CC12 Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Lines and Mouse Xenograft Model



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
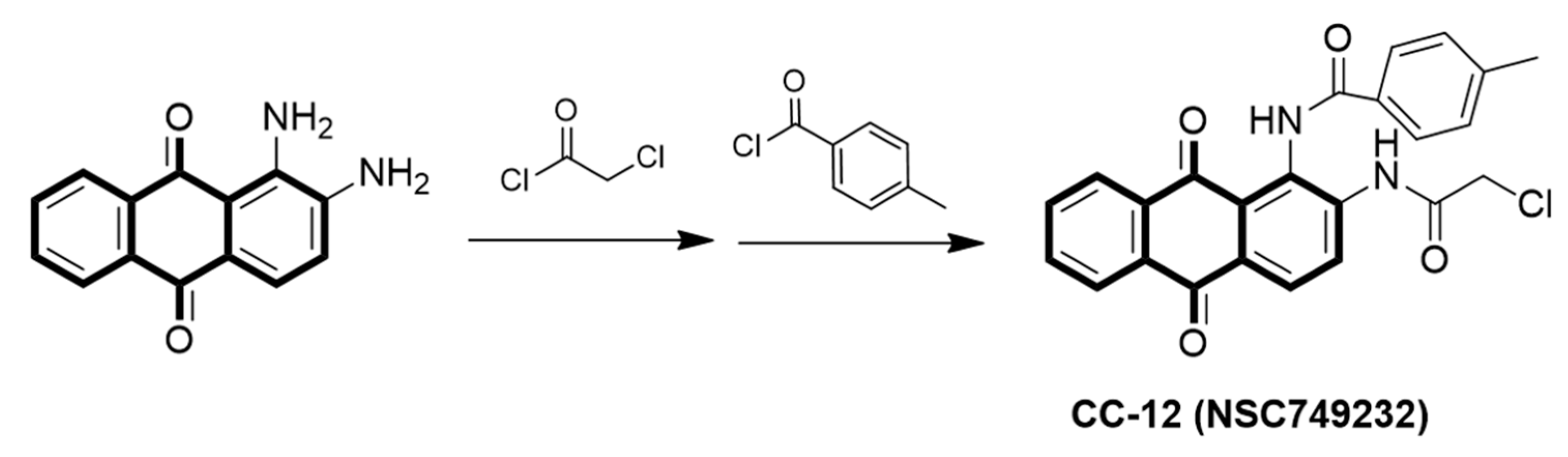
2.1. Preparation of CC12 and GBM Cell Lines
2.2. MTT Assay
2.3. Western Blot Analysis
2.4. Flow Cytometry-Based Apoptosisdetections
2.5. Mouse Xenograft Model and Positron Emission Tomography (PET) Scan Analysis
2.6. Data Analysis
3. Results
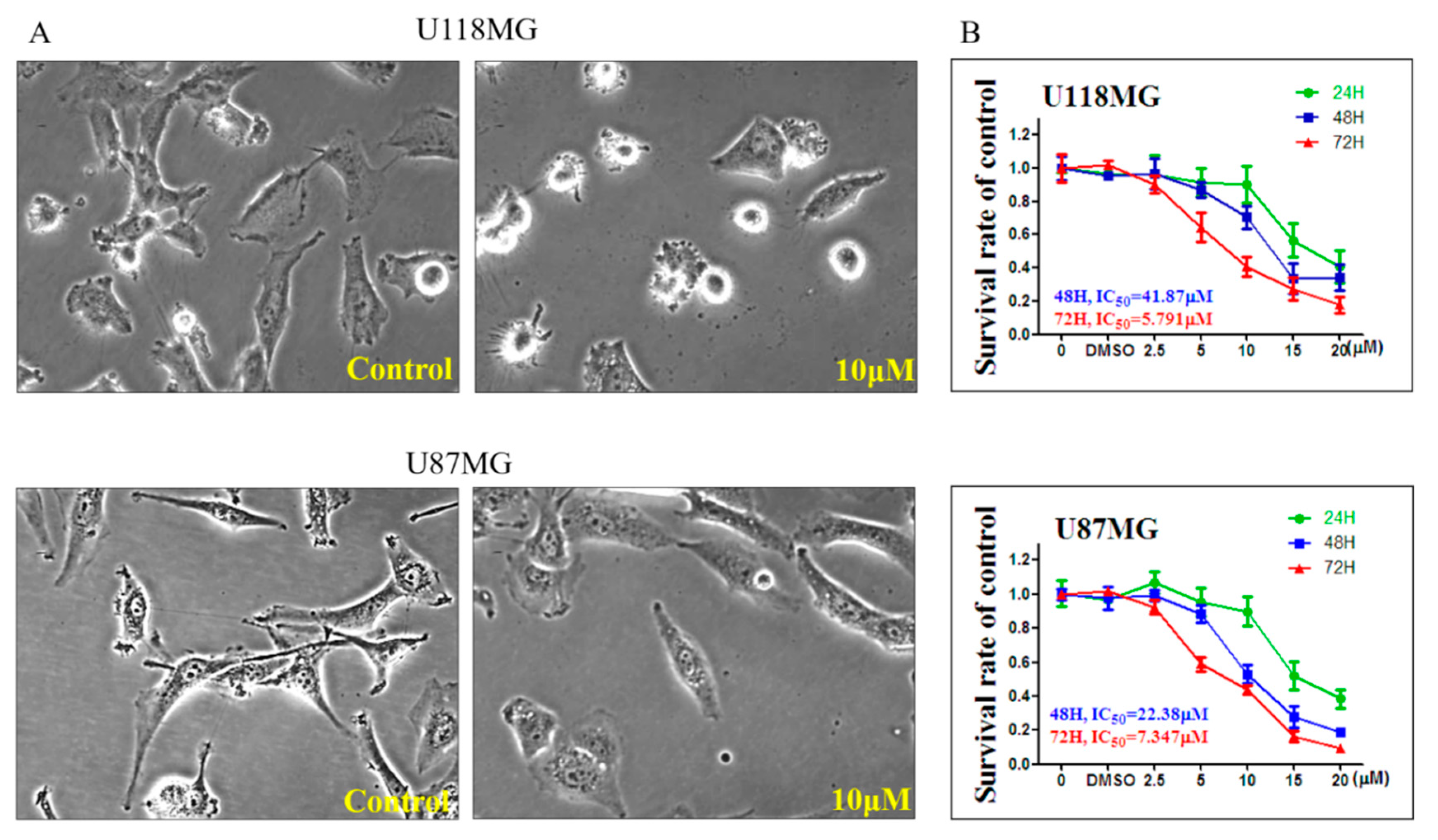
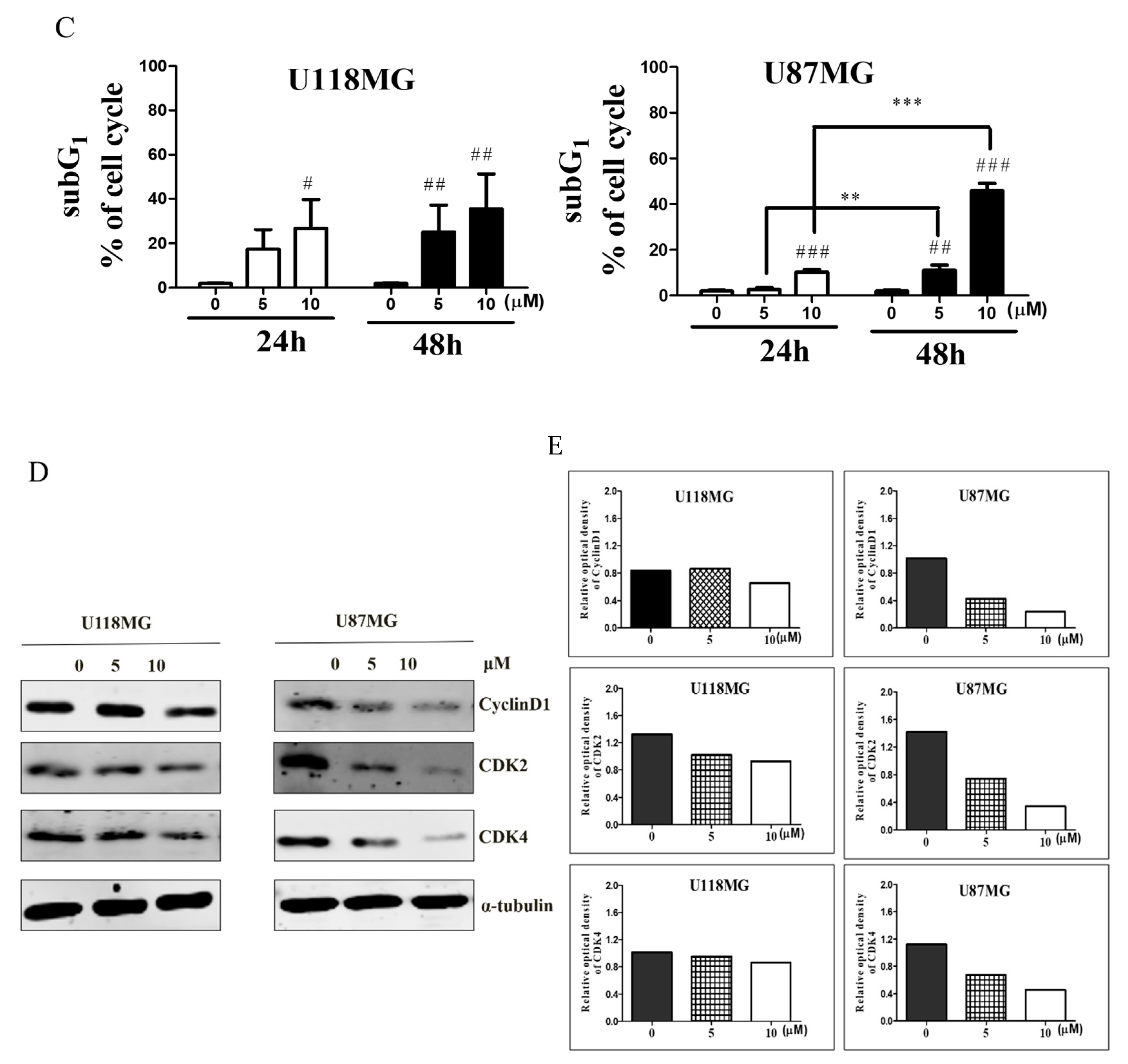
3.1. CC12 Induced Tumor Cell Death and Inhibited Tumor Cell Proliferation
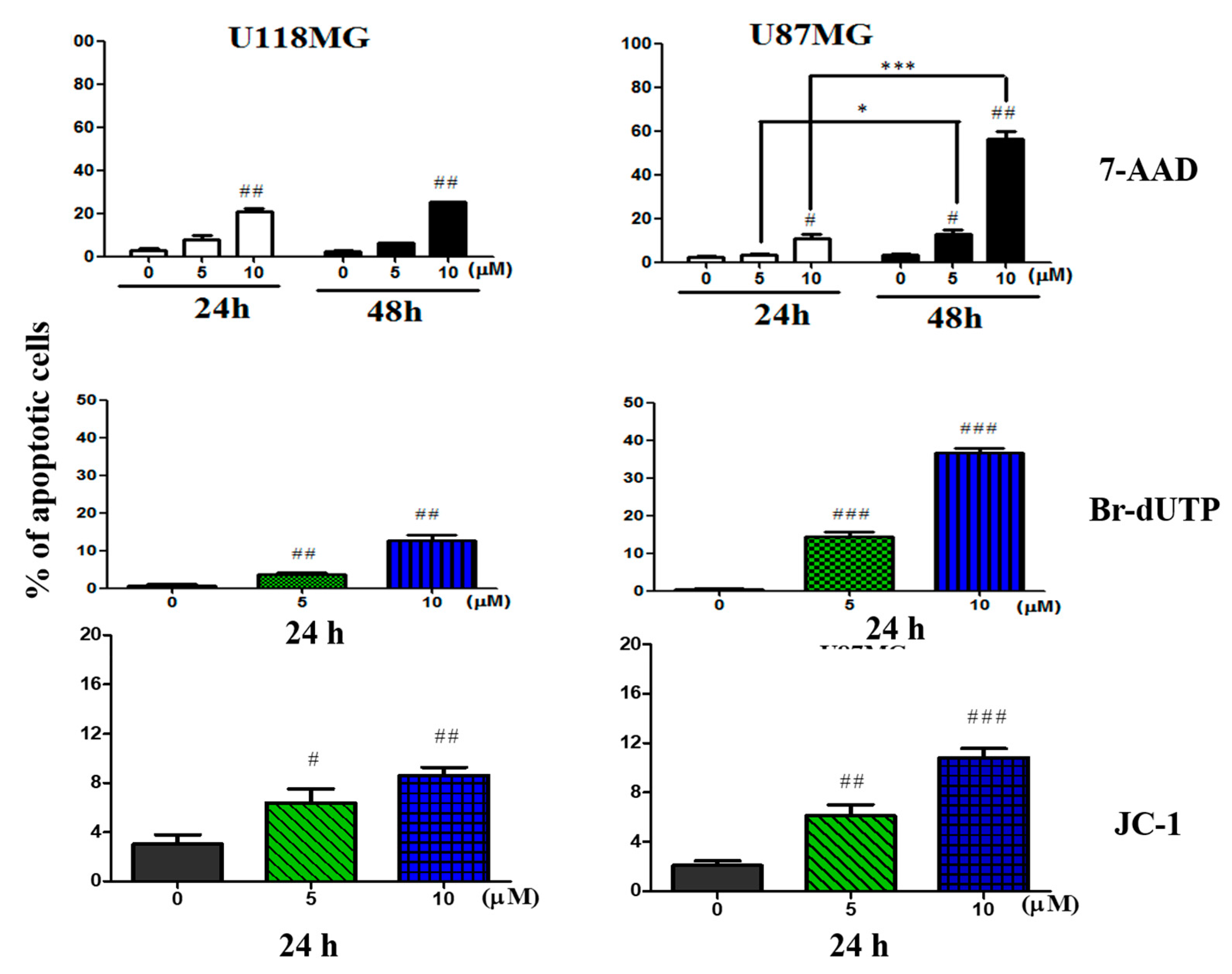
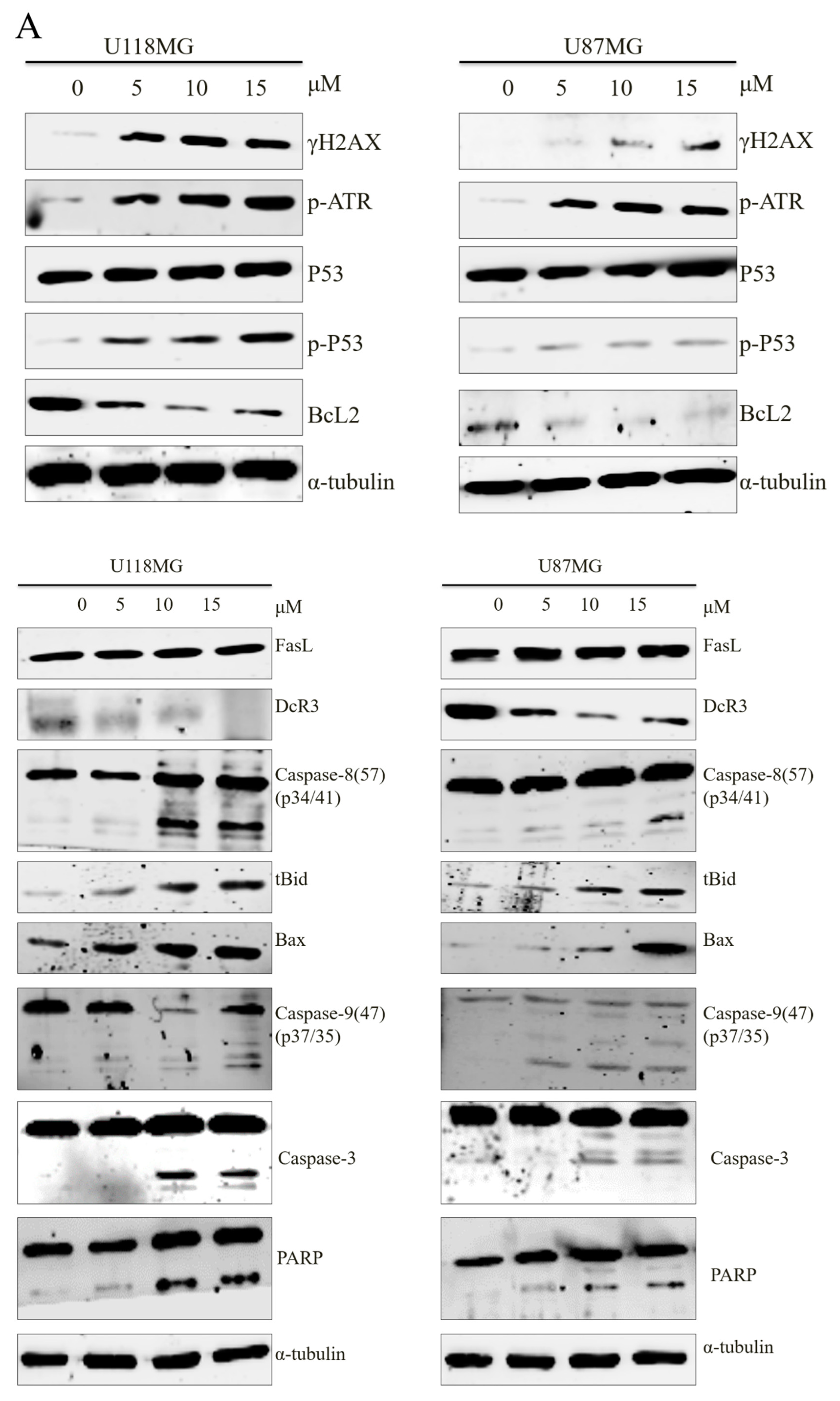
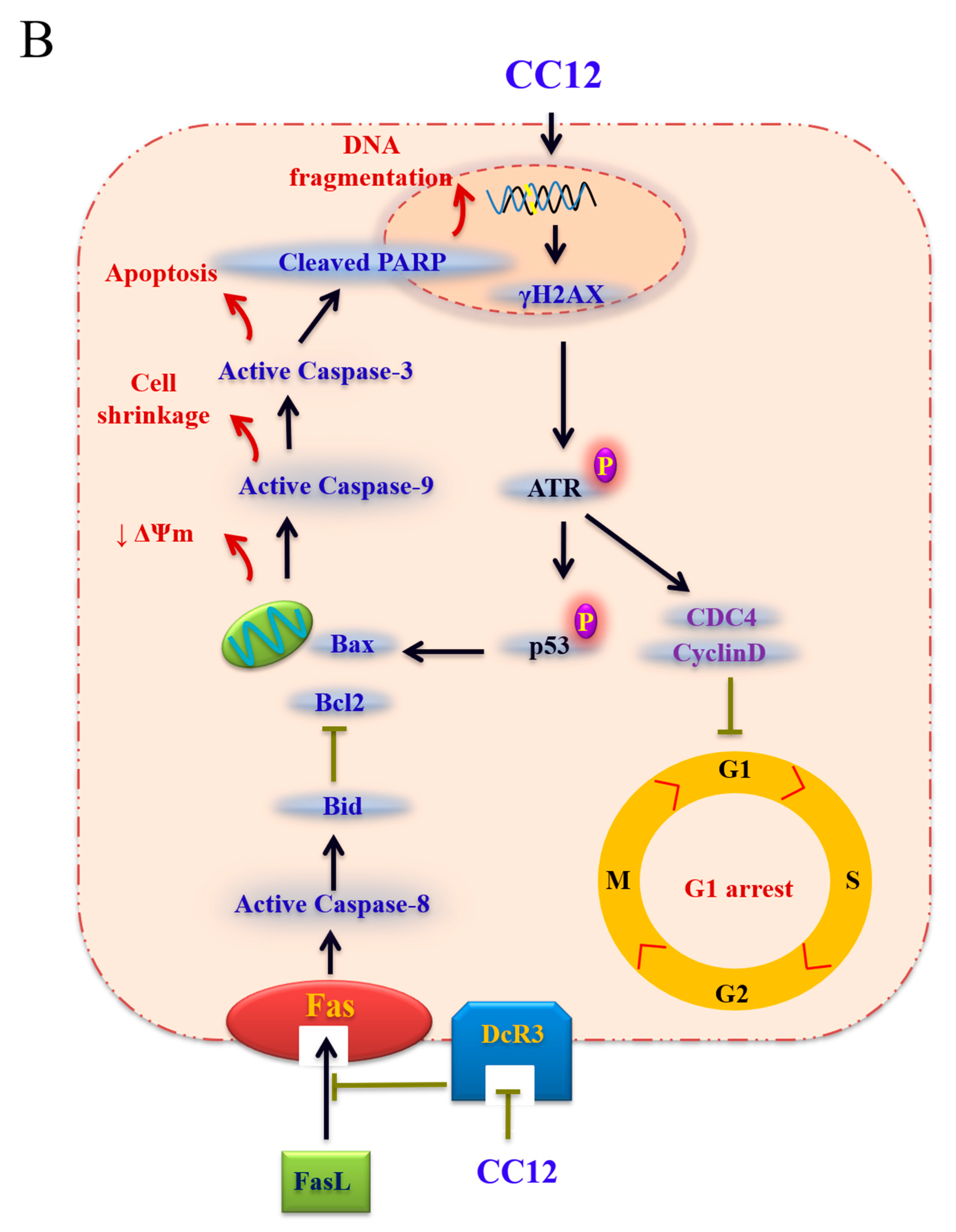
3.2. CC12 Induced Apoptosis of Tumor Cells
