
Total Syntheses of Marrubiin and Related Labdane Diterpene Lactones


Graduate School of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya 467-8603, Japan
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(7), 1610; https://doi.org/10.3390/molecules25071610
Submission received: 6 March 2020
/
Revised: 27 March 2020
/
Accepted: 30 March 2020
/
Published: 1 April 2020
(This article belongs to the Special Issue Total Synthesis of Biologically Active Product)
Abstract
:Total syntheses of the labdane diterpene lactones marrubiin, marrulibacetal, desertine, marrulibacetal A, marrubasch F, cyllenine C, marrulanic acid, and marrulactone are described. The trans-decalin moiety of these molecules was constructed in a stereoselective manner by a Pauson-Khand reaction, and the resultant cyclopentenone was oxidatively cleaved for formation of the lactone ring. Elongation of the side chain at C9 was achieved by an epoxide-opening reaction with a variety of nucleophiles, and the functional group manipulations completed the syntheses of these natural products. Stereochemistries of desertine could be established by the transformations.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
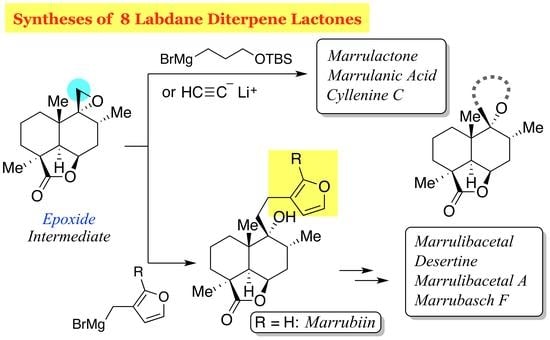
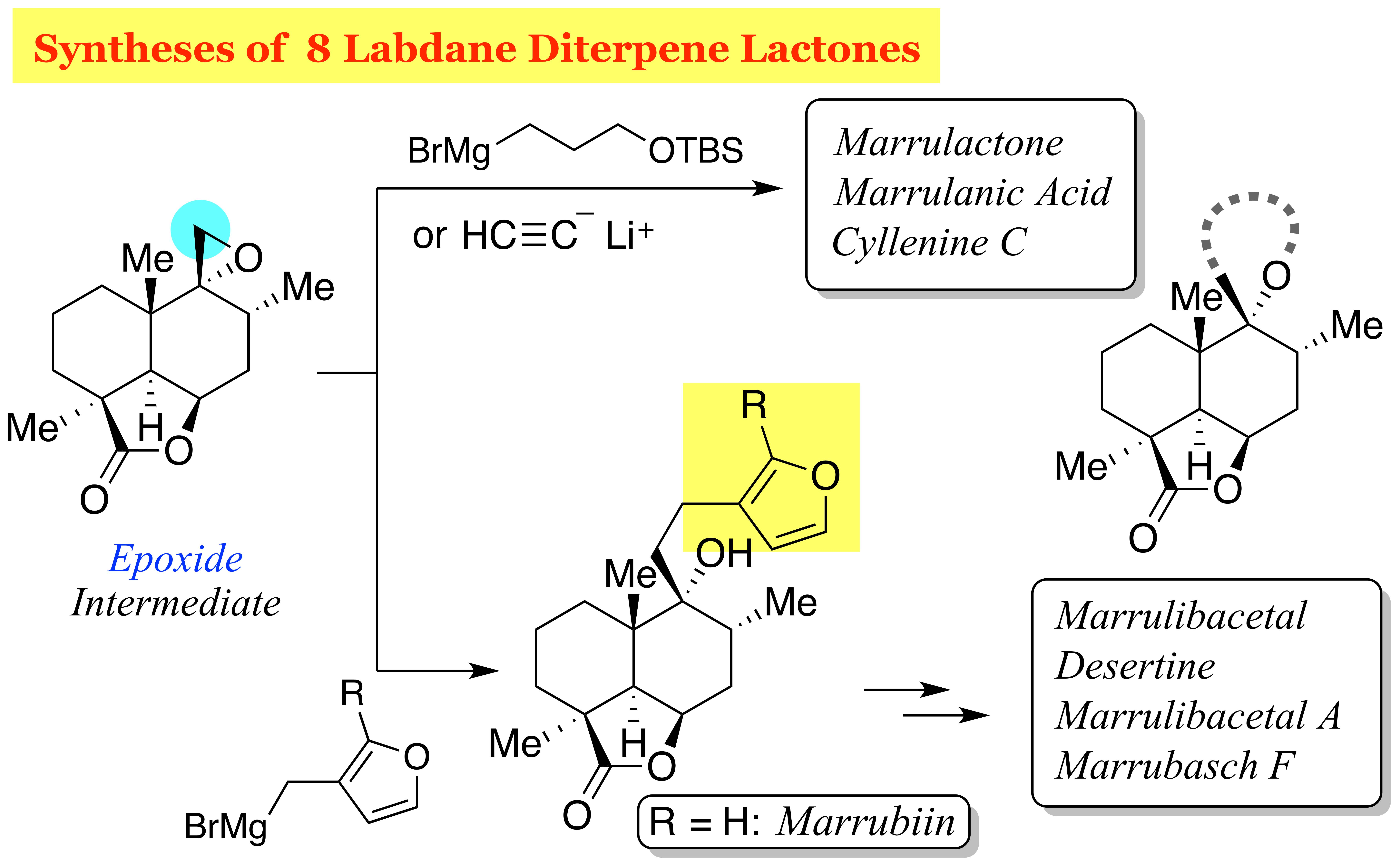
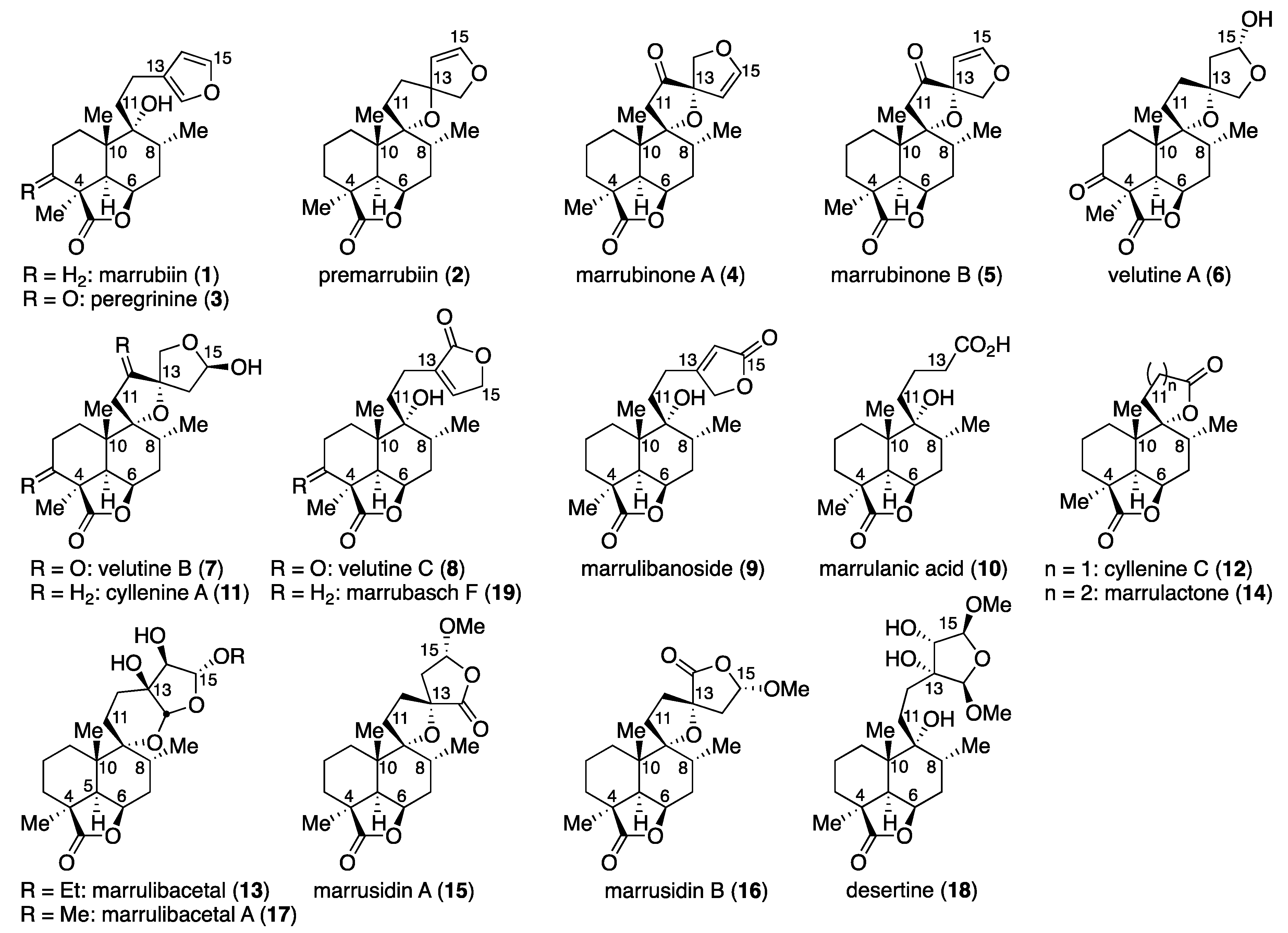
Flowering plants of the genus Marrubium (Lamiaceae) are distributed in the Mediterranean and temperate regions of the Eurasian zone, and most of the plants are used in folk medicine [1]. The therapeutic properties of these herbs, including anti-inflammatory, hypoglycemic, analgesic, antispasmodic, vasorelaxant, and anti-diabetic effects, are attributed in part to marrubiin (1), which was first isolated in 1842 from Marrubium vulgare and is a prominent member of the labdane diterpene lactones [2] (Figure 1). This furanoid natural product, formed from premarrubiin (2), was also reported to inhibit KCl-induced contraction of the rat aorta in a concentration-dependent manner [3]. The important pharmacological action of this family of mints prompted phytochemical analysis, leading to the isolation and characterization of a number of labdane diterpene lactones [4]: peregrinine (3) [5], marrubinones A (4) and B (polyodonine, 5) [6,7], velutines A (6), B (7) and C (8) [8], marrulibanoside (9) [9], marrulanic acid (10) [10], cyllenines A (11) and C (12) [11], marrulibacetal (13), marrulactone (14) [12], marrusidins A (15) and B (16) [13], marrulibacetal A (17), desertine (18) [14], and marrubasch F (19) [15] have been reported to date [16]. These natural products are biosynthesized from (E,E,E)-geranylgeranyl diphosphate (GGPP) through peregrinol diphosphate synthase (CPS1)-catalyzed bicyclization, followed by 9,13-epoxylabd-14-ene synthase (ELS)-catalyzed formation of tetrahydropyran and regiospecific oxygenations with P450s [17,18]. Despite their remarkable biological activities, only a total synthesis of marrubiin (1) in a racemic form [19] and semi-syntheses of premarrubiin (2), marrulibanoside (9), marrubasch F (19) and (13R)-9α,13α-epoxylabda-6β(19),16 (15)-diol dilactone from marrubiin (1) have been reported by Mangoni and co-workers [20].
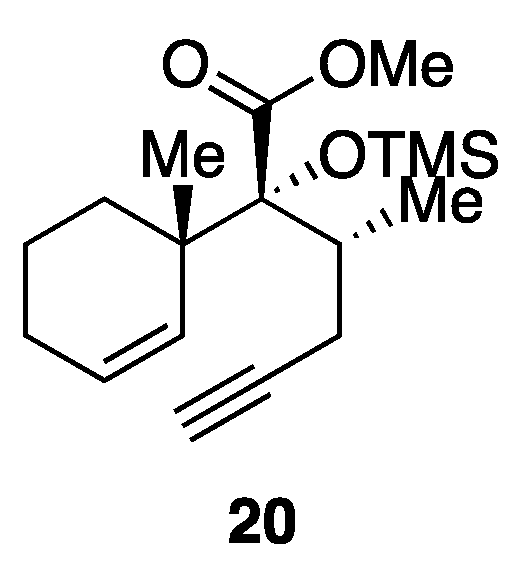
Recently, we have developed a method for stereocontrolled preparation of enyne 20 (Figure 2, TMS = trimethylsilyl) by exploiting a ring-contractive coupling between an α-bromo-δ-valerolactone and a secondary alcohol, and subsequent Ireland−Claisen rearrangement [21,22]. Since compound 20 embodies the C8−C10 stereotriad of C9-oxygenated labdane diterpenoids, compound 20 can serve as a useful chiral building block for the synthesis of pharmacologically interesting, marrubiin-related natural products. In this article, we describe the total syntheses of members of the marrubiin family including marrubiin (1), marrulanic acid (10), cyllenine C (12), marrulibacetal (13), marrulactone (14), marrulibacetal A (17), desertine (18), and marrubasch F (19) [23].
