
Strategy for Designing Selective Lysosomal Acid α-Glucosidase Inhibitors: Binding Orientation and Influence on Selectivity

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
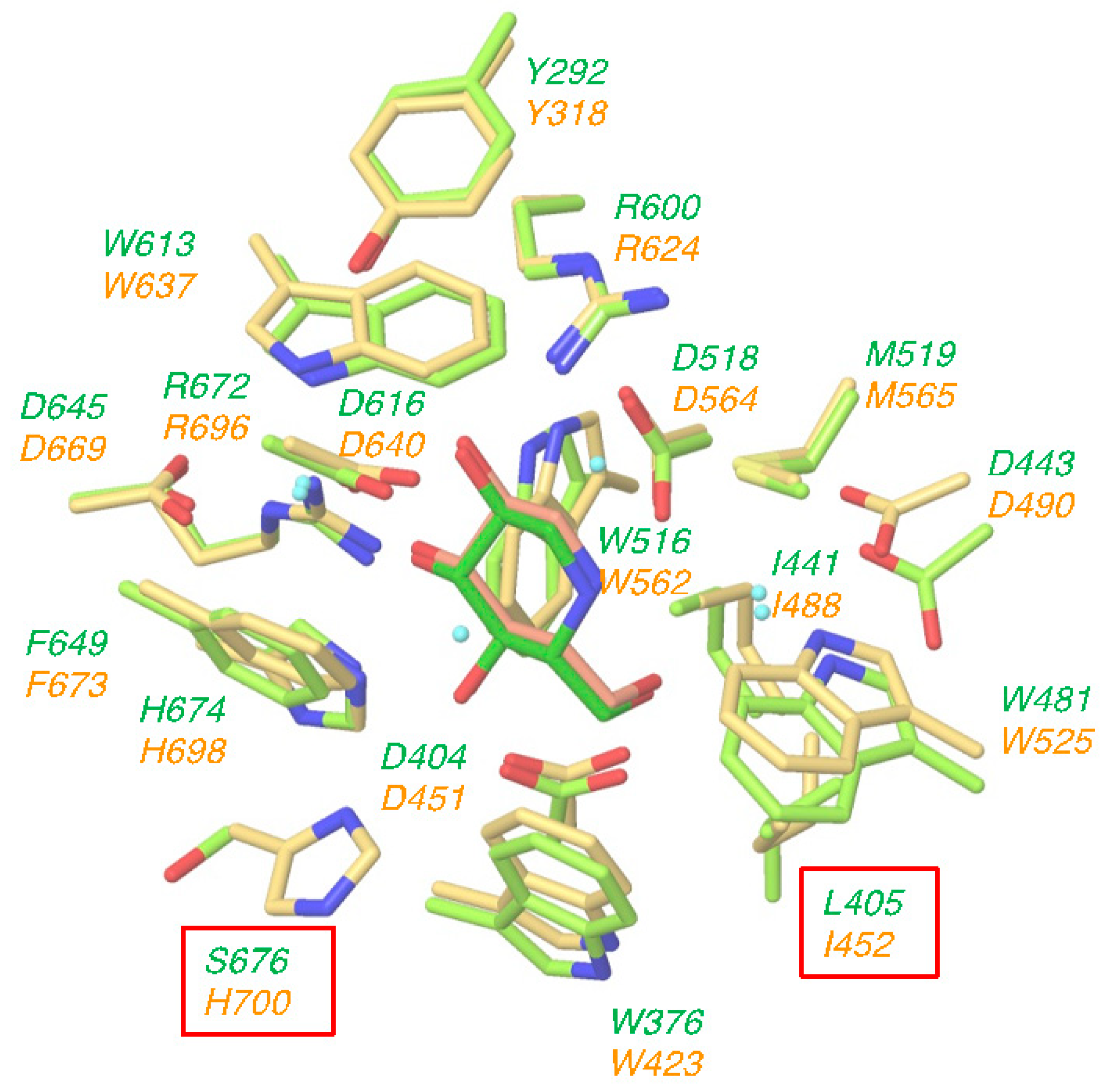
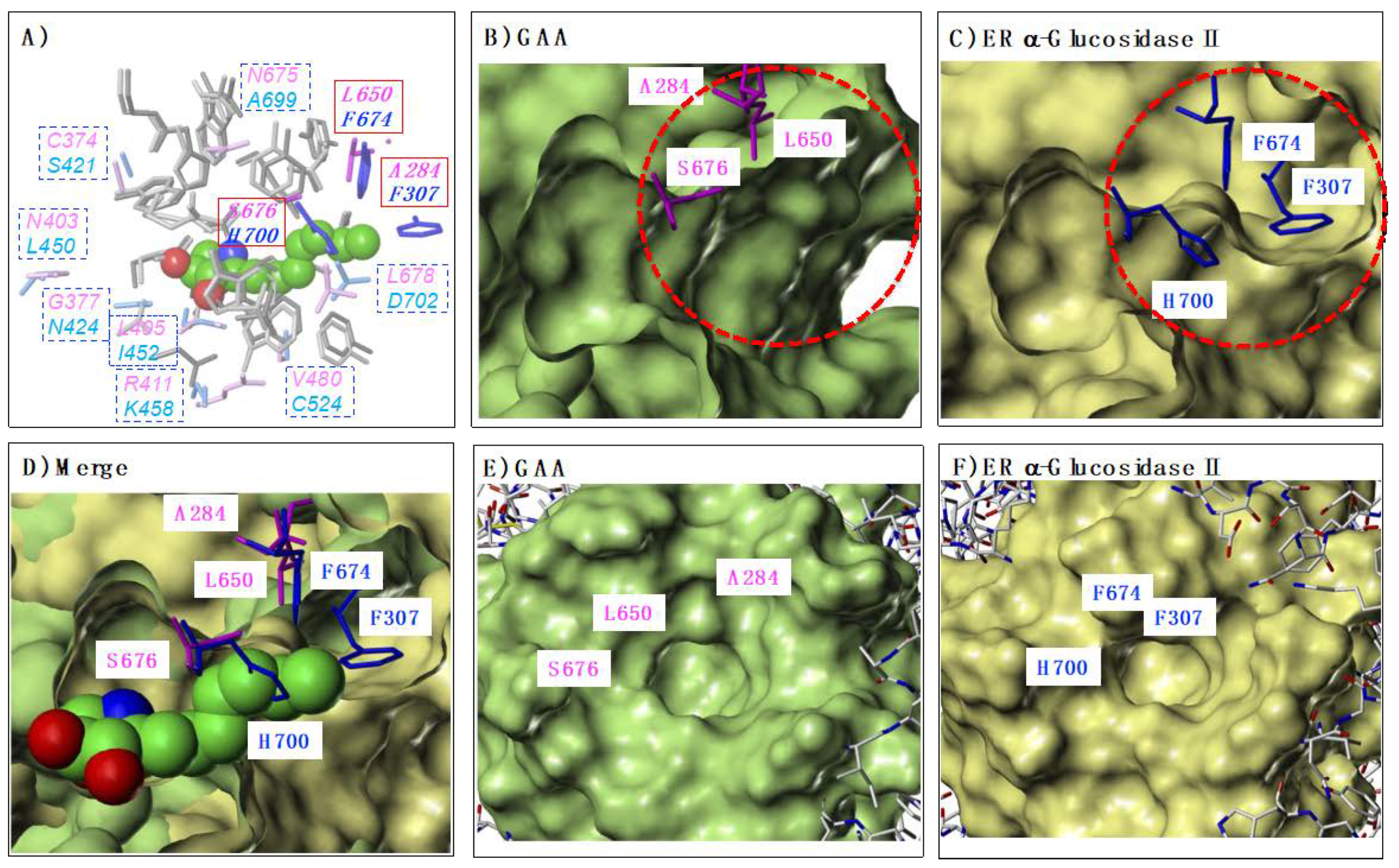
2.1. Structural Similarity of Active Site between GAA and ER α-Glucosidase II
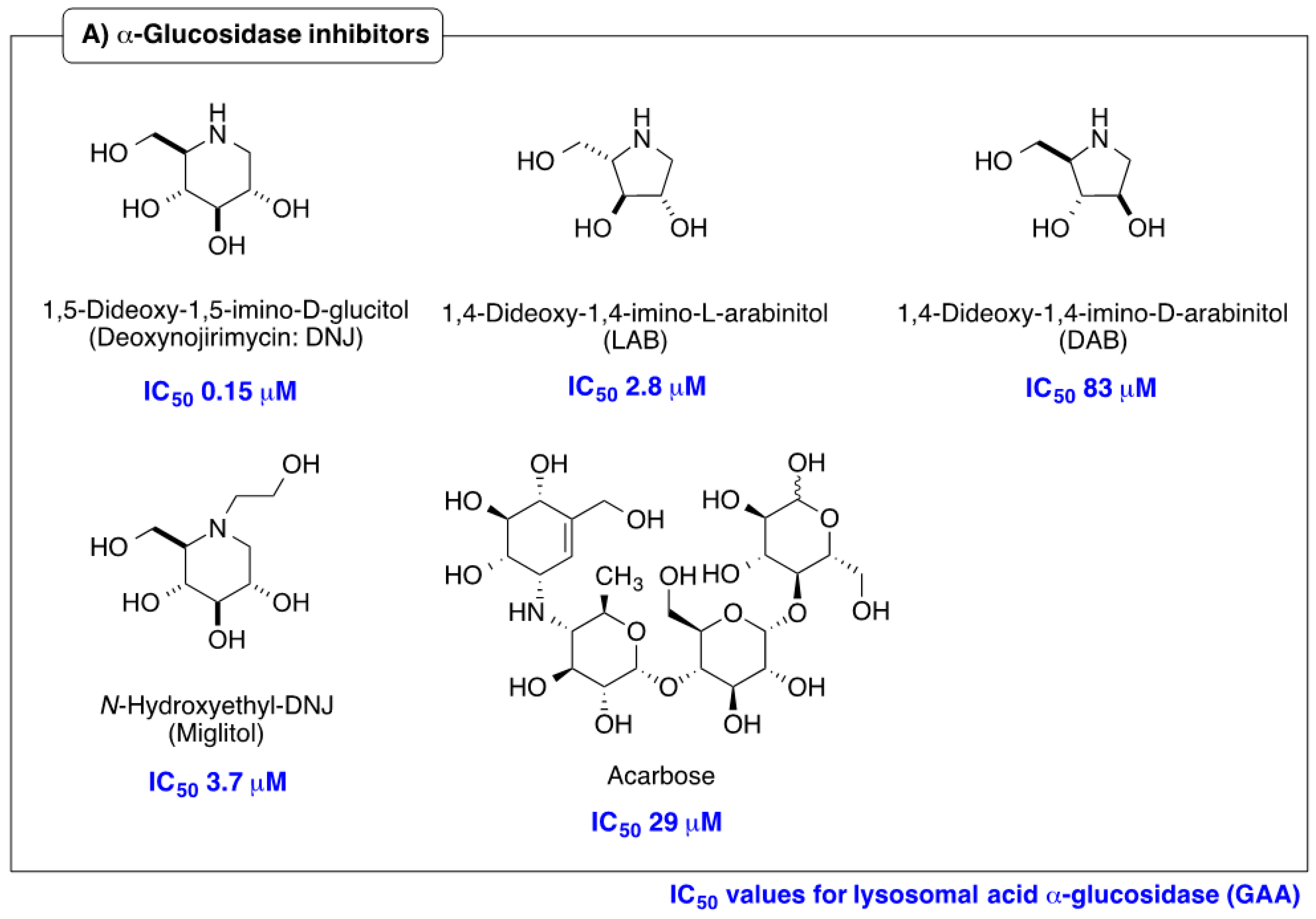
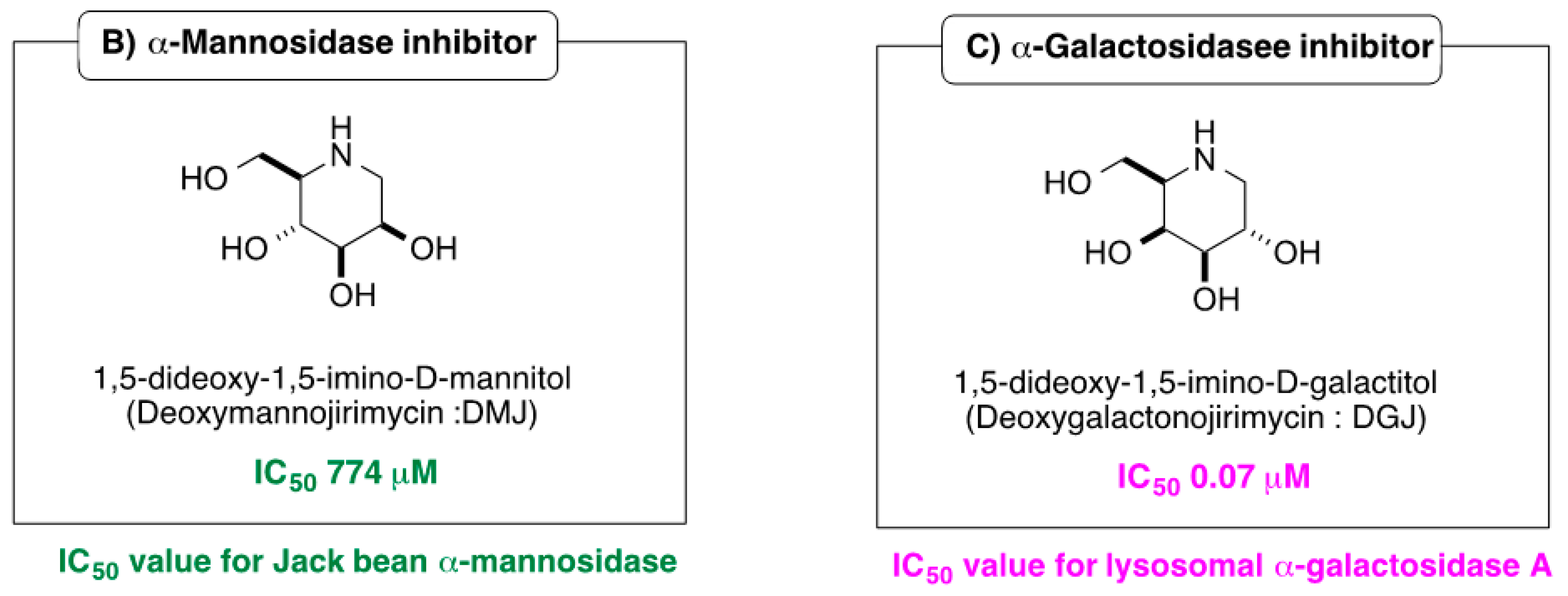
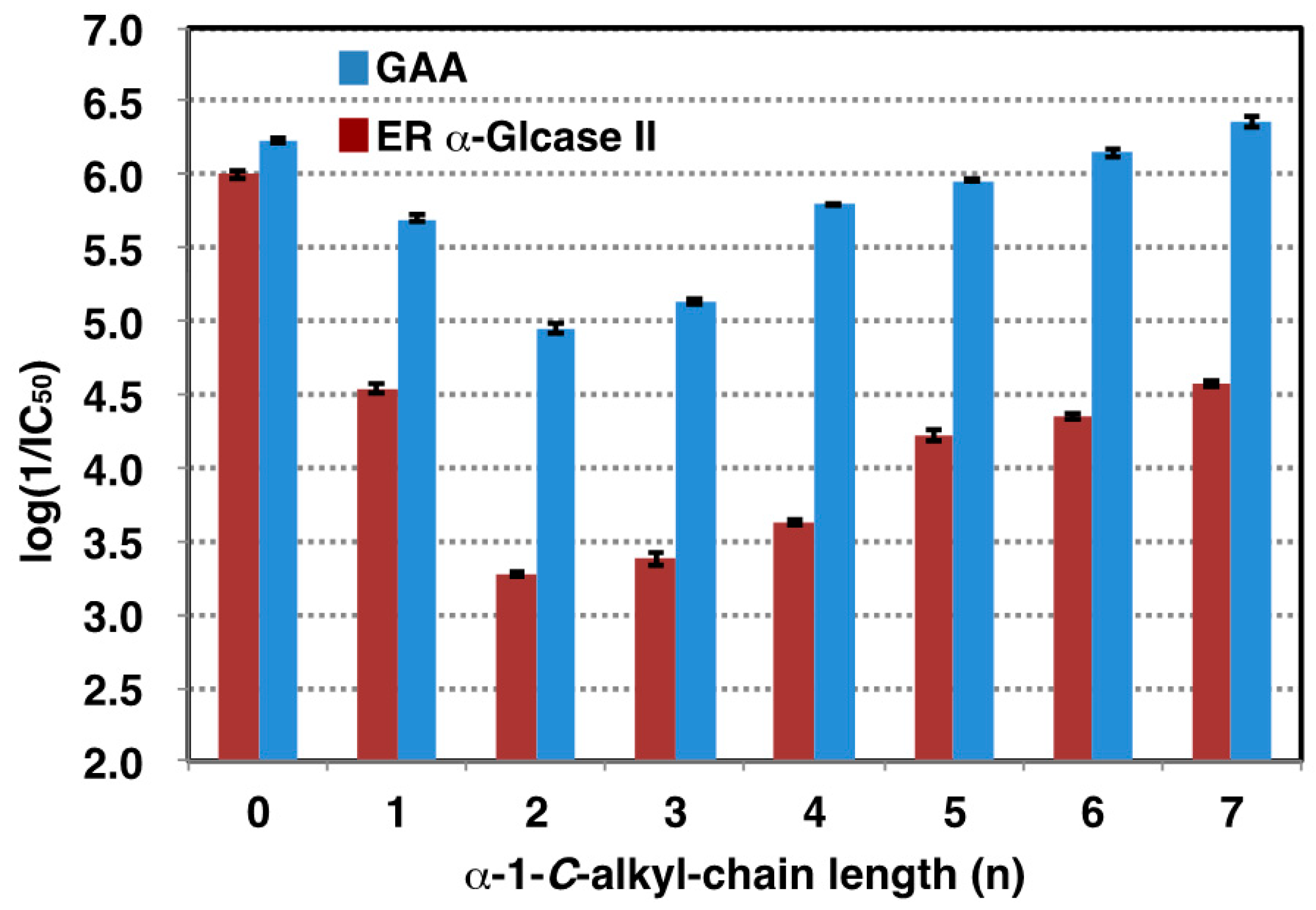
2.2. Influence of α-1-C-alkylation of LAB on Inhibition of GAA and ER α-Glucosidase II
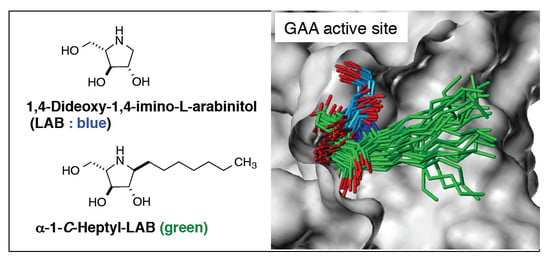
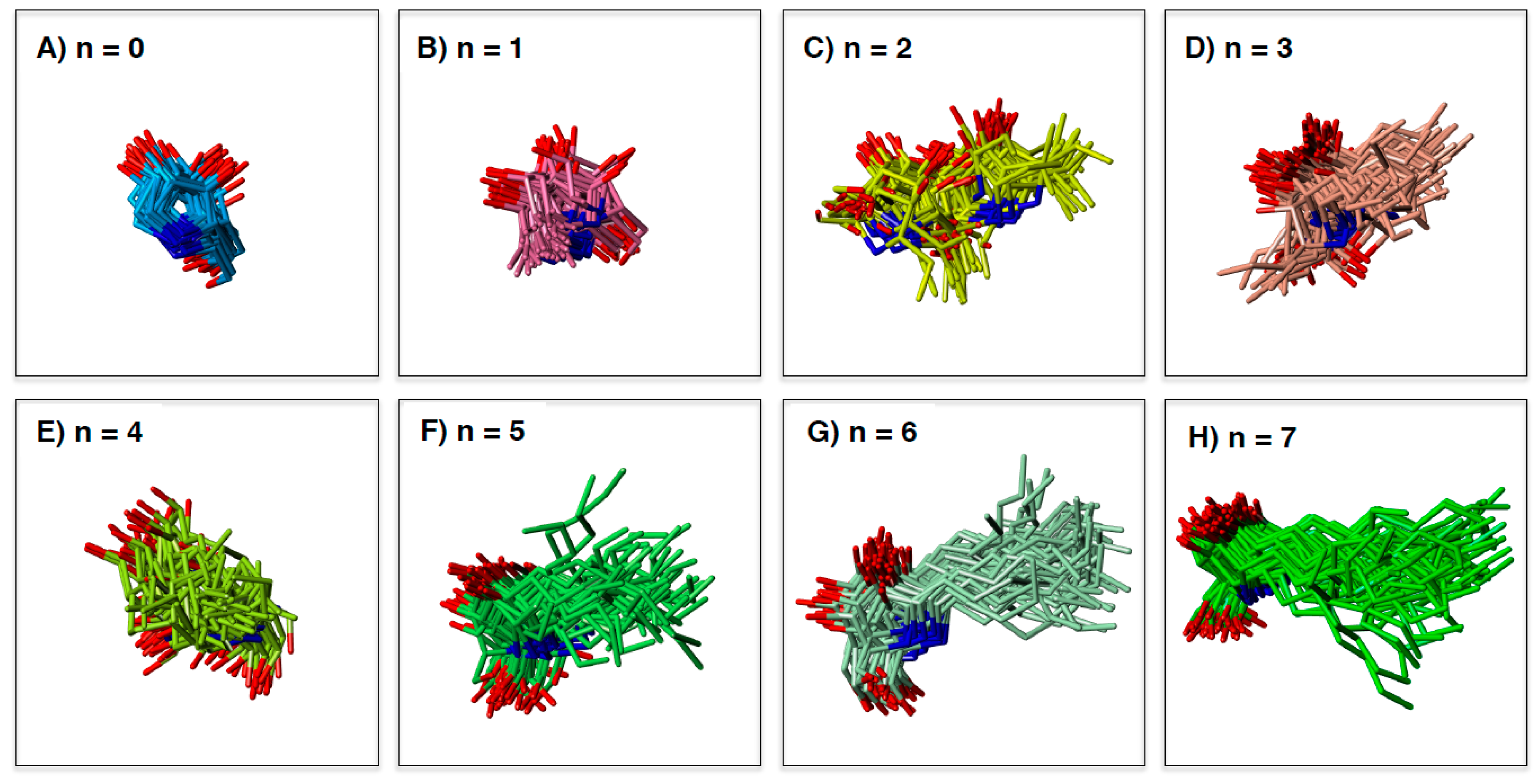
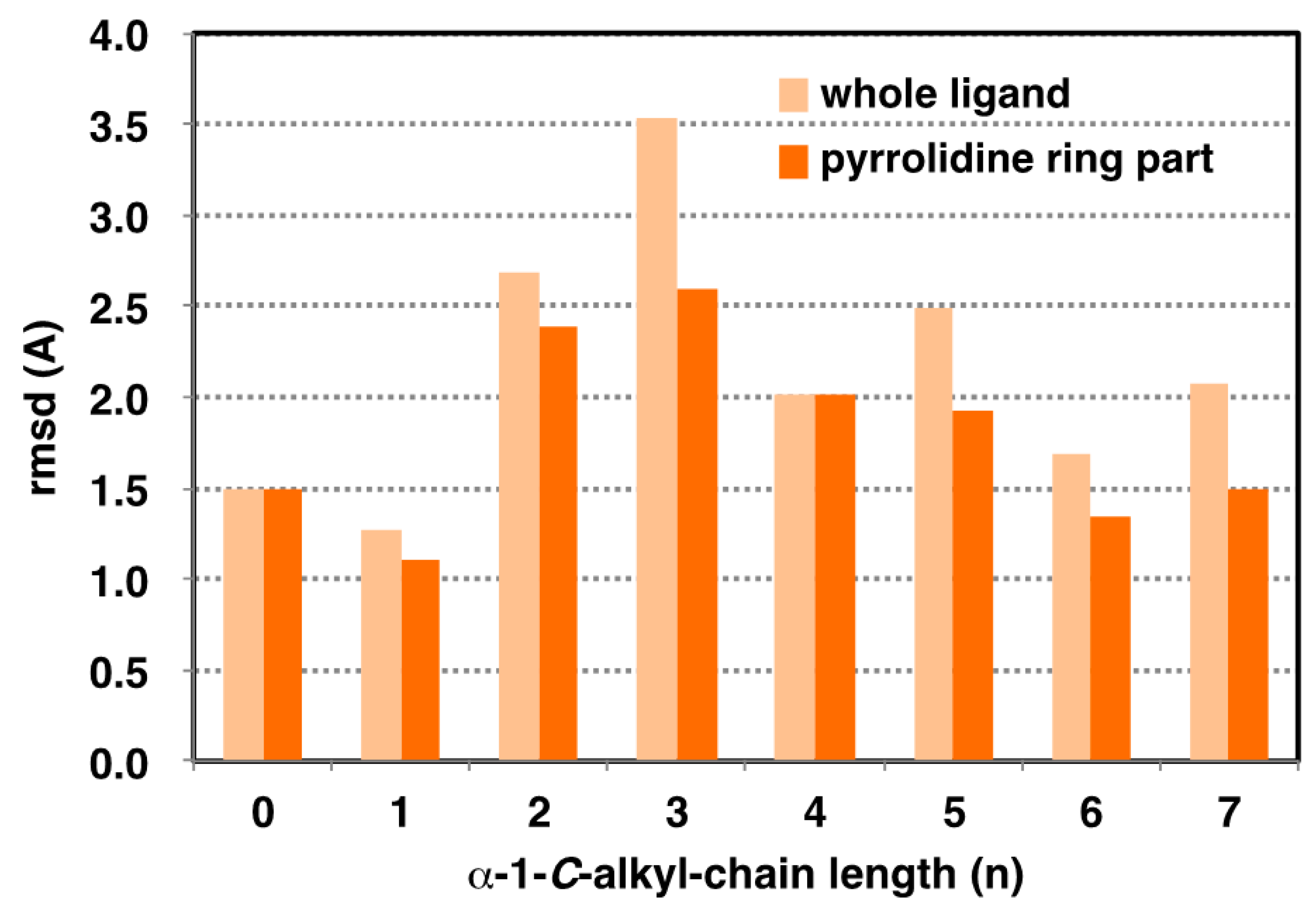
2.3. Analysis of GAA- Ligand Stable Binding Pose by Using MD Calculation
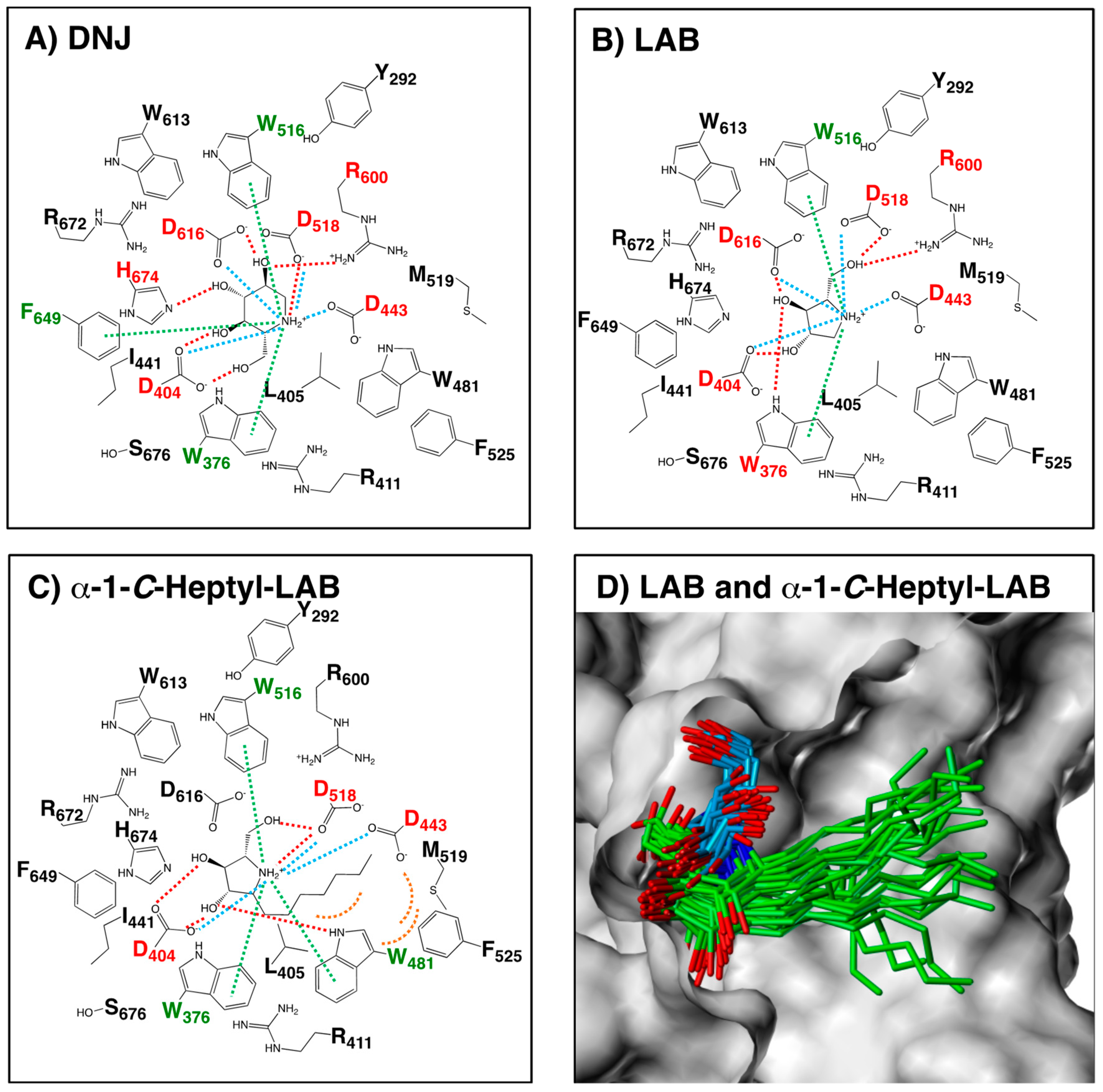
2.4. Hydrogen Bonding Interactions and Cation-π Interactions between GAA and DNJ, LAB, and α-1-C-heptyl-LAB
2.5. Comparison of GAA and ER α-Glucosidase II Focusing on LAB-alkyl Chain Storage Pocket
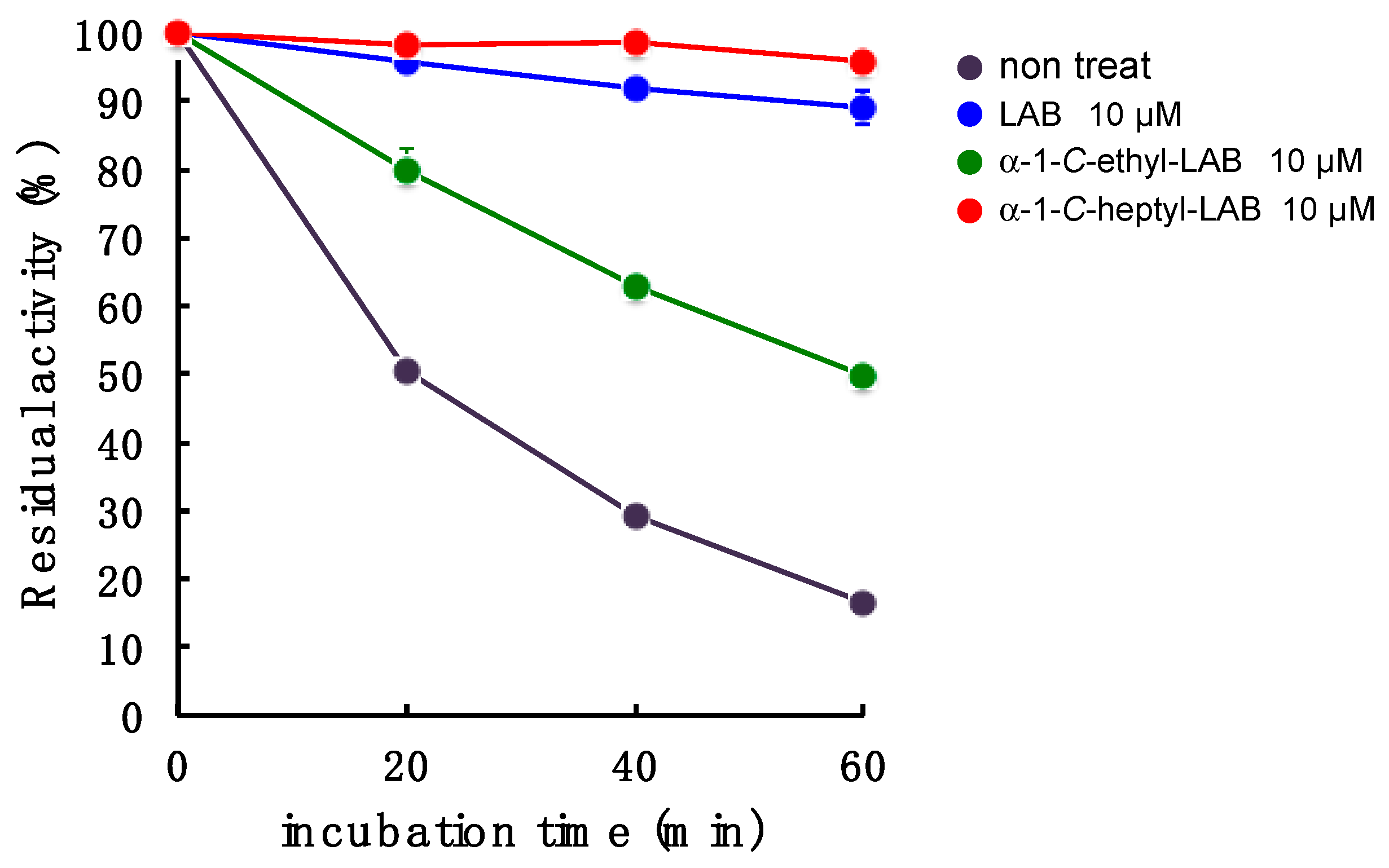
2.6. GAA Stabilization Effect of LAB, α-1-C-ethyl-LAB and α-1-C-heptyl-LAB
