-bromoalkenylphosphonates with arylboronic acids and lithium alkenylborates. Adv. Synth. Cat. 2004, 346, 1749–1757. [Google Scholar] [CrossRef]
- Kobayashi, Y.; William, A.; Tokoro, Y. Sharpless asymmetric dihydroxylation of trans-propenylphosphonate by using a modified AD-mix-α and the synthesis of fosfomycin. J. Org. Chem. 2001, 66, 7903–7906. [Google Scholar] [CrossRef]
- Trost, B.M.; Radinov, R. On the effect of a cation binding site in an asymmetric ligand for a catalyzed nucleophilic substitution reaction. J. Am. Chem. Soc. 1997, 119, 5962–5963. [Google Scholar] [CrossRef]
- Hall, R.G.; Riebli, P. Preparation of new phosphine oxide synthons: Synthesis of an analogue of muscarinic antagonists. Synlett 1999, 1633–1635. [Google Scholar] [CrossRef]
- Xie, J.-H.; Wang, L.-X.; Fu, Y.; Zhu, S.-F.; Fan, B.-M.; Duan, H.-F.; Zhou, Q.-L. Synthesis of spiro diphosphines and their application in asymmetric hydrogenation of ketones. J. Am. Chem. Soc. 2003, 125, 4404–4405. [Google Scholar] [CrossRef]
- Muthukumaran, K.; Loewe, R.S.; Ambroise, A.; Tamaru, S.-I.; Li, Q.; Mathur, G.; Bocian, D.F.; Misra, V.; Lindsey, J.S. Porphyrins bearing arylphosphonic acid tethers for attachment to oxide surfaces. J. Org. Chem. 2004, 69, 1444–1452. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Hervé, A.; Loiseau, F.; Virieux, D. Synthesis of new arylhydroxymethylphosphinic acids and derivatives. Synthesis 2003, 14, 2216–2220. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Z.; Xia, J.; Guo, H.; Huang, Y. Palladium-catalyzed synthesis of alkylarylphenylphosphine oxides. Synthesis 1984, 781–782. [Google Scholar] [CrossRef]
- Gooßen, L.J.; Dezfuli, M.K. Practical protocol for the palladium-catalyzed synthesis of arylphosphonates from bromoarenes and diethyl phosphite. Synlett 2005, 445–448. [Google Scholar] [CrossRef]
- Bessmertnykh, A.; Douaihy, C.M.; Guilard, R. Direct synthesis of amino-substituted aromatic phosphonates via palladium-catalyzed coupling of aromatic mono- and dibromides with diethyl phosphite. Chem. Lett. 2009, 38, 738–739. [Google Scholar] [CrossRef]
- Belabassi, Y.; Alzghari, S.; Montchamp, J.-L. Revisiting the Hirao cross-coupling: Improved synthesis of aryl and heteroaryl phosphonates. J. Organomet. Chem. 2008, 693, 3171–3178. [Google Scholar] [CrossRef] [Green Version]
- Bonnaventure, I.; Charette, A.B. Probing the importance of the hemilabile site of bis(phosphine) monoxide ligands in the copper-catalyzed addition of diethylzinc to N-phosphinoylimines: Discovery of new effective chiral ligands. J. Org. Chem. 2008, 73, 6330–6340. [Google Scholar] [CrossRef]
- Nandi, M.; Jin, J.; RajanBabu, T.V. Synergistic effects of hemilabile coordination and counterions in homogeneous catalysis: New tunable monophosphine ligands for hydrovinylation reactions. J. Am. Chem. Soc. 1999, 121, 9899–9900. [Google Scholar] [CrossRef]
- Yan, Y.-Y.; RajanBabu, T.V. Highly flexible synthetic routes to functionalized phospholanes from carbohydrates. J. Org. Chem. 2000, 65, 900–906. [Google Scholar] [CrossRef]
- Davies, L.H.; Stewart, B.; Harrington, R.W.; Clegg, W.; Higham, L.J. Air-stable, highly fluorescent primary phosphanes. Agnew. Chem. Int. Ed. 2012, 51, 4921–4924. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.P.; Savchenko, J.; Fanwick, P.E.; Kharlamova, A.; Ren, T. Diruthenium alkynyl compounds with phosphonate capping groups. Organometallics 2013, 32, 1129–1132. [Google Scholar] [CrossRef]