2. Results and Discussion
2.1. Retrosynthetic Analysis
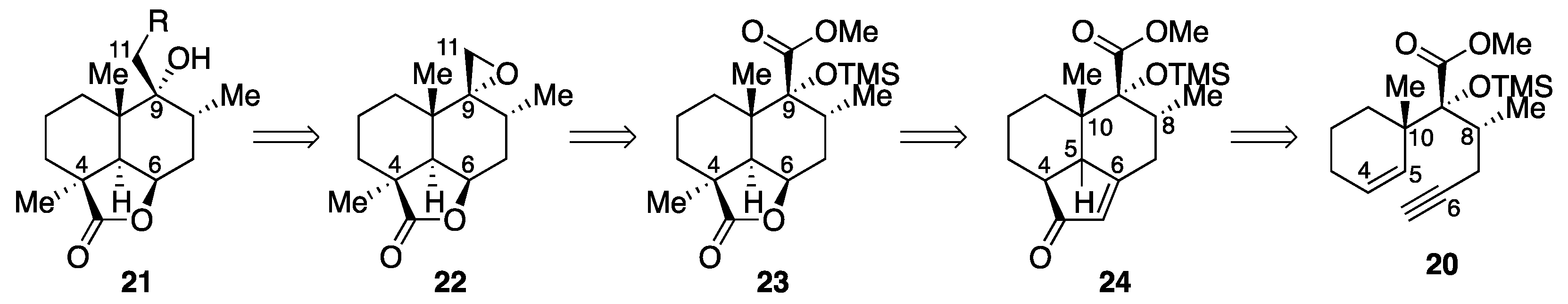
Our retrosynthetic analysis of marrubiin-related natural products is depicted in Scheme 1. Since the difference between the target molecules lies in the variation in the side chain at C9, we planned to install the full C9 side chain late in the synthesis by a nucleophilic ring-opening reaction of advanced epoxide intermediate 22, which could be derived from ester 23 through a chemoselective reduction of the ester functionality in the presence of the lactone moiety. While the methyl group at C4 would be introduced from the less-hindered diastereoface by an enolate alkylation, C5−C6 bond formation, carboxylation at C4, and oxidative cleavage of the C−C bond at C6 were required for the conversion of enyne 20 to lactone 23. In light of these requirements, we chose to use the intramolecular Pauson−Khand reaction (PKR) [24,25] to fashion the six-membered ring and introduce the carbonyl group at C4. Although some concern arose over the formation of cis-fused isomer cis-24 from enyne 20 considering that [RhCl(CO)2]2-catalyzed PKRs of substituted 5-(pent-4-ynyl)cyclohexa-1,3-dienes are precedented to afford cis-decalin derivatives stereoselectively [26], we expected that the stereocenter at C5 would be epimerized after oxidative cleavage of the double bond at C6.
2.2. Synthesis of Advanced Intermediate 22
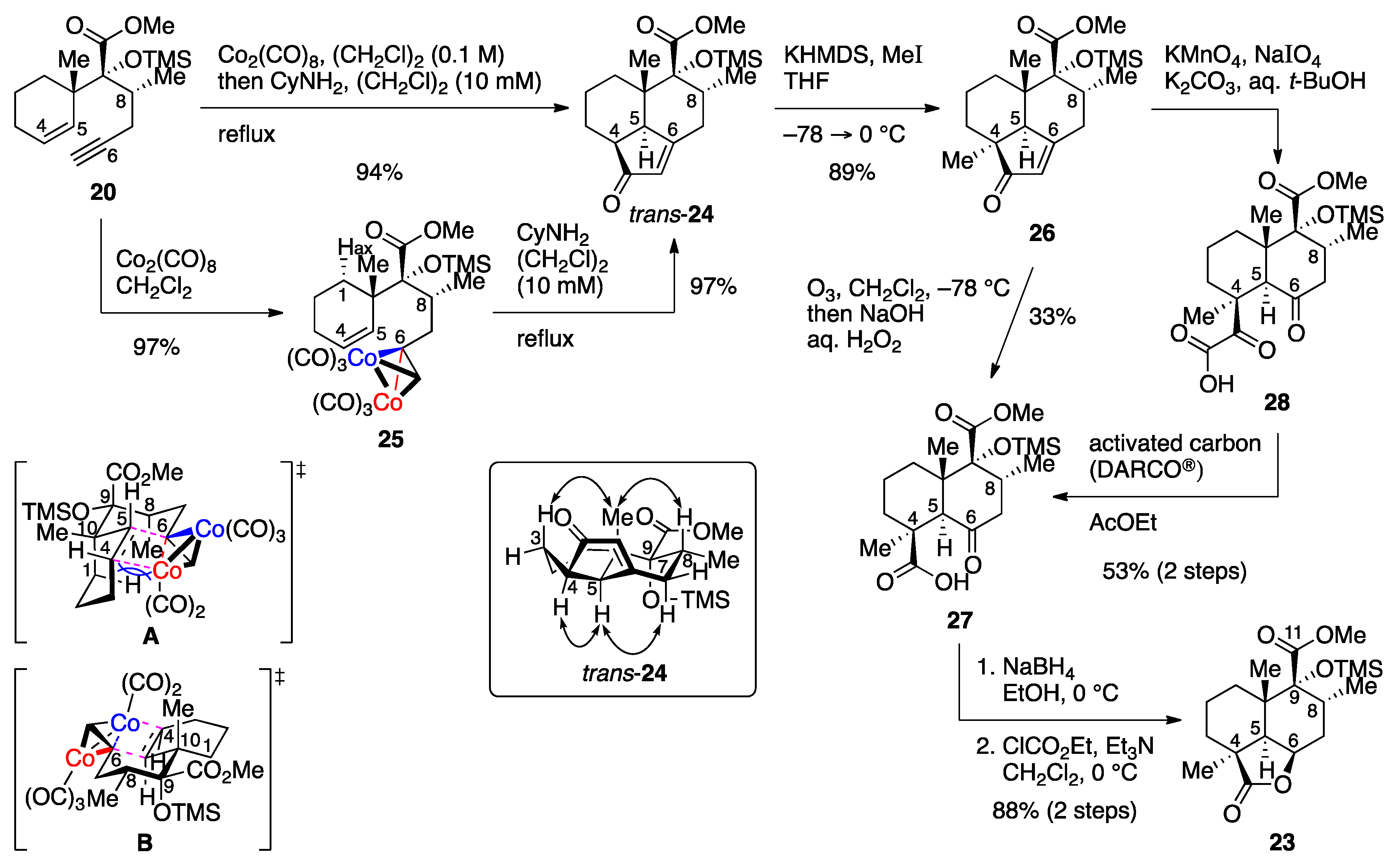
The synthesis of advanced intermediate 22 commenced with the key PKR of enyne 20 (Scheme 2). Initial attempts to cyclize dicobalt complex 25, prepared by treatment of enyne 20 with Co2(CO)8 in CH2Cl2 (97% yield), upon heating in refluxing acetonitrile failed to produce any of the PKR products, leading to decomplexation. After considerable experimentation, we found that the desired tricyclic compound 24 could be produced with the aid of a promoter, with CyNH2 (Cy = cyclohexyl) [27] being optimal for this purpose. Gratifyingly, the product, obtained in (CH2Cl)2 at a substrate concentration of 10 mM under optimized conditions in 97% yield, proved to be the desired stereoisomer trans-24 as confirmed by the absence of a cross-peak between C10-CH3 and C5-H in the nuclear Overhauser effect spectroscopy (NOESY) spectrum. It has been suggested on the basis of quantum mechanical studies that the stereochemistry of PKR is determined by the irreversible olefin insertion step [28]. Since the chair−chairlike transition state (TS) A suffers from steric repulsion between C8-CH3 and C1-Hax, the reaction proceeded through chair−boatlike TS B, leading to the exclusive formation of trans-decalin trans-24.
To enhance the synthetic utility of PKR, a number of catalytic methods have been reported, and a variety of metal complexes have been employed for this purpose [29]. However, our attempts to carry out the catalytic reaction with enyne 20 met with failure: the use of Krafft conditions [30] resulted in recovery of enyne 20 in 49% yield, whereas a complicated mixture was obtained when using 2-naphthaldehyde as a CO donor in the [RhCl(cod)]2-catalyzed reaction [31]. In contrast to these unsuccessful results, the transformation of enyne 20 to enone trans-24 could be refined to a one-pot procedure, wherein the cobalt complex was formed in (CH2Cl)2 and heated under reflux after addition of CyNH2 and 10-fold dilution with (CH2Cl)2. As anticipated, alkylation of the potassium enolate generated from trans-24 with MeI in THF took place exclusively from the less-hindered α-face to provide enone 26 in 89% yield.
With tricyclic compound 26 in hand, efforts were next focused on the transformation of the cyclopentenone moiety to the corresponding γ-butyrolactone. With regard to the oxidative cleavage of the cyclopentenone ring, it was found that desired γ-ketocarboxylic acid 27 could be obtained upon exposure of enone 26 to ozone in CH2Cl2 at −78 °C followed by either reductive (Me2S) or oxidative (H2O2, NaOH) workup [32], but the reaction suffered from low yield (33%) and reproducibility issues. While α-ketocarboxylic acid 28, detected as a byproduct, could be converged to γ-ketocarboxylic acid 27 upon treatment with H2O2 in aqueous NaOH/THF, no improvement in overall yield was observed. We then investigated a stepwise approach via α-ketocarboxylic acid 28. After an extensive screening of oxidants, the KMnO4/NaIO4 system proved to be effective for the conversion of enone 26 to 28. To our surprise, submission of crude α-ketocarboxylic acid 28 to activated carbon for removal of the residual Mn salt to avoid decomposition of H2O2 effected desired decarbonylation. As a consequence, γ-ketocarboxylic acid 27 could be obtained in 53% yield over two steps. The reason for the decarbonylation is unclear at present, but the possibility of involvement of contaminated Mn salt was excluded due to the fact that the reaction occurred from α-ketocarboxylic acid 28 produced by ozonolysis [33]. To the best of our knowledge, this is the first example of activated carbon-mediated decarbonylation of α-ketocarboxylic acid. Lactone formation from γ-ketocarboxylic acid 27 was achieved following the precedents of Wheeler [34] and Mangoni [19]: selective reduction of the carbonyl group at C6 with NaBH4 in EtOH at 0 °C was followed by lactonization through a mixed anhydride upon treatment with ClCO2Et in the presence of Et3N in CH2Cl2 at 0 °C to give lactone 23 in 88% yield over two steps [35].