3. Experimental Section
3.1. Chemistry
3.2. Enzyme Inhibition Assays
3.3. MD Calculations
3.4. Thermostability of Lysosomal Acid α-Glucosidase (GAA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Asano, N. Sugar-mimicking glycosidase inhibitors: Bioactivity and application. Cell. Mol. Life Sci. 2009, 66, 1479–1492. [Google Scholar] [CrossRef]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W.J. Iminosugars as therapeutic agents: Recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Nash, R.J.; Molyneux, R.J.; Fleet, G.W.J. Sugar-mimic glycosidase inhibitors: Natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Asymm. 2000, 11, 1645–1680. [Google Scholar] [CrossRef]
- Watson, A.A.; Fleet, G.W.J.; Asano, N.; Molyneux, R.J.; Nash, R.J. Polyhydroxylated alkaloids-natural occurrence and therapeutic applications. Phytochemistry 2001, 56, 265–295. [Google Scholar] [CrossRef]
- Kato, A.; Kato, N.; Kano, E.; Adachi, I.; Ikeda, K.; Yu, L.; Okamoto, T.; Banba, Y.; Ouchi, H.; Takahata, H.; et al. Biological properties of d- and l-1-deoxyazasugars. J. Med. Chem. 2005, 48, 2036–2044. [Google Scholar] [CrossRef]