- Reisinger, B.; Kuzmanovic, N.; Löffler, P.; Merkl, R.; König, B.; Sterner, R. Exploiting protein symmetry to design light-controllable enzyme inhibitors. Angew. Chem. Int. Ed. 2014, 53, 595–598. [Google Scholar] [CrossRef]
- Bazaga-García, M.; Colodrero, R.M.P.; Papadaki, M.; Garczarek, P.; Zoń, J.; Olivera-Pastor, P.; Losilla, E.R.; León-Reina, L.; Aranda, M.A.G.; Choquesillo-Lazarte, D.; et al. Guest molecule-responsive functional calcium phosphonate frameworks for tuned proton conductivity. J. Am. Chem. Soc. 2014, 136, 5731–5739. [Google Scholar] [CrossRef]
- Contrella, N.D.; Sampson, J.R.; Jordan, R.F. Copolymerization of ethylene and methyl acrylate by cationic palladium catalysts that contain phosphine-diethyl phosphonate ancillary ligands. Organometallics 2014, 33, 3546–3555. [Google Scholar] [CrossRef]
- Deal, E.L.; Petit, C.; Montchamp, J.-L. Palladium-catalyzed cross-coupling of H-phosphinate esters with chloroarenes. Org. Lett. 2011, 13, 3270–3273. [Google Scholar] [CrossRef]
- Berger, O.; Petit, C.; Deal, E.L.; Montchamp, J.-L. Phosphorus-Carbon Bond Formation: Palladium-Catalyzed Cross-Coupling of H-Phosphinates and Other P(O)H-Containing Compounds. Adv. Synth. Catal. 2013, 355, 1361–1373. [Google Scholar] [CrossRef]
- Villemin, D.; Jaffrès, P.-A.; Simèon, F. Rapid and efficient phosphonation of aryl halides catalysed by palladium under microwaves irradiation. Phosphorus Sulfur Silicon 1997, 130, 59–63. [Google Scholar] [CrossRef]
- Kalek, M.; Ziadi, A.; Stawinski, J. Microwave-assisted palladium-catalyzed cross-coupling of aryl and vinyl halides with H-phosphonate diesters. Org. Lett. 2008, 10, 4637–4640. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Allan, G.; Fiordeliso, J.J.; Linton, O.; Tannenbaum, P.; Xu, J.; Zhu, P.; Gunnet, J.; Demarest, K.; Lundeen, S.; et al. New progesterone receptor antagonists: Phosphorus-containing 11 β-aryl-substituted steroids. Bioorg. Med. Chem. 2006, 14, 6726–6732. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, J.; Sävmarker, J.; Sjöberg, P.J.R.; Larhed, M. Microwave-promoted palladium(II)-catalyzed C-P bond formation by using arylboronic acids or aryltrifluoroborates. Chem. Eur. J. 2009, 15, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Ananthnag, G.S.; Mague, J.T.; Balakrishna, M.S. A cyclodiphosphazane based pincer ligand, [2,6-{μ-(tBuN)2P(tBuHN)PO}2C6H3I]: NiII, PdII, PtII and CuI complexes and catalytic studies. Dalton Trans. 2015, 44, 3785–3793. [Google Scholar] [CrossRef] [PubMed]
- Rummelt, S.M.; Ranocchiari, M.; van Bokhoven, J.A. Synthesis of water-soluble phosphine oxides by Pd/C-catalyzed P-C coupling in water. Org. Lett. 2012, 14, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Jablonkai, E.; Keglevich, G. P-Ligand-free, microwave-assisted variation of the Hirao reaction under solvent-free conditions; the P–C coupling reaction of >P(O)H species and bromoarenes. Tetrahedron Lett. 2013, 54, 4185–4188. [Google Scholar] [CrossRef]
- Keglevich, G.; Jablonkai, E.; Balázs, L.B. A “green” variation of the Hirao reaction: The P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions. RSC Adv. 2014, 4, 22808–22816. [Google Scholar] [CrossRef] [Green Version]