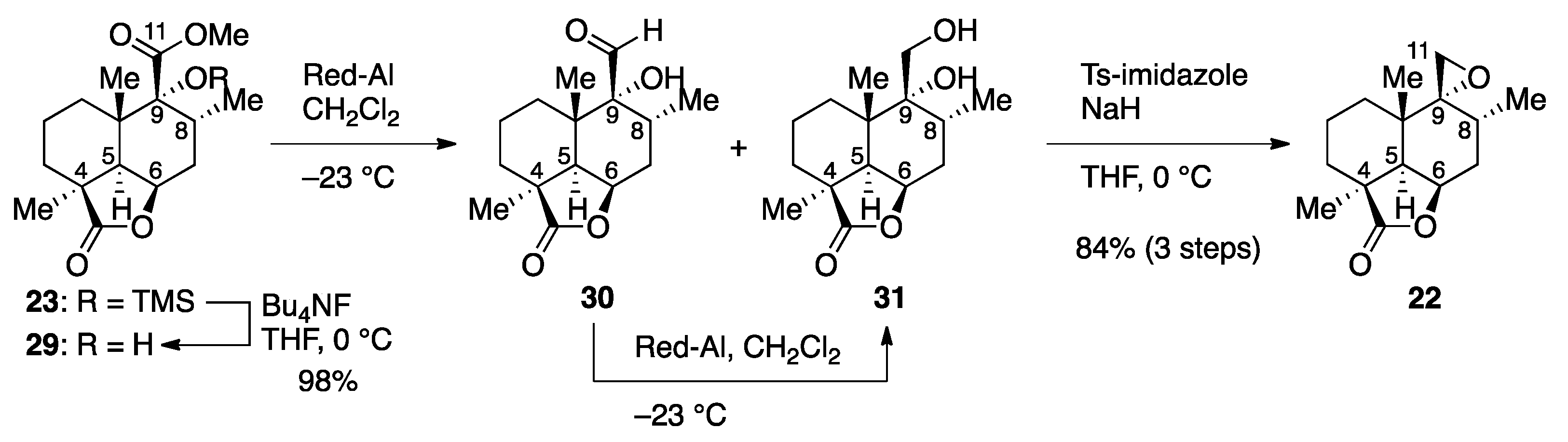
The remaining operation necessary for the synthesis of advanced intermediate 22 involved chemoselective reduction of the ester functionality in the presence of the lactone moiety (Scheme 3). As a prelude to the conversion, TMS ether 23 was converted to α-hydroxyester 29 by exposure to Bu4NF in THF at 0 °C (98% yield). With regard to the reduction, the use of NaBH4 or LiBH4 resulted in no reaction even at the reflux temperature, whereas the lactone moiety in 29 was selectively reduced with diisobutylaluminum hydride (DIBALH) in CH2Cl2 at −78 °C. We were gratified to find that the desired chemoselective reduction could be achieved by the use of sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al®) as a reducing agent. It should be noted that the hydroxyl-directed reduction was accompanied by some lactone reduction when performed using Et2O or THF as a solvent or when performed at temperatures above −20 °C. The use of CH2Cl2 proved optimal in terms of chemoselectivity and solubility of substrate 29, but the collapse of the five-membered aluminate intermediate was retarded under the optimal conditions, resulting in the formation of a mixture of aldehyde 30 and 1,2-diol 31 after aqueous workup. Thus, the mixture needed to be subjected again to Red-Al® in CH2Cl2 at −23 °C for the full reduction to 1,2-diol 31. The synthesis of advanced intermediate 22 was completed upon treatment of 1,2-diol 31 with p-toluenesulfonylimidazole and NaH in THF at 0 °C (84% yield over three steps).
2.3. Total Syntheses of Marrubiin and Related Labdane Diterpene Lactones
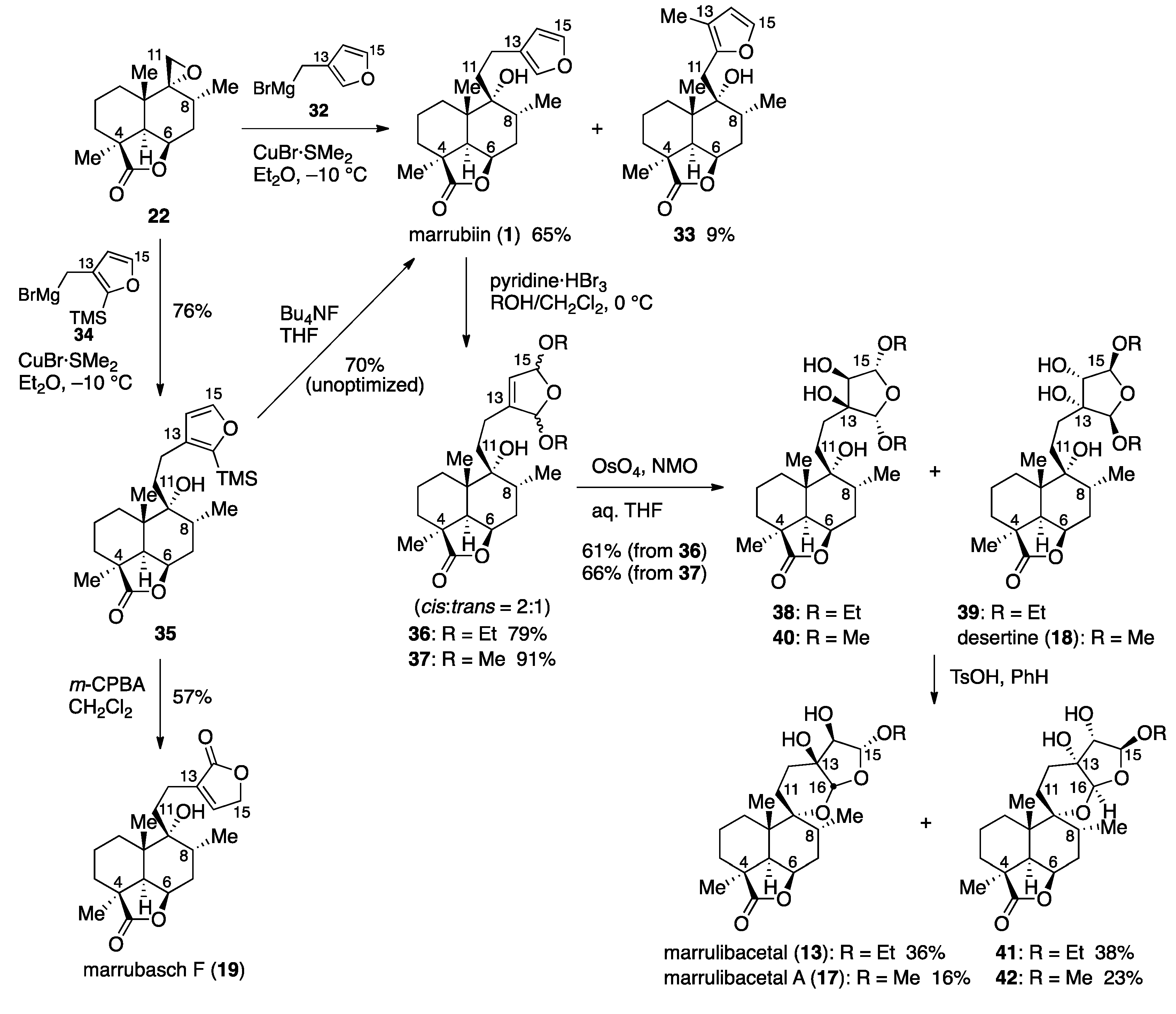
Having established a route to epoxide 22, the stage was now set for elongation of the side chain for total syntheses. In this regard, Welch and co-workers reported the synthesis of isomarrubiin (C9-epi-marrubiin) by a CuI-catalyzed epoxide ring-opening reaction of the C9-epimer of epoxide 22 in Et2O at room temperature (40% yield) [36]. With this reaction serving as a reference, we first examined Cu(I)-catalyzed epoxide-opening reaction with Grignard reagent 32 [37] (Scheme 4). After some experimentation, we found that the use of a stoichiometric amount of CuBr·SMe2 was more effective, providing marrubiin (1), [α +34.4 (c 1.04, CHCl3) [lit. [38], [α +35.8 (c 3.1, CHCl3)] in 65% yield. However, Grignard reagent 32 is prone to isomerization due to the high acidity of the furan 2-position, resulting in the formation of isomer 33 in 9% yield [39]. This problem was circumvented by the use of Grignard reagent 34 [40], the TMS group of which was uneventfully removed with Bu4NF in THF [41] after the epoxide-opening reaction.
An inspection of the structures of marrulibacetal (13) and marrulibacetal A (17) revealed that the highly oxidized tetrahydrofuran ring of these molecules could be formed by oxidation of the furan moiety in marrubiin (1) followed by internal acetalization. While unprecedented in the transformation of the marrubiin class labdane diterpenoids, successive oxidations of a furan ring have been documented in semi-synthesis of the neoclerodane diterpene natural products salvinicins A and B by Prisinzano and co-workers [42]. Furthermore, Frontana-Uribe and co-workers reported construction of the [6,6,5,5]-tetracyclic framework, in which 4,5,6,7a-tetrahydro-2H-furo[2,3-b]pyran is spirolinked to a trans-decalin ring system, by an electrochemical oxidation of hispanolone [43]. When marrubiin (1) was exposed to pyridine tribromide in EtOH/CH2Cl2 at 0 °C, oxidative acetalization occurred to give bisacetal 36 in 79% yield with a cis/trans ratio of 2:1, albeit with no sign of internal acetalization in contrast to Frontana-Uribe’s work. This result is attributed to the conformational constraint imposed by the lactone ring. The chemical yield was improved to 84% in the presence of K2CO3 as an acid scavenger. As expected from the precedent [44], the reaction rate of bisacetal 36 with OsO4/4-methylmorpholine N-oxide (NMO) in aqueous THF was influenced by the substrate structure: the reaction of a 1:1 mixture of diastereomers with the cis relative configuration at room temperature proceeded to completion within 1 h to give diols 38 and 39 in a stereoselective manner, whereas the trans-isomers were almost recovered unchanged under these conditions. Although the remaining trans-isomers could be consumed at an elevated temperature after prolonged reaction times, the lower π-facial selectivity (desired:undesired = 1:1.2), together with the fact that recovered trans-isomers could be isomerized to cis-isomers upon exposure to pyridinium p-toluenesulfonate (PPTS) in EtOH for 1 h, prompted us to perform the reaction at room temperature. Submission of an inseparable mixture of diols 38 and 39 to TsOH in benzene effected internal transacetalization, affording marrulibacetal (13), [α−21.7 (c 1.16, CHCl3) [lit. [12], [α−13.1 (c 0.29, CHCl3)], in 36% yield along with isomer 41 (38% yield) and other diastereomers (1:1, 18% combined yield). It is noteworthy that the dehydration of commercially available benzene with 3 Å molecular sieves prior to use affected the transacetalization, leading to the decomposition of substrates, although the reason is unclear at present.
Following the same reaction sequence using MeOH instead of EtOH, marrulibacetal A (17), [α−14.0 (c 1.69, CHCl3) [lit. [14], [α−10.77 (the solvent and concentration were not reported)], could be synthesized from marrubiin (1). It should be mentioned that one of two diastereomers, obtained as an inseparable mixture in a ratio of 1:1 by dihydroxylation of bisacetal 37, matched by 1H-NMR with desertine (18), although the chemical correlation to establish the stereochemistry will be presented later (vide infra). The low chemical yield (16%) of 17 was due to the formation of two isomers with the trans H15/H16 stereochemistry in 38% yield.
Kuwajima and Urabe reported that 2-(trimethylsilyl)furans could be oxidized regioselectively with peracetic acid [45], and the method was successfully applied for the synthesis of substituted Δ2-butenolides by Goldsmith, Liotta, and co-workers [46]. On the basis of these precedents, we next examined the oxidation of 2-(trimethylsilyl)furan 35 for the conversion to marrubasch F (19). Screening of peracids revealed that the use of m-chloroperoxybenzoic acid (m-CPBA) (57% yield) and peracetic acid (52% yield) afforded 19, mp 195−196 °C, [α + 41.1 (c 0.53, CHCl3) [lit. [20], mp 191−193 °C, [α +41.5 (c 1.00, CHCl3)], with m-CPBA being the optimal oxidizing agent, whereas only desilylation occurred to give marrubiin (1) with either performic acid or magnesium monoperoxyphthalate (MMPP).