- Arh, H.J.; Boberg, M.; Brendei, E.; Krause, H.P.; Steinke, W. Pharmacokinetics of miglitol. Absorption, distribution, metabolism, and excretion following administration to rats, dogs, and man. Arzneimittelforschung 1997, 47, 734–745. [Google Scholar]
- Kato, A.; Hayashi, E.; Miyauchi, S.; Adachi, I.; Imahori, T.; Natori, Y.; Yoshimura, Y.; Nash, R.J.; Shimaoka, H.; Nakagome, I.; et al. α-1-C-Butyl-1,4-dideoxy-1,4-imino-l-arabinitol as a second-generation iminosugar-based oral α-glucosidase inhibitor for improving postprandial hyperglycemia. J. Med. Chem. 2012, 55, 10347–10362. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Plotz, P.; Byrne, B.J. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr. Mol. Med. 2002, 2, 145–166. [Google Scholar] [CrossRef]
- Dardis, A.; Zanin, I.; Zampieri, S.; Stuani, C.; Pianta, A.; Romanello, M.; Baralle, F.E.; Bembi, B.; Buratti, E. Functional characterization of the common c.-32-13T>G mutation of GAA gene: Identification of potential therapeutic agents. Nucleic Acids Res. 2014, 42, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.Q.; Ishii, S. Active-site-specific chaperone therapy for Fabry disease. FEBS J. 2007, 274, 4962–4971. [Google Scholar] [CrossRef]
- Ishii, S. Pharmacological chaperone therapy for Fabry disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Flanagan, J.J.; Feng, J.; Soska, R.; Frascella, M.; Pellegrino, L.J.; Lun, Y.; Guillen, D.; Lockhart, D.J.; Valenzano, K.J. The pharmacological chaperone AT2220 increases recombinant human acid α-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS ONE 2012, 7, e40776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, R.; Powe, A.C.; Lun, Y.; Soska, R.; Feng, J.; Dhulipala, R.; Frascella, M.; Garcia, A.; Pellegrino, L.J.; Xu, S.; et al. The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid α-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS ONE 2014, 9, e102092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumiya, T.; Kroos, M.A.; Vliet, L.V.; Takeuchi, H.; Van der Ploeg, A.T.; Reuser, A.J. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol. Genet. Metab. 2007, 90, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Zuppaldi, A.; Gabriela Pittis, M.; Rosaria Tuzzi, M.; Annunziata, I.; Meroni, G.; Porto, C.; Donaudy, F.; Rossi, B.; Rossi, M.; et al. Pharmacological enhancement of mutated α-glucosidase activity in fibroblasts from patients with Pompe disease. Mol. Ther. 2007, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R.; Neville, D.C.A.; Harvey, D.J.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Cellular effects of deoxynojirimycin analogues: Inhibition of N-linked oligosaccharide processing and generation of free glucosylated oligosaccharides. Biochem. J. 2004, 381, 867–875. [Google Scholar] [CrossRef]
- Fleet, G.W.J.; Nicholas, S.J.; Smith, P.W.; Evans, S.V.; Fellows, L.E.; Nash, R.J. Potent competitive-inhibition of α-galactosidase and α-glucosidase activity by 1,4-dideoxy-1,4-iminopentitols - syntheses of 1,4-dideoxy-1,4-imino-d-lyxitol and of both enantiomers of 1,4-dideoxy-1,4-iminoarabinitol. Tetrahedron Lett. 1985, 26, 3127–3130. [Google Scholar] [CrossRef]
- Carmona, A.T.; Whightman, R.H.; Robina, I.; Vogel, P. Synthesis and glycosidase inhibitory activity of 7-deoxycasuarine. Helv. Chim. Acta. 2003, 86, 3066–3073. [Google Scholar] [CrossRef]
- Kato, A.; Zhang, Z.L.; Wang, H.Y.; Jia, Y.M.; Yu, C.Y.; Kinami, K.; Hirokami, Y.; Tsuji, Y.; Adachi, I.; Nash, R.J.; et al. Design and synthesis of labystegines, hybrid iminosugars from LAB and calystegine, as inhibitors of intestinal α-glucosidases: Binding conformation and interaction for ntSI. J. Org. Chem. 2015, 80, 4501–4515. [Google Scholar] [CrossRef]
- Merino, P.; Delso, I.; Tejero, T.; Cardona, F.; Marradi, M.; Faggi, E.; Parmeggiani, C.; Goti, A. Nucleophilic additions to cyclic nitrones en route to iminocyclitols - total syntheses of DMDP, 6-deoxy-DMDP, DAB-1, CYB-3, nectrisine, and radicamine B. Eur. J. Org. Chem. 2008, 17, 2929–2947. [Google Scholar] [CrossRef]