- Amaya, T.; Abe, Y.; Inada, Y.; Hirao, T. Synthesis of self-doped conducting polyaniline bearing phosphonic acid. Tetrahedron Lett. 2014, 55, 3976–3978. [Google Scholar] [CrossRef]
- Li, J.; Bi, X.; Wang, H.; Xiao, J. Palladium-catalyzed desulfitative C-P coupling of arylsulfinate metal salts and H-phosphonates. RSC Adv. 2014, 4, 19214–192217. [Google Scholar] [CrossRef]
- Henyecz, R.; Oroszy, R.; Keglevich, G. Microwave-assisted Hirao reaction of heteroaryl bromides and >P(O)H reagents using Pd(OAc)2 as the catalyst precursor in the absence of added P-ligands. Curr. Org. Chem. 2019, 23, 1151–1157. [Google Scholar] [CrossRef]
- Henyecz, R.; Huszár, B.; Grenitzer, V.; Keglevich, G. A study on the reactivity of monosubstituted benzenes in the MW-assisted Pd(OAc)2-catalyzed Hirao reaction with Ph2P(O)H and (EtO)2P(O)H reagents. Curr. Org. Chem. 2020, 24, 1048–1054. [Google Scholar] [CrossRef]
- Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N.Z. The palladium acetate-catalyzed microwave-assisted Hirao reaction without an added phosphorus ligand as a “green” protocol: A quantum chemical study on the mechanism. Adv. Synth. Catal. 2017, 359, 4322–4331. [Google Scholar] [CrossRef] [PubMed]
- Henyecz, R.; Mucsi, Z.; Keglevich, G. Palladium-catalyzed microwave-assisted Hirao reaction utilizing the excess of the diarylphosphine oxide reagent as the P-ligand; a study on the activity and formation of the “PdP2” catalyst. Pure Appl. Chem. 2019, 91, 121–134. [Google Scholar] [CrossRef]
- Jablonkai, E.; Balázs, L.B.; Keglevich, G. A P-ligand-free nickel-catalyzed variation of the Hirao reaction under microwave conditions. Curr. Org. Chem. 2015, 19, 197–202. [Google Scholar] [CrossRef]
- Henyecz, R.; Mucsi, Z.; Keglevich, G. A surprising mechanism lacking the Ni(0) state during the Ni(II)-catalyzed P–C cross-coupling reaction performed in the absence of a reducing agent—An experimental and a theoretical study. Pure Appl. Chem. 2020, 92, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Liu, H.Z.; Hu, X.M.; Tang, G.; Zhu, J.; Zhao, Y.F. Ni(II)/Zn Catalyzed reductive coupling of aryl halides with diphenylphosphine oxide in water. Org. Lett. 2011, 13, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Kinbara, A.; Ito, M.; Abe, T.; Yamagishi, T. Nickel-catalyzed C–P cross-coupling reactions of aryl iodides with H-phosphinates. Tetrahedron 2015, 71, 7614–7619. [Google Scholar] [CrossRef]
- Shen, C.R.; Yang, G.Q.; Zhang, W.B. Nickel-catalyzed C–P coupling of aryl mesylates and tosylates with H(O)PR1R2. Org. Biomol. Chem. 2012, 10, 3500–3505. [Google Scholar] [CrossRef]
- Keglevich, G.; Henyecz, R.; Mucsi, Z. Experimental and theoretical study on the “2,2′-bipiridyl-Ni-catalyzed” Hirao reaction of >P(O)H reagents and halobenzenes: A Ni(0) → Ni(II) or a Ni(II) → Ni(IV) mechanism? J. Org. Chem. 2020, 85, 14486–14495. [Google Scholar] [CrossRef] [PubMed]
- Jablonkai, E.; Keglevich, G. Catalyst-free P–C coupling reaction of halobenzoic acids and secondary phosphine oxides under microwave irradiation in water. Tetrahedron Lett. 2015, 56, 1638–1640. [Google Scholar] [CrossRef]
Scheme 1.
[4 + 2]Cycloadditions of a 1,2-dihydrophosphinine oxide (1) with dienophiles.