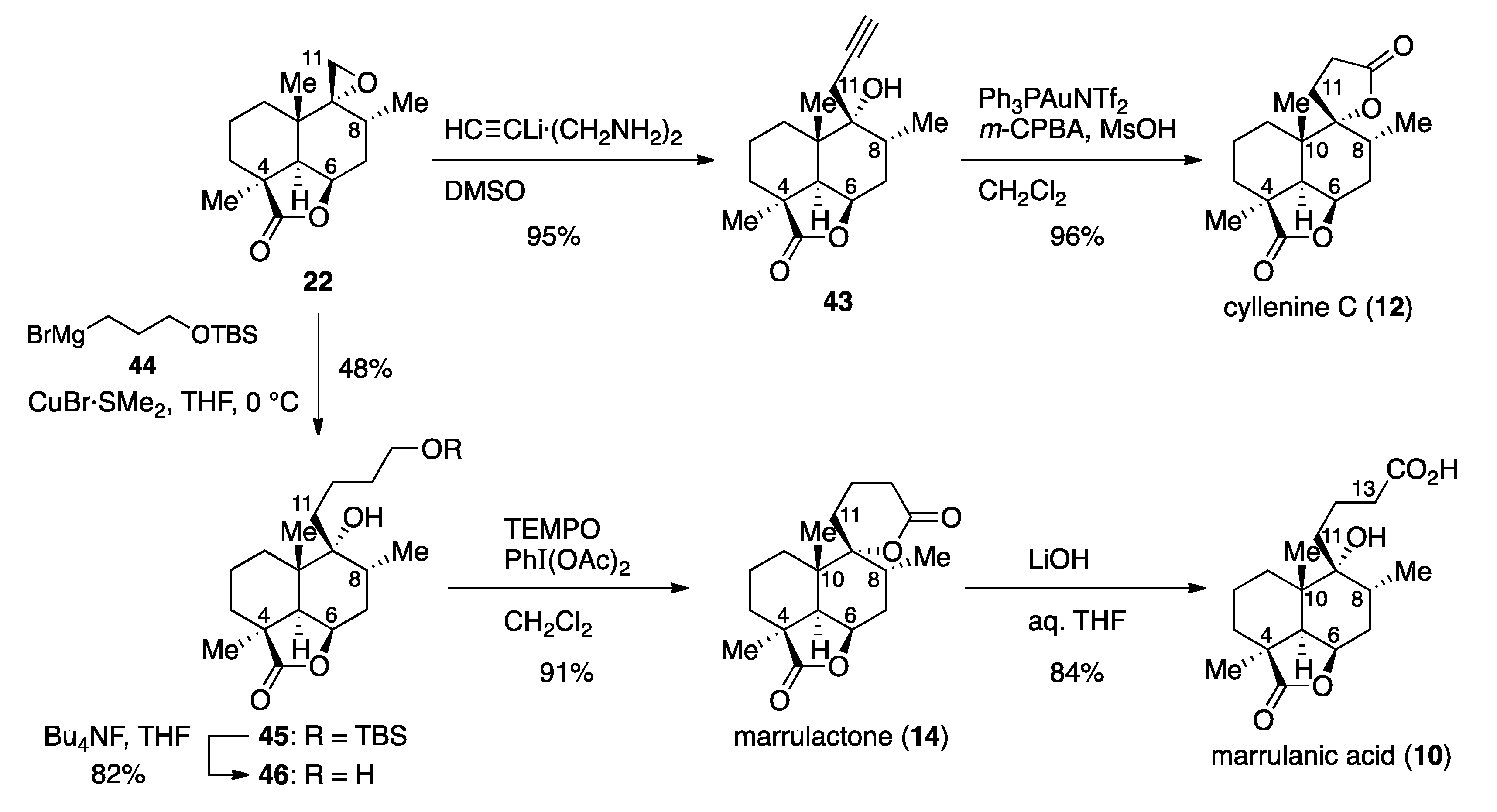
Having synthesized marrubiin (1) and natural products possessing the same carbon framework in a higher oxidation state, we next addressed the conversion of epoxide 22 to natural products 12 and 14, which required two- and three-carbon nucleophiles, respectively (Scheme 5). Fortunately, epoxide 22 underwent nucleophilic ring-opening reaction with commercially available lithium acetylide ethylenediamine complex, affording alcohol 43 in 95% yield. Oxidative lactonization of homopropargyl alcohol 43 could be attained by the gold(I)-catalyzed cycloisomerization/oxidation sequence under Ye conditions [47], completing the synthesis of cyllenine C (12). On the other hand, the use of Grignard reagent 44 [48] as a three-carbon nucleophile under conditions identical to those with 32 gave alcohol 45 (48% yield), which was desilylated with Bu4NF in THF to furnish 1,5-diol 46 in 82% yield. δ-Lactone could be formed upon oxidation of 1,5-diol 46 with 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO)/PhI(OAc)2 in CH2Cl2 according to the Forsyth protocol [49] to give marrulactone (14) in 91% yield. Since the γ-lactone in 14 proved more reluctant to hydrolyze, regioselective saponification could be realized with LiOH in THF, providing marrulanic acid (10) in 84% yield. Although synthetic compounds 12, 14, and 10 would be identical to natural products as judged by 1H and 13C-NMR analysis (see Supplementary Materials), their specific rotations [[α + 22.1 (c 0.69, CH2Cl2), [α − 11.6 (c 0.61, CHCl3), and [α + 25.4 (c 0.53, CHCl3), respectively] were inconsistent with those observed for natural cyllenine C (12), marrulactone (14), and marrulanic acid (10) [[α + 11.82 (c 0.33, CH2Cl2) [11], [α − 23.80 (c 0.22, CHCl3) [12], and [α − 10.8 (c 1.2, CHCl3) [12], respectively]. This result is attributed to contamination of the impurity in these natural products, as detected by NMR spectroscopy.
Of the eight natural products synthesized, desertine (18) could not be purified due to the difficulty of separation from its diastereomer 40. With regard to this natural product, stereochemistries were determined by an NOESY experiment by Dijoux-Franca and co-workers, who indicated two structures differing in the configuration at C15 [14]. Furthermore, evidence that supports the stereochemical relationship between the decalin and tetrahydrofuran moieties, separated by the C11−C12 two-carbon bridge, was not provided in their report. Therefore, we felt compelled to perform experiments to determine the stereochemistries of 18, and we found that exposure of marrulibacetal A (17) and its diastereomer 42 to TsOH in refluxing MeOH to effect transacetalization resulted in the formation of triol 40 and desertine (18), respectively, albeit in low yields (Scheme 6). These results suggest that the stereocenters at C13 and C14 of 18 have configurations opposite to those of 17. Together with our previous observation that desertine was produced from cis-37, the stereochemistries of this natural product can be established as shown for 18.
3. Materials and Methods
Chemistry
3.1.1. General Information
Optical rotations were recorded on a digital polarimeter with a sodium lamp (589 nm). Infrared (IR) spectra were recorded on an FT-IR spectrophotometer and absorbance bands are reported in wavenumber (cm−1). Proton nuclear magnetic resonance (1H-NMR) spectra were recorded with tetramethylsilane (δH 0.00), CHCl3 (δH 7.26), or CH2Cl2 (δH 5.32) as an internal standard. Coupling constants (J) are reported in hertz (Hz). Abbreviations of multiplicity are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Data are presented as follows: chemical shift, multiplicity, coupling constants, integration, and assignment. Marrulibacatal numbering is used for proton assignments of all intermediates. Carbon nuclear magnetic resonance (13C-NMR) spectra were recorded with CDCl3 (δC 77.0), CD2Cl2 (δC 53.84), or acetone-d6 (δC 29.84) as an internal standard. High-resolution mass spectra (HRMS) were recorded either by electrospray ionization (ESI) using a time-of-flight (TOF) analyzer, or by electron impact (EI) using a magnetic sector analyzer. Column chromatography was carried out on silica gel 60 N (63−210 μm or 40−50 μm). Analytical thin layer chromatography (TLC) was carried out with 0.25 mm silica gel plates. Visualization was accomplished with ultraviolet light and anisaldehyde stain, followed by heating. Reagents and solvents were purified by standard means or used as received unless otherwise noted. Dehydrated dichloromethane (CH2Cl2) and tetrahydrofuran (THF, stabilizer free) were purchased. Cyclohexylamine was distilled from calcium hydride. 4 Å molecular sieves was finely ground in mortar and heated in vacuo at 180 °C for 4 h. p-Toluenesulfonylimidazole [50], (3-furylmethyl)magnesium bromide [37], CuBr·SMe2 [51], [2-(trimethylsilyl)furan-3-yl]methanol [52], and 3-(tert-butyldimethylsilyl)oxypropylmagnesium bromide (44) [48] were prepared according to literature procedures. All reactions were conducted under an argon atmosphere unless otherwise noted.
3.1.2. Experimental Procedures and Compound Data
Methyl [2R,2(1S),3R]-3-methyl-2-(1-methylcyclohex-2-en-1-yl)-2-(trimethylsilyl)oxyhex-5-ynoate dicobalt hexacarbonyl complex (25). Co2(CO)8 (663 mg, 1.94 mmol) was added to an ice-cooled (0 °C) solution of enyne 20 (522 mg, 1.62 mmol) in CH2Cl2 (16 mL). After 1.5 h of stirring at room temperature, the reaction mixture was concentrated in vacuo, and the reddish brown residue was chromatographed twice (silica gel 38 g, 20:1 n-hexane/AcOEt) to give dicobalt complex 25 (958 mg, 97%) as a dark red oil. Rf 0.51 (20:1 n-hexane/AcOEt); IR (neat) 2951, 2091, 2048, 2016, 1748, 1250, 1175, 1138, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.19 (s, 9H, Si(CH3)3), 0.87 (d, J = 6.6 Hz, 3H, C8-CH3), 1.00 (s, 3H, C10-CH3), 1.42 (br d, J = 13.8 Hz, 1H, one of C1-H2), 1.63 (m, 1H, one of C2-H2), 1.71 (m, 1H, one of C2-H2), 1.89−2.03 (m, 3H, one of C1-H2, C3-H2), 2.28 (ddq, J = 2.7, 11.4, 6.6 Hz, 1H, C8-H), 2.60 (ddd, J = 1.0, 11.4, 14.7 Hz, 1H, one of C7-H2), 3.49 (br d, J = 14.7 Hz, 1H, one of C7-H2), 3.71 (s, 3H, CO2CH3), 5.71 (ddd, J = 1.7, 5.2, 10.2 Hz, 1H, C4-H), 5.82 (br d, J = 10.2 Hz, 1H, C5-H), 6.01 (br t, J = 1.0 Hz, 1H, C6-CH); 13C-NMR (125.7 MHz, CDCl3) δ 2.8 (CH3), 16.1 (CH3), 20.0 (CH2), 24.4 (CH2), 25.1 (CH3), 31.5 (CH2), 36.9 (CH2), 41.3 (CH), 42.4 (C), 51.5 (CH3), 74.2 (C), 88.5 (C), 95.8 (CH), 126.8 (CH), 132.8 (CH), 175.1 (C), 200.0 (C).
Methyl (1S,6R,7R,8S,12R)-6,8-dimethyl-2-oxo-7-(trimethylsilyl)oxytricyclo[6.3.1.04,12]dodec-3-ene-7-carboxylate (trans-24). A solution of dicobalt complex 25 (150 mg, 0.247 mmol) in 1,2-dichloroethane (5 mL plus 2 × 2.5 mL rinse) was added to a refluxing solution of cyclohexylamine (0.17 mL, 1.49 mmol) in 1,2-dichloroethane (15 mL), and the resulting mixture was refluxed for 1.5 h. After cooling, the mixture was filtered through a Celite pad, and the filtrate was concentrated in vacuo. Purification of the residue (503 mg) by column chromatography (silica gel 30 g, 3:1 n-hexane/AcOEt) gave enone trans-24 (84.0 mg, 97%) as a white solid. Rf 0.42 (3:1 n-hexane/AcOEt); mp 75−76 °C (colorless plates from n-hexane); [α + 62.1 (c 1.34, CHCl3); IR (KBr) 2945, 1736, 1694, 1620, 1458, 1260, 1184, 1096, 1034, 982, 843 cm−1; 1H-NMR (500 MHz, CD2Cl2) δ 0.20 (s, 9H, Si(CH3)3), 0.62 (s, 3H, C10-CH3), 0.91 (d, J = 6.2 Hz, 3H, C8-CH3), 1.01 (dt, J = 13.4, 7.4 Hz, 1H, one of C1-H2), 1.51−1.63 (m, 2H, one of C2-H2, one of C3-H2), 1.69 (m, 1H, one of C2-H2), 1.80 (m, 1H, one of C3-H2), 1.89 (ddd, J = 4.5, 8.9, 13.4 Hz, 1H, one of C1-H2), 2.16 (t, J = 13.9 Hz, 1H, one of C7-H2), 2.47 (ddd, J = 7.3, 8.5, 9.5 Hz, 1H, C4-H), 2.56−2.61 (m, 2H, one of C7-H2, C8-H), 3.16 (d, J = 7.3 Hz, 1H, C5-H), 3.70 (s, 3H, CO2CH3), 5.80 (s, 1H, =CHCO); 13C-NMR (125.7 MHz, CD2Cl2) δ 2.6 (CH3), 18.3 (CH3), 19.7 (CH2), 19.9 (CH2), 21.1 (CH3), 28.8 (CH2), 34.9 (CH2), 37.0 (CH), 43.7 (CH), 45.0 (CH), 45.8 (C), 51.8 (CH3), 86.1 (C), 126.0 (CH), 172.9 (CH), 179.4 (C), 211.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H30O4SiNa 373.1806; found 373.1823.