- Hino, Y.; Rothman, J.E. Glucosidase II, a glycoprotein of the endoplasmic reticulum membrane. Proteolytic cleavage into enzymatically active fragments. Biochemistry 1985, 24, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraesm, M.A.; Sacerdotim, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Presented at the 2006 ACM/IEEE conference on Supercomputing; Tampa, FL, USA: 11–17 November 2006.
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the alpha-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1,4-dideoxy-1,4-imino-L-arabinitol (LAB) and deoxynojirimycin (DNJ) are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | Selectivity Index | ||||
|---|---|---|---|---|---|
| n = | Compounds | ER α-Glcase II | GAA | ER α-Glcase II/GAA | |
 | 0 | LAB | 2.8 ± 0.22 | 0.52 ± 0.032 | 5.4 |
| 1 | α-1-C-Methyl-LAB | 26 ± 2.0 | 2.0 ± 0.12 | 13.0 | |
| 2 | α-1-C-Ethyl-LAB | 541 ± 26 | 11 ± 1.1 | 49.2 | |
| 3 | α-1-C-Propyl-LAB | 413 ± 50 | 7.4 ± 0.31 | 55.8 | |
| 4 | α-1-C-Butyl-LAB | 228 ±13 | 1.6 ± 0.040 | 142.5 | |
| 5 | α-1-C-Pentyl-LAB | 180 ± 19 | 1.1 ± 0.088 | 163.6 | |
| 6 | α-1-C-Hexyl-LAB | 100 ± 6.2 | 0.71 ± 0.029 | 140.8 | |
| 7 | α-1-C-Heptyl-LAB | 74 ± 3.7 | 0.44 ± 0.044 | 168.2 | |