Scheme 2.
The inverse Wittig-type reaction of a P-aryl 1,2-dihydrohosphine oxide with dimethyl acetylenedicarboxylate (DMAD).
Scheme 2.
The inverse Wittig-type reaction of a P-aryl 1,2-dihydrohosphine oxide with dimethyl acetylenedicarboxylate (DMAD).

Scheme 3.
The inverse Wittig-type reaction of P-aryl ring phosphine oxides with DMAD.

Scheme 4.
The MW-assisted catalytic direct esterification 1-hydroxy-3-methyl-3-phospholene 1-oxide (8).
Scheme 4.
The MW-assisted catalytic direct esterification 1-hydroxy-3-methyl-3-phospholene 1-oxide (8).

Scheme 5.
MW-promoted direct esterification of different phenyl-phosphinic acids (18, 20, and 22).

Scheme 6.
The preparation of dialkyl phenylphosphonates (25) via alkyl phenyl-H-phosphinates (19).

Scheme 7.
The new protocol elaborated for the synthesis of dialkyl phenylphosphonates.

Scheme 8.
MW-assisted hydrolysis of alkyl diphenylphosphinates (29).

Scheme 9.
MW-assisted phospha-Mannich condensations.

Scheme 10.
The bis(phospha-Mannich) reaction.

Scheme 11.
MW-promoted synthesis of α-hydroxyphosphonates and α-hydroxyphosphine oxides by the Pudovik reaction.
Scheme 11.
MW-promoted synthesis of α-hydroxyphosphonates and α-hydroxyphosphine oxides by the Pudovik reaction.

Scheme 12.
The classical P–C coupling reactions.

Scheme 13.
The first P–C coupling realized on MW irradiation.

Scheme 14.
The derivation of 11β-aryl-substituted steroids with the help of the Hirao reaction.

Scheme 15.
Hirao reaction in water using Pd/C on MW irradiation.

Scheme 16.
MW-promoted P–C couplings applying Pd(OAc)2 without added P-ligands.

Scheme 17.
An additional Pd(OAc)2-catalyzed “P-ligand-free” P–C coupling.

Scheme 18.
Hirao reactions applying NiCl2 as the catalyst without added P-ligands.

Scheme 19.
NiCl2-catalyzed P–C couplings of bromoarenes with diethyl phosphite.

Scheme 20.
A catalyst-free Hirao reaction in aqueous medium.