One-pot reaction. Co2(CO)8 (4.08 g, 11.9 mmol) was added to an ice-cooled (0 °C) solution of enyne 20 (3.20 g, 9.93 mmol) in 1,2-dichloroethane (100 mL), and the mixture was stirred at room temperature for 3 h. The resulting dark red suspension was diluted with 1,2-dichloroethane (900 mL), and cyclohexylamine (8.0 mL, 70.7 mmol) was added. After 5 h of heating at reflux, the reaction mixture was cooled to room temperature, and the volatile elements were removed in vacuo. The residue was suspended in 4:1 n-hexane/AcOEt and filtered through a Celite pad. Evaporation of the filtrate in vacuo furnished the crude product (5.75 g), which was purified by column chromatography (silica gel 200 g, 25:1 → 10:1 → 4:1 → 1:1 n-hexane/AcOEt) to give enone trans-24 (3.26 g, 94%) as a white solid.
Methyl (1S,6R,7R,8S,12R)-1,6,8-trimethyl-2-oxo-7-(trimethylsilyl)oxytricyclo[6.3.1.04,12]dodec-3-ene-7-carboxylate (26). KHMDS in toluene (0.5 M, 6.2 mL, 3.10 mmol) was added to a cooled (−78 °C) solution of enone trans-24 (988 mg, 2.82 mmol) in THF (30 mL). After 40 min of stirring, iodomethane (0.23 mL, 3.69 mmol) was added, and the resulting mixture was stirred at 0 °C for 40 min. The reaction was quenched with saturated aqueous NH4Cl (30 mL), and the mixture was extracted with AcOEt (3 × 60 mL). The combined organic extracts were washed with brine (2 × 30 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (1.16 g), which was purified by column chromatography (silica gel 30 g, 6:1 n-hexane/AcOEt) to give methylated product 26 (915 mg, 89%) as a yellow oil. Rf 0.40 (4:1 n-hexane/AcOEt); [α +72.2 (c 0.95, CHCl3); IR (neat) 3433, 2953, 1738, 1703, 1624, 1458, 1252, 1186, 1103, 1042, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.20 (s, 9H, Si(CH3)3), 0.71 (s, 3H, C10-CH3), 0.91 (d, J = 6.3 Hz, 3H, C8-CH3), 1.02 (ddd, J = 7.4, 9.2, 13.5 Hz, 1H, one of C1-H2), 1.11 (s, 3H, C4-CH3), 1.43 (m, 1H, one of (CH2)2), 1.60 (m, 1H, one of (CH2)2), 1.64−1.75 (m, 2H, two of (CH2)2), 1.92 (ddd, J = 2.8, 10.2, 13.5 Hz, 1H, one of C1-H2), 2.12 (m, 1H, one of C7-H2), 2.54−2.62 (m, 2H, one of C7-H2, C8-H), 2.67 (br s, 1H, C5-H), 3.73 (s, 3H, CO2CH3), 5.78 (t, J = 1.4 Hz, 1H, =CHCO); 13C-NMR (125.7 MHz, CDCl3) δ 2.5 (CH3), 17.7 (CH2), 18.0 (CH3), 21.1 (CH3), 26.6 (CH2), 27.3 (CH3), 28.0 (CH2), 34.8 (CH2), 36.4 (CH), 45.80 (C), 45.84 (C), 51.5 (CH3), 52.3 (CH), 85.7 (C), 123.8 (CH), 172.6 (C), 178.5 (C), 215.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H32O4SiNa 387.1962; found 387.1950.
(1S,4aS,5R,6R,8aR)-5-(Methoxycarbonyl)-1,4a,6-trimethyl-8-oxo-5-(trimethylsilyl)oxydecahydronaphthalene-1-carboxylic acid (27). KMnO4 (4.6 mg, 29.1 μmol) was added to a solution of NaIO4 (270 mg, 1.26 mmol) in H2O (7.0 mL), and the mixture was stirred for 30 min. To the mixture was added K2CO3 (23.0 mg, 0.166 mmol), followed by a solution of enone 26 (50.3 mg, 0.138 mmol) in t-BuOH (1.4 mL plus 2 × 1 mL rinse). After 24 h of stirring, the reaction was quenched with NaHSO3 (507 mg, 4.87 mmol), and the resulting mixture was extracted with AcOEt (3 × 20 mL). The combined organic extracts were washed with brine (2 × 15 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (70.9 mg), which was used without further purification.
Activated carbon (708 mg) was added to an ice-cooled (0 °C) solution of the crude α-ketocarboxylic acid 28 (70.9 mg) in AcOEt (14 mL). After 63 h of stirring at room temperature, the resulting suspension was filtered through a Celite pad, and the filtrate was evaporated in vacuo. Purification of the crude product (74.7 mg) by column chromatography (silica gel 5.2 g, 7:1 n-hexane/AcOEt) gave γ-ketocarboxylic acid 27 (28.3 mg, 53%) as an amorphous solid. Rf 0.60 (1:1 n-hexane/AcOEt); [α + 62.5 (c 1.01, CHCl3); IR (neat) 2953, 1732, 1717, 1674, 1456, 1252, 1182, 1153, 1090, 843 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.24 (s, 9H, Si(CH3)3), 0.82 (dt, J = 3.5, 13.4 Hz, 1H, one of C3-H2), 0.88 (d, J = 6.4 Hz, 3H, C8-CH3), 0.97 (s, 3H, C10-CH3), 1.04 (d, J = 13.2 Hz, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.51 (m, 1H, one of C2-H2), 1.85 (m, 1H, one of C2-H2), 1.92 (dt, J = 4.3, 13.2 Hz, 1H, one of C1-H2), 2.26 (d, J = 13.4 Hz, 1H, one of C3-H2), 2.30 (dd, J = 4.3, 13.0 Hz, 1H, one of C7-H2), 2.51 (t, J = 13.0 Hz, 1H, one of C7-H2), 2.72 (ddq, J = 4.3, 13.0, 6.4 Hz, 1H, C8-H), 3.13 (s, 1H, C5-H), 3.75 (s, 3H, CO2CH3), 12.62 (br s, 1H, CO2H); 13C-NMR (125.7 MHz, CDCl3) δ 2.8 (CH3), 17.3 (CH3), 17.5 (CH3), 18.4 (CH2), 28.0 (CH3), 32.7 (CH2), 37.0 (CH), 38.7 (CH2), 43.6 (C), 46.4 (CH2), 49.6 (C), 52.0 (CH3), 60.3 (CH), 86.1 (C), 171.9 (C), 176.0 (C), 218.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H32O6SiNa 407.1860; found 407.1847.
Methyl (1S,4R,6R,7R,8S,12R)-1,6,8-trimethyl-2-oxo-7-(trimethylsilyl)oxy-3-oxatricyclo[6.3.1.04,12]dodecane-7-carboxylate (23). A solution of γ-ketocarboxylic acid 27 (38.6 mg, 0.10 mmol) in EtOH (0.25 mL plus 2 × 0.25 mL rinse) was added to an ice-cooled (0 °C) solution of NaBH4 (4.3 mg, 0.11 mmol) in EtOH (0.5 mL). After 30 min of stirring at 0 °C, NaBH4 (3.7 mg, 0.098 mmol) was added, and the mixture was stirred at 0 °C for another 30 min. The reaction was quenched with saturated aqueous NH4Cl (5 mL), and the resulting mixture was partitioned between AcOEt (10 mL) and H2O (3 mL). The aqueous layer was extracted with AcOEt (5 × 10 mL), and the combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (42.7 mg), which was used without further purification.
Ethyl chloroformate (50 μL, 0.523 mmol) was added to an ice-cooled (0 °C) mixture of crude γ-hydroxycarboxylic acid (42.7 mg) and Et3N (0.10 mL, 0.717 mmol) in CH2Cl2 (1.7 mL), and the mixture was stirred for 30 min. The reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the mixture was partitioned between n-hexane/AcOEt (10:1, 22 mL) and H2O (5 mL). The aqueous layer was extracted with n-hexane/AcOEt (10:1, 22 mL), and the combined organic extracts were washed with brine (20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo followed by column chromatography (silica gel 2.5 g, 9:1 n-hexane/AcOEt) afforded lactone 23 (32.6 mg, 88% for two steps) as a colorless oil. Rf 0.44 (4:1 n-hexane/AcOEt); [α + 24.6 (c 1.07, CHCl3); IR (neat) 2953, 1771, 1732, 1456, 1250, 1180, 1142, 1105, 1053, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.15 (s, 9H, Si(CH3)3), 0.79 (d, J = 6.4 Hz, 3H, C8-CH3), 0.85 (dt, J = 13.0, 8.5 Hz, 1H, one of C1-H2), 1.10 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.42−1.56 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.69 (m, 1H, one of C2-H2), 1.77 (dd, J = 11.5, 13.0 Hz, 1H, one of C1-H2), 2.10−2.16 (m, 2H, one of C3-H2, C5-H), 2.22 (dd, J = 6.0, 16.2 Hz, 1H, one of C7-H2), 2.56 (m, 1H, C8-H), 3.73 (s, 3H, CO2CH3), 4.68 (br t, J = 5.3 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, acetone-d6) δ 2.6 (CH3), 18.38 (CH3), 18.40 (CH2), 22.3 (CH3), 23.4 (CH3), 29.0 (CH2), 29.4 (CH2), 30.6 (CH), 31.9 (CH2), 40.4 (C), 44.1 (C), 45.6 (CH), 52.0 (CH3), 76.0 (CH), 86.1 (C), 173.6 (C), 183.2 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H32O5SiNa 391.1911; found 391.1917.
Methyl (1S,4R,6R,7R,8S,12R)-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane-7-carboxylate (29). Bu4NF in THF (1.0 M, 2.0 mL, 2.0 mmol) was added to an ice-cooled (0 °C) solution of TMS ether 23 (488 mg, 1.32 mmol) in THF (14 mL). After 1 h of stirring at 0 °C, the mixture was partitioned between AcOEt (40 mL) and H2O (15 mL), and the aqueous layer was extracted with AcOEt (40 mL). The combined organic extracts were washed with brine (2 × 20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (634 mg), which was purified by column chromatography (silica gel 10 g, 10:1 → 3:1 n-hexane/AcOEt) to give α-hydroxyester 29 (383 mg, 98%) as a white solid. Rf 0.45 (2:1 n-hexane/AcOEt); mp 142−143 °C (colorless needles from n-hexane); [α + 25.0 (c 1.04, CHCl3); IR (KBr) 3455, 2959, 1761, 1732, 1466, 1375, 1236, 1144, 1098, 1045, 932 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.78 (d, J = 6.4 Hz, 3H, C8-CH3), 0.87 (dt, J = 13.0, 8.5 Hz, 1H, one of C1-H2), 1.22 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.44−1.71 (m, 5H, one of C1-H2, C2-H2, one of C3-H2, one of C7-H2), 2.15 (dt, J = 4.5, 13.4 Hz, 1H, one of C3-H2), 2.28 (dd, J = 6.3, 16.4 Hz, 1H, one of C7-H2), 2.32 (d, J = 4.6 Hz, 1H, C5-H), 2.53 (ddq, J = 6.3, 11.7, 6.4 Hz, 1H, C8-H), 3.33 (s, 1H, C9-OH), 3.82 (s, 3H, CO2CH3), 4.74 (dd, J = 4.6, 6.8 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 17.6 (CH2), 21.6 (CH3), 23.0 (CH3), 27.9 (CH2), 28.1 (CH2), 29.0 (CH), 30.7 (CH2), 38.9 (C), 43.4 (C), 44.7 (CH), 52.8 (CH3), 75.9 (CH), 80.8 (C), 175.9 (C), 183.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C16H24O5Na 319.1516; found 319.1519; Anal. Calcd for C16H24O5: C, 64.84; H, 8.16. Found: C, 64.82; H, 8.06.