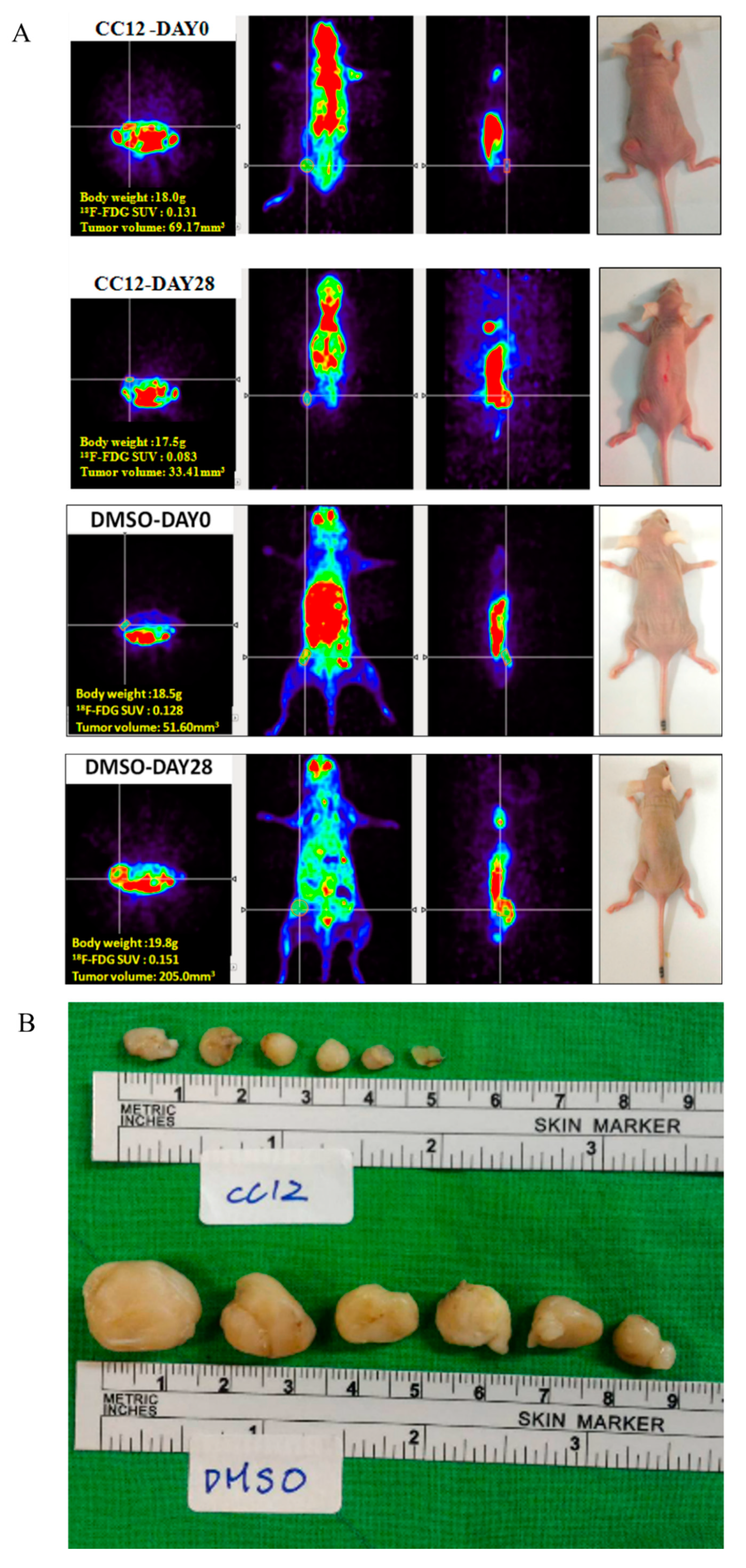
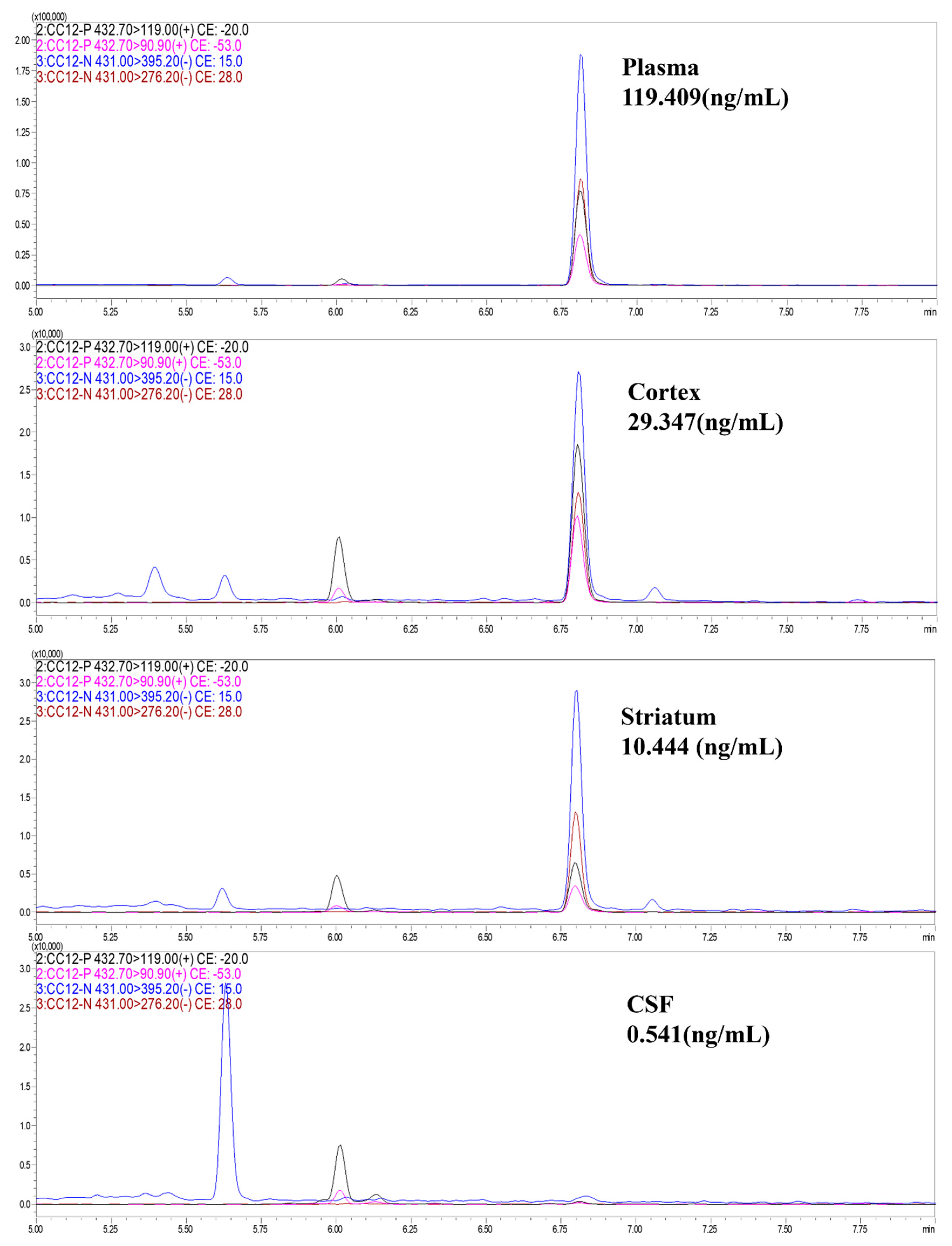
3.3. CC12 Showed Tumor Elimination Effects In Vivo
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro-Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, A.P. Postoperative irradiation of glioblastomas. Results in a randomized series. Acta Radiol. Oncol. Radiat. Phys. Biol. 1978, 17, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA: Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Huang, H.S.; Chen, T.C.; Chen, R.H.; Huang, K.F.; Huang, F.C.; Jhan, J.R.; Chen, C.L.; Lee, C.C.; Lo, Y.; Lin, J.J. Synthesis, cytotoxicity and human telomerase inhibition activities of a series of 1,2-heteroannelated anthraquinones and anthra[1, 2-d]imidazole-6,11-dione homologues. Bioorg. Med. Chem. 2009, 17, 7418–7428. [Google Scholar] [CrossRef]