{kind=link}
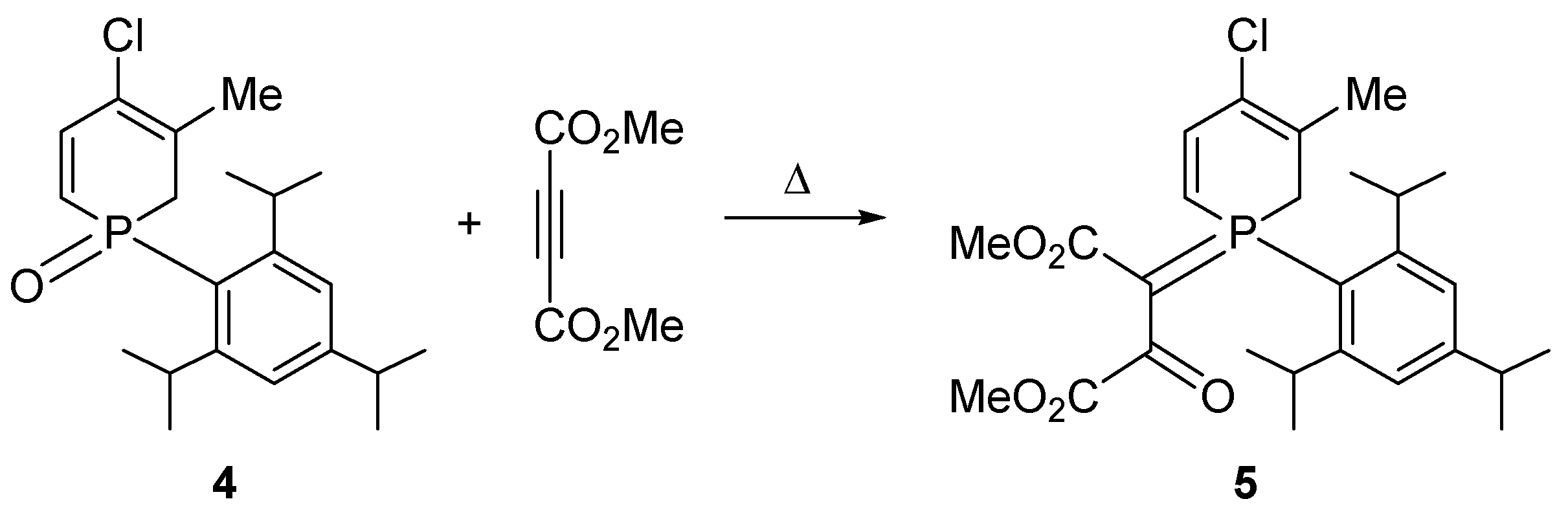{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
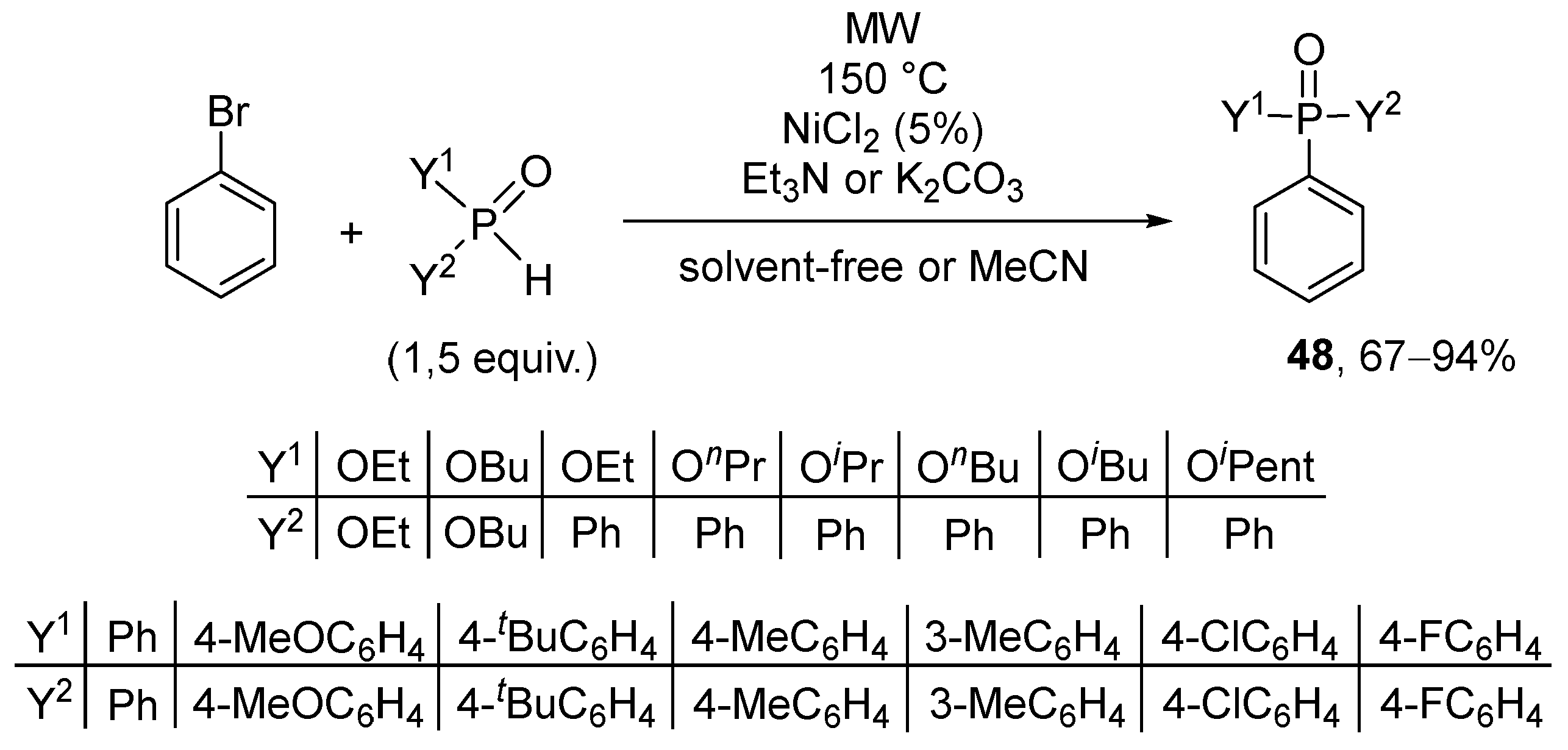{kind=link}
{kind=link}
{kind=link}
Table 1.
Esterification with other alcohols.