(2′R,2aS,5aS,6R,7R,8aR)-2a,5a,7-Trimethyloctahydrospiro[6H-naphtho[1,8-bc]furan-6,2′-oxiran]-2(2aH)-one (22). Sodium bis(2-methoxyethoxy)aluminum hydride in toluene (3.3 M, 1.1 mL, 3.6 mmol) was diluted with CH2Cl2 (5.6 mL), and the solution was cooled to −78 °C. A solution of α-hydroxyester 29 (255 mg, 0.86 mmol) in CH2Cl2 (2 mL plus 2 × 0.5 mL rinse) was added, and the mixture was stirred at −23 °C for 22 h. The reaction was quenched by addition of MeOH (3 mL) followed by 10% aqueous potassium sodium tartrate (12 mL). After 3 h of stirring, the resulting mixture was extracted with AcOEt (2 × 40 mL), and the combined organic extracts were washed with brine (2 × 20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (235 mg), which was used without further purification.
This sequence was repeated employing bis(2-methoxyethoxy)aluminum hydride in toluene (3.3 M, 0.16 mL, 0.53 mmol) and CH2Cl2 (4 mL) with the reaction time of 16 h at −23 °C. The crude product (216 mg) was used without further purification.
A solution of crude diol 31 (216 mg) in THF (2 mL plus 1 mL and 2 × 0.5 mL rinse) was added dropwise to an ice-cooled (0 °C) suspension of NaH (60% in oil, 240 mg, 6.01 mmol) in THF (4.6 mL). After 30 min of stirring at 0 °C, p-toluenesulfonyl imidazole (577 mg, 2.59 mmol) was added, and the mixture was stirred at 0 °C for 11 h. The reaction was quenched with saturated aqueous NH4Cl (10 mL), and the resulting mixture was extracted with AcOEt (2 × 40 mL). The combined organic extracts were washed with brine (2 × 20 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (563 mg), which was purified by column chromatography (silica gel 15 g, 40:1 → 10:1 n-hexane/AcOEt) to give epoxide 22 (182 mg, 84% for three steps) as a white solid. Rf 0.53 (2:1 n-hexane/AcOEt); mp 83−84 °C (colorless needles from 8:1 n-hexane/Et2O); [α +13.7 (c 1.21, CHCl3); IR (KBr) 2941, 1761, 1464, 1393, 1354, 1209, 1105, 997 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.76 (d, J = 6.6 Hz, 3H, C8-CH3), 0.97 (dt, J = 12.4, 8.5 Hz, 1H, one of C1-H2), 1.24 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.35 (dd, J = 10.1, 12.4 Hz, 1H, one of C1-H2), 1.45−1.51 (m, 2H, one of C2-H2, one of C3-H2), 1.66 (ddd, J = 4.7, 11.8, 16.0 Hz, 1H, one of C7-H2), 1.72 (m, 1H, one of C2-H2), 1.90 (d, J = 4.7 Hz, 1H, C5-H), 2.14 (dt, J = 5.4, 15.5 Hz, 1H, one of C3-H2), 2.37 (dd, J = 6.1, 16.0 Hz, 1H, one of C7-H2), 2.55 (ddq, J = 6.1, 11.8, 6.6 Hz, 1H, C8-H), 2.63 (d, J = 3.9 Hz, 1H, one of C11-H2), 2.71 (d, J = 3.9 Hz, 1H, one of C11-H2), 4.79 (br t, J = 4.7 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.0 (CH3), 17.3 (CH2), 22.4 (CH3), 24.9 (CH), 25.0 (CH3), 26.8 (CH2), 28.1 (C), 32.5 (CH2), 34.6 (C), 44.1 (C), 45.9 (CH2), 47.5 (CH), 63.7 (C), 76.0 (CH), 183.3 (C); HRMS (ESI) m/z [M + Na]+ calcd for C15H22O3Na 273.1461; found 273.1469.
Marrubiin (1). To a cooled (−10 °C) suspension of CuBr·SMe2 (65.1 mg, 0.316 mmol) in Et2O (1.5 mL) was added a 0.14 M solution of (3-furylmethyl)magnesium bromide (32) in Et2O (3.35 mL, 0.69 mmol) [prepared from 3-(bromomethyl)furan (563 mg, 3.37 mmol) and magnesium (103 mg, 4.22 mmol) in Et2O (4 mL) at 0 °C], followed by addition of a solution of epoxide 22 (40.2 mg, 0.161 mmol) in Et2O (0.5 mL plus 2 × 0.5 mL rinse). After 2 h, the reaction was quenched with saturated aqueous NH4Cl (5 mL), and the resulting mixture was extracted with AcOEt (3 × 10 mL). The combined organic extracts were washed with brine (20 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (200 mg), which was chromatographed twice (silica gel 5 g, 20:1 → 10:1 n-hexane/AcOEt) to give marrubiin (1, 34.7 mg, 65%) and isomer 33 (4.7 mg, 9%) as white solids. Rf 0.59 (1:1 n-hexane/AcOEt); mp 160−161 °C (colorless needles from 4:1 n-hexane/AcOEt) (lit. [38], mp 160 °C); [α +34.4 (c 1.04, CHCl3) [lit. [38], [α +35.8 (c 3.1, CHCl3)]; IR (KBr) 3466, 2940, 2870, 1740, 1468, 1395, 1356, 1304, 1269, 1200, 1153, 1101, 1024, 984 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.97 (d, J = 6.4 Hz, 3H, C8-CH3), 1.07 (s, 3H, C10-CH3), 1.26 (s, 1H, C9-OH), 1.29 (s, 3H, C4-CH3), 1.32 (dt, J = 12.8, 8.5 Hz, 1H, one of C1-H2), 1.43−1.55 (m, 2H, one of C2-H2, one of C3-H2), 1.66−1.79 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C11-H2), 1.90 (ddd, J = 7.2, 10.1, 14.4 Hz, 1H, one of C11-H2), 2.09−2.18 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.23 (d, J = 4.6 Hz, 1H, C5-H), 2.48−2.58 (m, 2H, C12-H2), 4.74 (br dd, J = 4.6, 6.5 Hz, 1H, C6-H), 6.27 (s, 1H, C14-H), 7.24 (s, 1H, C16-H), 7.37 (s, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.6 (CH3), 18.2 (CH2), 21.0 (CH2), 22.3 (CH3), 22.9 (CH3), 28.3 (CH2), 28.6 (CH2), 31.5 (CH2), 32.4 (CH), 35.1 (CH2), 39.7 (C), 43.8 (C), 44.8 (CH), 75.8 (C), 76.2 (CH), 110.7 (CH), 125.0 (C), 138.6 (CH), 143.1 (CH), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H28O4Na 355.1880; found 355.1867.
Data for (1S,4R,6R,7R,8S,12R)-7-[(3-methylfuran-2-yl)methyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (33): Rf 0.27 (3:1 n-hexane/AcOEt); mp 173−174 °C (colorless needles from 3:1 n-hexane/AcOEt); [α +12.0 (c 0.38, CHCl3); IR (KBr) 3453, 2955, 2928, 2874, 1738, 1456, 1393, 1352, 1300, 1279, 1198, 1152, 1138, 999, 982 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.85 (d, J = 6.4 Hz, 3H, C8-CH3), 1.07 (s, 3H, C10-CH3), 1.15 (m, 1H, one of C1-H2), 1.28 (s, 3H, C4-CH3), 1.41−1.52 (m, 2H, one of C1-H2, one of C2-H2), 1.66−1.74 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.98 (s, 3H, C13-CH3), 2.06−2.21 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.28 (d, J = 4.7 Hz, 1H, C5-H), 2.33 (s, 1H, C9-OH), 2.78 (d, J = 15.5 Hz, 1H, one of C11-H2), 2.88 (d, J = 15.5 Hz, 1H, one of C11-H2), 4.73 (ddd, J = 1.4, 4.7, 6.1 Hz, 1H, C6-H), 6.19 (d, J = 1.8 Hz, 1H, C14-H), 7.28 (d, J = 1.8 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 10.3 (CH3), 16.3 (CH3), 18.3 (CH2), 22.2 (CH3), 22.9 (CH3), 28.23 (CH2), 28.24 (CH2), 30.6 (CH2), 31.93 (CH), 31.94 (CH2), 39.6 (C), 43.8 (C), 44.6 (CH), 76.2 (CH), 76.9 (C), 113.3 (CH), 116.4 (C), 140.5 (CH), 148.0 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H28O4Na 355.1880; found 355.1871.
(1S,4R,6R,7R,8S,12R)-7-[2-[2-(Trimethylsilyl)furan-3-yl]ethyl]]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (35). Phosphorus tribromide (0.28 mL, 2.98 mmol) was added to an ice-cooled (0 °C) solution of [2-(trimethylsilyl)furan-3-yl]methanol (998 mg, 5.86 mmol) in Et2O (30 mL). After 30 min of stirring, the reaction was quenched with brine (30 mL), and the resulting mixture was extracted with Et2O (30 mL). The organic extract was dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale brown oil, which was purified by distillation to give 3-(bromomethyl)-2-(trimethylsilyl)furan (618 mg, 45%) as a pale brown oil.
A solution of 3-(bromomethyl)-2-(trimethylsilyl)furan (584 mg, 2.50 mmol) in Et2O (1 mL) was added to a cooled (−10 °C) suspension of magnesium tuning (102 mg, 4.20 mmol) in Et2O (1 mL), and the reaction mixture was stirred for 1.5 h. The 0.23 M solution of [2-(trimethylsilyl)furan-3-yl]methylmagnesium bromide (34) in Et2O (1.5 mL, 0.345 mmol) thus obtained was added to a cooled (−10 °C) suspension of CuBr·SMe2 (69.6 mg, 0.339 mmol) in Et2O (0.6 mL), followed by addition of a solution of epoxide 22 (42.6 mg, 0.170 mmol) in Et2O (1.0 mL). After 4 h of stirring, the reaction was quenched with saturated aqueous NH4Cl (3 mL), and the resulting mixture was extracted with AcOEt (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale brown oil (206 mg), which was purified by flash column chromatography (silica gel 4 g, n-hexane → 40:1 → 20:1 → 5:1 n-hexane/AcOEt) to give silylated marrubiin 35 (52.0 mg, 76%) as a colorless solid. Rf 0.32 (3:1 n-hexane/AcOEt); [α +29.3 (c 2.05, acetone); IR (neat) 3479, 3417, 2954, 2870, 1749, 1633, 1568, 1464, 1387, 1248, 1197, 1149, 1089, 1045, 1020, 991, 914, 840 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.29 (s, 9H, Si(CH3)3), 0.99 (d, J = 6.5 Hz, 3H, C8-CH3), 1.06 (s, 3H, C10-CH3), 1.29 (s, 3H, C4-CH3), 1.30 (m, 1H, one of C1-H2), 1.45 (m, 1H, one of C3-H2), 1.52 (m, 1H, one of C2-H2), 1.68−1.75 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C11-H2), 1.87 (ddd, J = 7.1, 10.3, 14.5 Hz, 1H, one of C11-H2), 2.09−2.19 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.23 (d, J = 4.6 Hz, 1H, C5-H), 2.56−2.75 (m, 2H, C12-H2), 4.74 (dd, J = 4.6, 6.3 Hz, 1H, C6-H), 6.27 (d, J = 1.2 Hz, 1H, C14-H), 7.55 (d, J = 1.2 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ −0.9 (CH3), 16.6 (CH3), 18.2 (CH2), 21.7 (CH2), 22.3 (CH3), 22.9 (CH3), 28.3 (CH2), 28.7 (CH2), 31.5 (CH2), 32.4 (CH), 36.2 (CH2), 39.7 (C), 43.8 (C), 44.9 (CH), 75.8 (C), 76.2 (CH), 110.7 (CH), 135.0 (C), 146.4 (CH), 154.3 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C23H36O4SiNa 427.2275; found 427.2259.