| DNJ | 8.0 ± 0.55 | 0.15 ± 0.0032 | 53.3 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, A.; Nakagome, I.; Hata, M.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Yoshimura, Y.; Adachi, I.; Hirono, S. Strategy for Designing Selective Lysosomal Acid α-Glucosidase Inhibitors: Binding Orientation and Influence on Selectivity. Molecules 2020, 25, 2843. https://doi.org/10.3390/molecules25122843
Kato A, Nakagome I, Hata M, Nash RJ, Fleet GWJ, Natori Y, Yoshimura Y, Adachi I, Hirono S. Strategy for Designing Selective Lysosomal Acid α-Glucosidase Inhibitors: Binding Orientation and Influence on Selectivity. Molecules. 2020; 25(12):2843. https://doi.org/10.3390/molecules25122843
Chicago/Turabian StyleKato, Atsushi, Izumi Nakagome, Mizuki Hata, Robert J. Nash, George W. J. Fleet, Yoshihiro Natori, Yuichi Yoshimura, Isao Adachi, and Shuichi Hirono. 2020. "Strategy for Designing Selective Lysosomal Acid α-Glucosidase Inhibitors: Binding Orientation and Influence on Selectivity" Molecules 25, no. 12: 2843. https://doi.org/10.3390/molecules25122843