| Entry | R | [bmim][PF6] | T (°C) | t (h) | Conversion (%) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Pr | – | 180 | 4 | 40 | 30 |
| 2 | Pr | 10% | 180 | 3 | 98 | 68 |
| 3 | Pent | – | 220 | 2.5 | 100 | 82 |
| 4 | Pent | 10% | 180 | 0.5 | 100 | 94 |
| 5 | iPent | – | 235 | 3 | 100 * | 76 |
| 6 | iPent | 10% | 180 | 0.5 | 100 | 95 |
| 7 | Oct | – | 220 | 2 | 95 * | 71 |
| 8 | Oct | 10% | 180 | 0.33 | 100 | 85 |
| 9 | iOct | – | 220 | 1 | 100 | 76 |
| 10 | iOct | 10% | 180 | 0.33 | 100 | 84 |
| 11 | Dodecyl | – | 230 | 2 | 100 | 95 |
| 12 | Dodecyl | 10% | 180 | 0.33 | 100 | 94 |
* Estimated value.
Table 2.
Extension of the MW-assisted IL-catalyzed direct esterification to other cyclic phosphinic acids.
Table 2.
Extension of the MW-assisted IL-catalyzed direct esterification to other cyclic phosphinic acids.
| Model Reaction | [bmim][PF6] | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|
 | – | 235 | 3 | 67 |
| 10% | 200 | 1 | 72 | |
 | – | 235 | 3 | 79 |
| 10% | 220 | 1 | 89 | |
 | – | 235 | 5 | 60 |
| 10% | 220 | 2 | 84 | |
 | – | 220 | 4 | 31 |
| 10% | 200 | 2 | 42 |
Table 3.
MW-promoted direct esterification of phenylphosphonic acid (26).