Marrubiin (1). Bu4NF in THF (1.0 M, 20 μL, 20 μmol) was added to a solution of silylated marrubiin 35 (0.7 mg, 1.7 μmol) in THF (0.2 mL). After 4 h of stirring, the mixture was partitioned between AcOEt (8 mL) and H2O (8 mL), and the aqueous layer was extracted with AcOEt (8 mL). The combined organic extracts were washed with brine (8 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (0.5 mg), which was purified by column chromatography (silica gel 1 g, 4:1 → 1:1 n-hexane/AcOEt) to give marrubiin (1, 0.4 mg, 70%) as a white solid.
(1S,4R,6R,7R,8S,12R)-7-[2-(2,5-Diethoxy-2,5-dihydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (36). Pyridinium tribromide (11.0 mg, 34 μmol) was added to an ice-cooled (0 °C) solution of marrubiin (1, 10.4 mg, 31 μmol) in CH2Cl2/EtOH (1:1, 0.6 mL). After 10 min of stirring, the reaction was quenched with a mixture of saturated aqueous NaHCO3 (3 mL) and 1 M aqueous Na2S2O3 (2 mL), and the resulting mixture was extracted with AcOEt (2 × 20 mL). The combined organic extracts were washed with brine (30 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (14.7 mg), which was purified by column chromatography (silica gel 2.5 g, 3:1 → 1:1 n-hexane/AcOEt) to give bisacetals 36 (10.4 mg, 79%, dr = 2:2:1:1) as a colorless oil. Rf 0.58 (2:3 n-hexane/AcOEt); [α +33.9 (c 1.19, CHCl3); IR (neat) 3516, 2972, 2930, 1767, 1755, 1458, 1373, 1346, 1198, 1101, 1045, 1018, 984 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.92 (d, J = 6.3 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.21−1.34 (m, 11H), 1.43−1.56 (m, 2H), 1.66−1.77 (m, 4H), 1.86 (m, 1H, one of CH2), 2.05−2.32 (m, 6H), 3.53−3.82 (m, 4H, 2 × OCH2CH3), 4.74 (m, 1H, C6-H), 5.48 (s, 0.65H, C15-H), 5.60 (s, 0.65H, C16-H), 5.67 (s, 1H, C14-H), 5.75 (br d, J = 3.6 Hz, 0.35H, C16-H), 5.85 (br d, J = 3.6 Hz, 0.35H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.3 (CH3), 15.4 (CH3), 16.50 (CH3), 16.51 (CH3), 16.54 (CH3), 18.09 (CH2), 18.12 (CH2), 22.22 (CH3), 22.23 (CH3), 22.26 (CH3), 22.28 (CH3), 22.31 (CH2), 22.4 (CH2), 22.49 (CH2), 22.52 (CH2), 22.87 (CH3), 22.89 (CH3), 22.96 (CH3), 22.97 (CH3), 28.27 (CH2), 28.28 (CH2), 28.59 (CH2), 28.61 (CH2), 28.63 (CH2), 31.4 (CH2), 31.46 (CH2), 31.47 (CH2), 32.0 (CH2), 32.11 (CH2), 32.15 (CH), 32.22 (CH), 32.29 (CH2), 32.33 (CH2), 32.5 (CH), 39.69 (C), 39.70 (C), 39.71 (C), 39.73 (C), 43.71 (C), 43.72 (C), 43.76 (C), 44.7 (CH), 44.8 (CH), 62.3 (CH2), 62.61 (CH2), 62.62 (CH2), 62.8 (CH2), 63.1 (CH2), 63.41 (CH2), 63.42 (CH2), 63.5 (CH2), 75.37 (C), 75.388 (C), 75.394 (C), 76.06 (CH), 76.12 (CH), 76.13 (CH), 105.95 (CH), 105.97 (CH), 106.937 (CH), 106.943 (CH), 107.5 (CH), 107.6 (CH), 108.2 (CH), 108.3 (CH), 123.8 (CH), 124.0 (CH), 124.1 (CH), 124.3 (CH), 145.3 (C), 145.78 (C), 145.81 (C), 183.7 (C), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H38O6Na 445.2561; found 445.2550.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(2,5-Diethoxy-3,4-dihydroxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (38) and [1S,4R,6R,7R,7(2R,3R,4S,5S),8S, 12R]-7-[2-(2,5-diethoxy-3,4-dihydroxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatri cyclo[6.3.1.04,12]dodecane (39). A 0.157 M solution of OsO4 in t-BuOH (0.06 mL, 9.4 μmol) was added to a solution of bisacetals 36 (21.7 mg, 51 μmol) and NMO (4.8 M in H2O, 0.04 mL, 0.19 mmol) in THF/H2O (10:1, 0.55 mL). After 1 h of stirring, the reaction was quenched with 1 M aqueous Na2S2O3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (15 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (29.1 mg), which was purified by flash column chromatography (silica gel 2.3 g, 1:1 n-hexane/AcOEt) to give a 1:1 mixture of triols 38 and 39 (14.3 mg, 61%) as a colorless oil, along with recovered bisacetals 36 (8.2 mg, 38%) as a colorless oil. Rf 0.49 (1:4 n-hexane/AcOEt); [α +29.1 (c 0.56, CHCl3); IR (neat) 3462, 2930, 1751, 1458, 1389, 1375, 1101, 978 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.5 Hz, 1.5H, C8-CH3), 0.92 (d, J = 6.4 Hz, 1.5H, C8-CH3), 1.04 (s, 3H, C10-CH3), 1.207 (t, J = 7.1 Hz, 1.5H, OCH2CH3), 1.214 (t, J = 7.1 Hz, 1.5H, OCH2CH3), 1.23 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.29 (s, 3H, C4-CH3), 1.42−1.52 (m, 2H, CH2), 1.60−1.97 (m, 8H, 4 × CH2), 2.03−2.15 (m, 3.5H, CH2, C8-H, OH), 2.24 (d, J = 4.7 Hz, 1H, C5-H), 2.33 (br s, 0.5H, OH), 2.99 (d, J = 6.9 Hz, 0.5H, OH), 3.05 (m, 1H, OH), 3.44−3.57 (m, 2.5H, OCH2CH3, OH), 3.76−3.84 (m, 2H, OCH2CH3), 3.96 (m, 1H, C14-H), 4.73 (br s, 1H, C6-H), 4.85 (s, 0.5H, C16-H), 4.86 (s, 0.5H, C16-H), 4.97 (d, J = 4.3 Hz, 0.5H, C15-H), 4.98 (d, J = 4.3 Hz, 0.5H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 14.88 (CH3), 14.93 (CH3), 15.2 (CH3), 16.5 (CH3), 16.8 (CH3), 18.1 (CH2), 22.2 (CH3), 22.3 (CH3), 22.85 (CH3), 22.93 (CH3), 27.4 (CH2), 28.1 (CH2), 28.27 (CH2), 28.28 (CH2), 28.31 (CH2), 28.4 (CH2), 28.5 (CH2), 28.6 (CH2), 31.5 (CH2), 31.6 (CH2), 32.47 (CH), 32.49 (CH), 39.97 (C), 40.03 (C), 43.88 (C), 43.91 (C), 44.90 (CH), 44.93 (CH), 63.07 (CH2), 63.10 (CH2), 64.55 (CH2), 64.57 (CH2), 75.30 (C), 75.33 (C), 76.3 (CH), 76.4 (CH), 80.3 (CH), 80.4 (CH), 81.2 (C), 81.3 (C), 106.5 (CH), 106.7 (CH), 109.17 (CH), 109.19 (CH), 184.10 (C), 184.12 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H40O8Na 479.2615; found 479.2631.
Marrulibacetal (13). TsOH (2.9 mg, 17 μmol) was added to a mixture of triols 38 and 39 (12.5 mg, 27 μmol) in benzene (1 mL), and the mixture was stirred for 1.5 h. The reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (15 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (19.2 mg), which was purified by flash column chromatography (silica gel 2 g, 2:1 n-hexane/AcOEt) to give a mixture of marrulibacetal (13) and its diastereomers. The mixture was flash chromatographed (silica gel 4.5 g, 30:1 CHCl3/ acetone) to provide a mixture of marrulibacetal (13) and C13,14,15,16-epimer 41, along with a 1:1 mixture of C15-epimer and C13,14,16-epimer (2.0 mg, 18%). Separation of marrulibacetal (13) and 41 by flash column chromatography (silica gel 4.5 g, 50:1 CHCl3/acetone) yielded marrulibacetal (13, 4.1 mg, 36%) and C13,14,15,16-epimer 41 (4.3 mg, 38%) as white solids. Rf 0.44 (4:1 CH2Cl2/acetone); mp 177−179 °C (colorless needles from n-hexane/benzene); [α −21.7 (c 1.16, CHCl3) [lit. [12], [α −13.1 (c 0.29, CHCl3)]; IR (neat) 3435, 2961, 2928, 1773, 1740, 1458, 1389, 1244, 1200, 1111, 1053, 935 cm−1; 1H-NMR (500 MHz, CDCl3) δ 1.03 (s, 3H, C10-CH3), 1.11 (d, J = 6.8 Hz, 3H, C8-CH3), 1.19 (m, 1H, one of C1-H2), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.28 (s, 3H, C4-CH3), 1.42 (m, 1H, one of C3-H2), 1.49 (m, 1H, one of C2-H2), 1.71−1.78 (m, 2H, one of C2-H2, one of C11-H2), 1.80−1.90 (m, 3H, one of C7-H2, one of C11-H2, one of C12-H2), 1.98 (m, 1H, one of C1-H2), 2.05−2.20 (m, 4H, one of C3-H2, one of C7-H2, C8-H, one of C12-H2), 2.36 (d, J = 4.7 Hz, 1H, C5-H), 2.60 (s, 1H, C13-OH), 2.61 (d, J = 6.2 Hz, 1H, C14-OH), 3.51 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 3.82 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 3.95 (dd, J = 2.0, 6.2 Hz, 1H, C14-H), 4.77 (br dd, J = 4.7, 5.9 Hz, 1H, C6-H), 5.04 (d, J = 2.0 Hz, 1H, C15-H), 5.46 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.0 (CH3), 17.9 (CH2), 19.5 (CH3), 21.1 (CH2), 22.1 (CH3), 23.1 (CH3), 27.8 (CH2), 28.2 (CH2), 29.6 (CH2), 32.3 (CH2), 33.6 (CH), 40.9 (C), 43.9 (C), 44.6 (CH), 63.9 (CH2), 75.6 (C), 76.5 (CH), 78.5 (CH), 80.4 (C), 105.3 (CH), 108.7 (CH), 184.0 (C); HRMS (EI) m/z [M+] calcd for C22H34O7 410.2305; found 410.2300.
Data for (2S,2′aS,3S,3aR,5′aS,6R,7′R,7aR,8′aR,8′bR)-2-ethoxy-3,3a-dihydroxy-2′a,5′a,7′-trimethyltetradecahydrospiro[6H-furo[2,3-b]pyran-6,6′-[6H]naphtho[1,8-bc]furan]-2′(2′aH)-one (41). Rf 0.56 (4:1 CH2Cl2/acetone); [α +57.2 (c 1.26, CHCl3); IR (KBr) 3458, 2930, 1771, 1749, 1466, 1389, 1260, 1198, 1153, 1094, 1063, 1040, 989, 941 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.96 (d, J = 6.1 Hz, 3H, C8-CH3), 1.01 (s, 3H, C10-CH3), 1.21 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.30 (s, 3H, C4-CH3), 1.39−1.54 (m, 4H, one of C1-H2, one of C2-H2, one of C3-H2, one of C11-H2), 1.70−1.83 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C12-H2), 1.95−2.11 (m, 4H, one of C3-H2, one of C7-H2, C8-H, one of C11-H2), 2.20 (m, 1H, one of C12-H2), 2.34 (d, J = 4.5 Hz, 1H, C5-H), 2.75 (d, J = 4.8 Hz, 1H, C14-OH), 2.79 (s, 1H, C13-OH), 3.46 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 3.74 (d, J = 4.8 Hz, 1H, C14-H), 3.86 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 4.78 (br dd, J = 4.5, 7.7 Hz, 1H, C6-H), 5.01 (s, 1H, C15-H), 5.50 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 14.8 (CH3), 16.1 (CH3), 18.1 (CH2), 21.5 (CH2), 23.2 (CH3), 23.9 (CH3), 28.3 (CH2), 30.4 (CH2), 30.9 (CH2), 31.3 (CH2), 35.0 (CH), 40.8 (C), 43.9 (C), 45.5 (CH), 63.7 (CH2), 75.9 (C), 76.7 (CH), 80.0 (CH), 80.5 (C), 106.1 (CH), 108.2 (CH), 183.9 (C); HRMS (EI) m/z [M+] calcd for C22H34O7 410.2305; found 410.2297.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(3,4-Dihydroxy-2,5-dimethoxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (40) and desertine (18). Pyridinium tribromide (32.9 mg, 0.102 mmol) was added to an ice-cooled (0 °C) solution of marrubiin (1, 30.5 mg, 91.7 μmol) in CH2Cl2/MeOH (1:1, 1.8 mL). After 20 min of stirring, the reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (2 × 10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the yellow oil (53.1 mg), which was purified by column chromatography (silica gel 3 g, 4:1 → 2:1 → 1:1 n-hexane/AcOEt) to give bisacetals 37 (33.1 mg, 91%, dr = 2:2:1:1) as a yellow oil.