- Alves, D.S.; Perez-Fons, L.; Estepa, A.; Micol, V. Membrane-related effects underlying the biological activity of the anthraquinones emodin and barbaloin. Biochem. Pharmacol. 2004, 68, 549–561. [Google Scholar] [CrossRef]
- Huang, H.S.; Huang, K.F.; Li, C.L.; Huang, Y.Y.; Chiang, Y.H.; Huang, F.C.; Lin, J.J. Synthesis, human telomerase inhibition and anti-proliferative studies of a series of 2,7-bis-substituted amido-anthraquinone derivatives. Bioorg. Med. Chem. 2008, 16, 6976–6986. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, T.C.; Lee, C.C.; Chen, C.L.; Ahmed Ali, A.A.; Tikhomirov, A.; Guh, J.H.; Yu, D.S.; Huang, H.S. Ring fusion strategy for synthesis and lead optimization of sulfur-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as promising scaffold of antitumor agents. Eur. J. Med. Chem. 2015, 102, 661–676. [Google Scholar] [CrossRef]
- Chen, T.C.; Wu, C.L.; Lee, C.C.; Chen, C.L.; Yu, D.S.; Huang, H.S. Structure-based hybridization, synthesis and biological evaluation of novel tetracyclic heterocyclic azathioxanthone analogues as potential antitumor agents. Eur. J. Med. Chem. 2015, 103, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Liu, F.L.; Lee, C.C.; Chen, T.C.; Chang, W.W.; Guh, J.H.; Ahmed Ali, A.A.; Chang, D.M.; Huang, H.S. Ring fusion strategy for the synthesis of anthra[2,3-d]oxazole-2-thione-5,10-dione homologues as DNA topoisomerase inhibitors and as antitumor agents. Eur. J. Med. Chem. 2014, 87, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zheng, X.; Rich, K.M. Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J. Neurochem. 2003, 84, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Bock, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar] [CrossRef] [Green Version]
- Cristofanon, S.; Fulda, S. ABT-737 promotes tBid mitochondrial accumulation to enhance TRAIL-induced apoptosis in glioblastoma cells. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.J.; Travers, J.; Pshenichnaya, I.; Kogera, F.A.; Barthorpe, S.; Mironenko, T.; Richardson, L.; Benes, C.H.; Stratton, M.R.; McDermott, U.; et al. Combinations of PARP Inhibitors with Temozolomide Drive PARP1 Trapping and Apoptosis in Ewing’s Sarcoma. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Franco, Y.E.M.; Okubo, M.Y.; Torre, A.D.; Paiva, P.P.; Rosa, M.N.; Silva, V.A.O.; Reis, R.M.; Ruiz, L.T.A.; Imamura, M.P.; de Carvalho, J.E.; et al. Coronarin D Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Line. Molecules 2019, 24, 4498. [Google Scholar] [CrossRef] [Green Version]
- Pitti, R.M.; Marsters, S.A.; Lawrence, D.A.; Roy, M.; Kischkel, F.C.; Dowd, P.; Huang, A.; Donahue, C.J.; Sherwood, S.W.; Baldwin, D.T.; et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 1998, 396, 699–703. [Google Scholar] [CrossRef]
- Roth, W.; Isenmann, S.; Nakamura, M.; Platten, M.; Wick, W.; Kleihues, P.; Bahr, M.; Ohgaki, H.; Ashkenazi, A.; Weller, M. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer Res. 2001, 61, 2759–2765. [Google Scholar]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Migone, T.S.; Zhang, J.; Luo, X.; Zhuang, L.; Chen, C.; Hu, B.; Hong, J.S.; Perry, J.W.; Chen, S.F.; Zhou, J.X.; et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 2002, 16, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Tai, S.K.; Chang, H.C.; Lan, K.L.; Lee, C.T.; Yang, C.Y.; Chen, N.J.; Chou, T.Y.; Tarng, D.C.; Hsieh, S.L. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J. Immunol. 2012, 188, 2464–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the CC12 are available from Hsu-Shan Huang, Graduate Institute of Cancer Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University. |










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fann, L.-Y.; Shih, J.-H.; Tseng, J.-H.; Huang, H.-S.; Hsiao, S.-H. CC12 Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Lines and Mouse Xenograft Model. Molecules 2020, 25, 1793. https://doi.org/10.3390/molecules25081793
Fann L-Y, Shih J-H, Tseng J-H, Huang H-S, Hsiao S-H. CC12 Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Lines and Mouse Xenograft Model. Molecules. 2020; 25(8):1793. https://doi.org/10.3390/molecules25081793
Chicago/Turabian StyleFann, Li-Yun, Jui-Hu Shih, Jen-Ho Tseng, Hsu-Shan Huang, and Sheng-Huang Hsiao. 2020. "CC12 Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Lines and Mouse Xenograft Model" Molecules 25, no. 8: 1793. https://doi.org/10.3390/molecules25081793