| R | T (°C) | t (h) | Conversion (%) | Composition | Yield of the Monoester (%) | |
|---|---|---|---|---|---|---|
| Monoester (%) | Diester (%) | |||||
| Bu | 180 | 0.75 | 100 | 95 | 5 | 82 |
| Et | 165 | 8 | 82 | 94 | 6 | 70 |
| Oct | 180 | 0.75 | 100 | 96 | 4 | 90 |
Table 4.
MW-promoted direct esterification of phenylphosphonic monoesters (27).

| R | T (°C) | t (h) | Conversion (%) |
|---|---|---|---|
| Bu | 220 | 6 | 45 |
| Oct | 235 | 3 | 72 |
Table 5.
Alkylating esterification of phenylphosphonic acid (26).

| BuBr (Equiv.) | T (°C) | t (h) | Conversion (%) | Composition | |
|---|---|---|---|---|---|
| Monoester (%) | Diester (%) | ||||
| 1 | 100 | 2 | 62 | 61 | 39 |
| 2.2 | 100 | 4 | 95 | 32 | 68 |
| 5 | 120 | 4 | 100 | 17 | 83 * |
* The diester was prepared in a yield of 69%.
Table 6.
Alkylation of phenylphosphonic monoesters (27)—synthesis of dialkyl phenylphosphonates (25).
Table 6.
Alkylation of phenylphosphonic monoesters (27)—synthesis of dialkyl phenylphosphonates (25).

| R | RX | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|
| Bu | BuBr | 85 * | 0.5 | 80 |
| Et | EtI | 85 | 0.5 | 92 |
| Oct | OctBr | 100 | 1 | 72 |
* The yield of the comparative thermal experiment was 57%.
Table 7.
The preparation of phenylphosphonates (28) with different alkyl groups.

| R | R’X | Yield of the “Mixed” Ester (%) |
|---|---|---|
| Bu | EtI | 71 |
| Bu | nPrBr | 68 |
| Bu | iPrBr | 48 |
| Et | nPrBr | 72 |
| Et | iPrBr | 54 |
| Et | BuBr | 72 |
Table 8.
Deoxygenation of triphenylphosphine oxide (31) using (EtO)2MeSiH with acid catalysts.

| Catalyst | Yield (%) |
|---|---|
| – | <1 |
| PhCOOH | 6 |
| (PhO)2P(O)OH | 75 |
| (4-NO2C6H4O)2P(O)OH | >99 |
| (4-CF3C6H4O)2P(O)OH | >99 |
Table 9.
Reductions with triethoxysilane.

| R | Yield (%) |
|---|---|
| Ph | 85 |
| CH2CH2P(O)Ph2 | 90 |
| Me | 99 |
| Et | 95 |
| iPr | 81 |
| iPr | 41 |
| Bn | 98 |
| CH2Bn | 83 |
Table 10.
The catalytic deoxygenation of triphenylphosphine oxide (31) applying 1,1,3,3-tetramethyldisiloxane (TMDS) as the reducing agent.
Table 10.
The catalytic deoxygenation of triphenylphosphine oxide (31) applying 1,1,3,3-tetramethyldisiloxane (TMDS) as the reducing agent.

| Equivalent of TMDS | Catalyst (mol%) | T (°C) | t (h) | Conversion (%) |
|---|---|---|---|---|
| 12 | – | 100 | 2 | <1 |
| 3 | Cu(OTf)2 (10) | 100 | 15 | 96 |
| 3 | (PhO)2P(O)OH (15) | 110 | 24 | 62 |
| 1.2 | Ti(OiPr)4 (10) | 100 | 24 | 86 |
| 3 | InBr3 (1) | 100 | 18 | >99 |
Table 11.
Deoxygenation of triphenylphosphine oxide (31) using TMDS in the absence of catalysts.

| Equivalent of TMDS | Mode of Heating | T (°C) | t (h) | Conversion (%) |
|---|---|---|---|---|
| 2 | Δ | 175 | 24 | 92 |
| 2 | MW | 200 | 6.5 | 100 |
Table 12.
Deoxygenation of phospholene oxides (35) applying TMDS.

| P=O | Equivalent of TMDS | Catalyst (mol%) | Solvent | Mode of Heating | T (°C) | t (h) | Conversion (%) |
|---|---|---|---|---|---|---|---|
| a | 3 | InBr3 (1) | PhMe | Δ | 100 | 40 | 95 |
| a | 2 | – | PhMe | Δ | 110 | 8 | 82 |
| b | 2 | – | – | Δ | 110 | 5 | 100 |
| b | 2 | – | – | MW | 110 | 3 | 100 |
Table 13.
Deoxygenation of triphenylphosphine oxide with polymethylhydrosiloxane (PMHS).

| Equivalent of PMHS | Catalyst (mol%) | Mode of Heating | Solvent | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 5 | – | Δ | – | 290 | 2 | 86 |
| 12 | – | Δ | PhMe | 100 | 2 | 0 |
| 6 | Cu(OTf)2 (10) | Δ | PhMe | 100 | 15 | 88 |
| 4 | (PhO)2P(O)OH (15) | Δ | PhMe | 110 | 24 | 35 |
| 2 | – | Δ | – | 175 | 17 | 87 |
| 2 | – | MW | – | 175 | 8 | 90 |
Table 14.
The deoxygenation of a series of phospholene oxides using PMHS.

| Phosphine Oxide | Silane (Equiv.) | Mode of Heating | Solvent | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|---|---|
| a | 5 | Δ | – | 250 | 2 | 88 |
| a | 2 | Δ | PhMe | 110 | 6 | 85 |
| b | 2 | Δ | – | 110 | 4 | 91 |
| b | 2 | MW | – | 110 | 2 | 92 |
| c | 5 | Δ | – | 250 | – | 35 |
| c | 2 | Δ | PhMe | 110 | 6 | 92 |
Table 15.
MW-assisted Hirao reaction of bromoarenes and diethyl phosphite applying Pd(OAc)2 as the catalyst precursor.
Table 15.
MW-assisted Hirao reaction of bromoarenes and diethyl phosphite applying Pd(OAc)2 as the catalyst precursor.

| Y | Pd(OAc)2 (%) | T (°C) | t (min) | Conversion (%) | Yield (46) (%) |
|---|---|---|---|---|---|
| H | 5 | 150 | 5 | 99 | 93 |
| 4-MeO | 10 | 200 | 2 | 80 | 69 |
| 3-MeO | 10 | 200 | 2 | 93 | 79 |
| 4-Pr | 10 | 200 | 2 | 86 | 71 |
| 4-Et | 10 | 175 | 15 | 93 | 85 |
| 4-Me | 10 | 175 | 10 | 86 | 73 |
| 4-Cl | 10 | 175 | 10 | 95 | 83 |
| 3-Cl | 10 | 175 | 10 | 95 | 87 |
| 4-F | 5 | 175 | 5 | 99 | 91 |
| 3-F | 5 | 175 | 10 | 100 | 88 |
| 4-CO2Et | 5 | 175 | 15 | 100 | 89 |
| 3-CO2Et | 10 | 200 | 2 | 93 | 81 |
| 4-C(O)Me | 5 | 175 | 5 | 96 | 71 |
| 3-C(O)Me | 10 | 175 | 5 | 100 | 92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Keglevich, G. Microwaves as “Co-Catalysts” or as Substitute for Catalysts in Organophosphorus Chemistry. Molecules 2021, 26, 1196. https://doi.org/10.3390/molecules26041196
AMA Style
Keglevich G. Microwaves as “Co-Catalysts” or as Substitute for Catalysts in Organophosphorus Chemistry. Molecules. 2021; 26(4):1196. https://doi.org/10.3390/molecules26041196
Chicago/Turabian StyleKeglevich, György. 2021. "Microwaves as “Co-Catalysts” or as Substitute for Catalysts in Organophosphorus Chemistry" Molecules 26, no. 4: 1196. https://doi.org/10.3390/molecules26041196