A 0.157 M solution of OsO4 in t-BuOH (0.08 mL, 12.5 μmol) was added to a solution of bisacetals 37 (28.3 mg, 71.7 μmol) and NMO (4.8 M in H2O, 0.06 mL, 0.29 mmol) in THF/H2O (1:1, 0.8 mL). After 1 h of stirring, the reaction was quenched with 1 M aqueous Na2S2O3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (2 × 10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the brown oil (40.7 mg), which was purified by column chromatography (silica gel 3.1 g, 2:1 → 1:1 n-hexane/AcOEt) to give a 1:1 mixture of desertine (18) and its diastereomer 40 (20.2 mg, 66%) as a brown oil, along with recovered bisacetals 37 (8.8 mg, 29%) as a colorless oil. Rf 0.30 (1:1 n-hexane/AcOEt); [α +12.9 (c 1.02, CHCl3); IR (neat) 3464, 2951, 1748, 1454, 1391, 1258, 1198, 1148, 1101, 1043, 989 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.5 Hz, 3H, C8-CH3), 1.04 (s, 1.5H, C10-CH3), 1.05 (s, 1.5H, C10-CH3), 1.28 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.45 (m, 1H, one of C3-H2), 1.51 (m, 1H, one of C2-H2), 1.65 (m, 1H, one of C7-H2), 1.60−1.98 (m, 6H, one of C1-H2, one of C2-H2, C11-H2, C12-H2), 2.07 (m, 1H, C8-H), 2.11 (m, 1H, one of C3-H2), 2.14 (m, 1H, one of C7-H2), 2.22 (d, J = 4.2 Hz, 0.5H, C5-H), 2.23 (d, J = 4.2 Hz, 0.5H, C5-H), 3.40 (s, 3H, C16-OCH3), 3.47 (s, 3H, C15-OCH3), 3.92 (d, J = 3.5 Hz, 0.5H, C14-H), 3.95 (d, J = 3.5 Hz, 0.5H, C14-H), 4.73 (m, 1H, C6-H), 4.76 (s, 0.5H, C16-H), 4.77 (s, 0.5H, C16-H), 4.89 (d, J = 3.5 Hz, 0.5H, C15-H), 4.90 (d, J = 3.5 Hz, 0.5H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 16.8 (CH3), 18.1 (CH2), 22.2 (CH3), 22.3 (CH3), 22.86 (CH3), 22.94 (CH3), 27.4 (CH2), 28.0 (CH2), 28.2 (CH2), 28.26 (CH2), 28.28 (CH2), 28.41 (CH2), 28.43 (CH2), 28.5 (CH2), 31.5 (CH2), 31.6 (CH2), 32.5 (CH), 32.6 (CH), 40.0 (C), 40.1 (C), 43.87 (C), 43.91 (C), 44.9 (CH), 45.0 (CH), 55.08 (CH3), 55.11 (CH3), 56.40 (CH3), 56.42 (CH3), 75.3 (C), 75.4 (C), 76.2 (CH), 76.3 (CH), 80.2 (CH), 80.3 (CH), 81.2 (C), 81.4 (C), 108.3 (CH), 108.6 (CH), 110.7 (CH), 110.8 (CH), 183.95 (C), 183.99 (C); HRMS (ESI) m/z [M + Na]+ calcd for C22H36O8Na 451.2302; found 451.2308.
Marrulibacetal A (17). TsOH (8.6 mg, 50 μmol) was added to a mixture of desertine (18) and its diastereomer 40 (29.6 mg, 69.0 μmol) in benzene (1.4 mL). After 5 h of stirring, an additional portion of TsOH (1.6 mg, 9.3 μmol) was added, and the reaction mixture was stirred for 3.5 h. The reaction was quenched with saturated aqueous NaHCO3 (2 mL), and the resulting mixture was extracted with AcOEt (3 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the brown oil (68.1 mg), which was purified by flash column chromatography (silica gel 5 g, 1:1 n-hexane/AcOEt) to give marrulibacetal A (17, 4.3 mg, 16%) and C13,14,15,16-isomer 42 (6.2 mg, 23%) as colorless amorphous solids. Rf 0.73 (AcOEt); [α −14.0 (c 1.69, CHCl3) (lit. [14], [α −10.77); IR (neat) 2953, 2928, 1769, 1748, 1456, 1259, 1198, 1117, 1053, 1015, 989, 935 cm−1; 1H-NMR (500 MHz, CDCl3) δ 1.03 (s, 3H, C10-CH3), 1.11 (d, J = 7.0 Hz, 3H, C8-CH3), 1.19 (m, 1H, one of C1-H2), 1.28 (s, 3H, C4-CH3), 1.42 (m, 1H, one of C3-H2), 1.49 (m, 1H, one of C2-H2), 1.71−1.92 (m, 5H, one of C2-H2, one of C7-H2, C11-H2, one of C12-H2), 1.96 (t, J = 10.6 Hz, 1H, one of C1-H2), 2.05−2.15 (m, 3H, one of C3-H2, C8-H, one of C12-H2), 2.20 (dd, J = 5.5, 16.0 Hz, 1H, one of C7-H2), 2.38 (d, J = 4.5 Hz, 1H, C5-H), 3.42 (s, 3H, OCH3), 3.89 (d, J = 1.2 Hz, 1H, C14-H), 4.79 (dd, J = 4.5, 6.3 Hz, 1H, C6-H), 4.93 (d, J = 1.2 Hz, 1H, C15-H), 5.48 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 17.9 (CH2), 19.4 (CH3), 20.7 (CH2), 22.0 (CH3), 23.1 (CH3), 27.8 (CH2), 28.2 (CH2), 30.0 (CH2), 32.2 (CH2), 33.5 (CH), 40.9 (C), 43.9 (C), 44.6 (CH), 55.5 (CH3), 75.8 (C), 76.5 (CH), 78.6 (CH), 80.3 (C), 105.7 (CH), 109.8 (CH), 184.1 (C); HRMS (ESI) m/z [M + Na]+ calcd for C21H32O7Na 419.2045; found 419.2037.
Data for (2S,2′aS,3S,3aR,5′aS,6R,7′R,7aR,8′aR,8′bR)-3,3a-dihydroxy-2-methoxy-2′a,5′a,7′-trimethyl tetradecahydrospiro[6H-furo[2,3-b]pyran-6,6′-[6H]naphtho[1,8-bc]furan]-2′(2′aH)-one (42): Rf 0.78 (AcOEt); [α +49.1 (c 0.75, CHCl3); IR (neat) 2955, 2928, 1749, 1541, 1506, 1456, 1265 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.94 (d, J = 6.1 Hz, 3H, C8-CH3), 1.01 (s, 3H, C10-CH3), 1.31 (s, 3H, C4-CH3), 1.42−1.56 (m, 4H, one of C1-H2, one of C2-H2, one of C3-H2, one of C11-H2), 1.70−1.83 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C12-H2), 1.98 (dt, J = 3.8, 14.2 Hz, 1H, one of C11-H2), 1.99−2.12 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.17 (dt, J = 4.4, 14.2 Hz, 1H, one of C12-H2), 2.35 (d, J = 4.5 Hz, 1H, C5-H), 2.87 (br s, 1H, OH), 3.41 (s, 3H, OCH3), 3.73 (s, 1H, C14-H), 4.79 (dd, J = 4.5, 6.2 Hz, 1H, C6-H), 4.91 (s, 1H, C15-H), 5.52 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.0 (CH3), 18.1 (CH2), 21.4 (CH2), 23.1 (CH3), 24.0 (CH3), 28.3 (CH2), 30.6 (CH2), 30.9 (CH2), 31.3 (CH2), 35.0 (CH), 40.8 (C), 43.9 (C), 45.5 (CH), 55.2 (CH3), 76.1 (C), 76.7 (CH), 79.8 (CH), 80.5 (C), 106.3 (CH), 109.4 (CH), 184.1 (C); HRMS (ESI) m/z [M + Na]+ calcd for C21H32O7Na 419.2045; found 419.2059.
Marrubasch F (19). m-CPBA (ca. 70%, 10.7 mg, 43.4 μmol) was azeotropically dried with benzene, and dissolved in CH2Cl2 (0.2 mL). The m-CPBA solution was added to an ice-cooled (0 °C) solution of silylated marrubiin 35 (8.1 mg, 20.0 μmol) in CH2Cl2 (0.8 mL). After 1.5 h of stirring, the reaction was quenched with a mixture of 1 M aqueous Na2S2O3 (2 mL) and saturated aqueous NaHCO3 (2 mL), and the resulting mixture was extracted with AcOEt (10 mL). The organic extract was washed with brine (4 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (28.4 mg), which was purified by preparative thin layer chromatography (200 mm × 100 mm × 0.25 mm preparative silica gel plate and elution with 1:1 n-hexane/AcOEt) to give marrubasch F (19, 3.9 mg, 56%) as a white solid. Rf 0.32 (1:1 n-hexane/AcOEt); mp 195−196 °C (pale yellow plates from 1:2 n-hexane/AcOEt) (lit. [20], mp 191−193 °C); [α +41.1 (c 0.534, CHCl3) [lit. [20], [α]D + 41.5 (c 1.00, CHCl3)]; IR (neat) 3536, 2926, 2873, 1747, 1456, 1199, 1139, 1095, 1076, 1041, 983 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.96 (d, J = 6.4 Hz, 3H, C8-CH3), 1.04 (s, 3H, C10-CH3), 1.25 (dt, J = 12.8, 8.5 Hz, 1H, one of C1-H2), 1.30 (s, 3H, C4-CH3), 1.45 (dt, J = 14.4, 4.8 Hz, 1H, one of C3-H2), 1.53 (m, 1H, one of C2-H2), 1.69−1.84 (m, 5H, one of C1-H2, one of C2-H2, one of C7-H2, C11-H2), 2.05−2.17 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.28 (d, J = 4.5 Hz, 1H, C5-H), 2.41−2.52 (m, 2H, C12-H2), 4.75 (dd, J = 4.5, 6.5 Hz, 1H, C6-H), 4.80 (br d, J = 1.4 Hz, 2H, C15-H2), 7.14 (t, J = 1.4 Hz, 1H, C14-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.7 (CH3), 18.1 (CH2), 21.2 (CH2), 22.3 (CH3), 22.9 (CH3), 28.3 (CH2), 28.6 (CH2), 31.6 (CH2), 32.4 (CH2), 32.6 (CH2), 39.9 (C), 43.8 (C), 44.9 (CH), 70.4 (CH2), 75.2 (C), 76.2 (CH), 134.7 (C), 144.3 (CH), 174.9 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H28O5Na 371.1830; found 371.1834.
(1S,4R,6R,7R,8S,12R)-7-Hydroxy-1,6,8-trimethyl-2-oxo-7-propargyl-3-oxatricyclo[6.3.1.04,12]dodecane (43). Lithium acetylide ethylene diamine complex (36.5 mg, 0.397 mmol) was added to a solution of epoxide 22 (10.7 mg, 42.7 μmol) in DMSO (0.2 mL). After 30 min of stirring, the reaction was quenched with H2O (1 mL), and the resulting mixture was extracted with AcOEt (3 × 5 mL). The combined organic extracts were washed with brine (2 × 5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow solid (13.1 mg), which was purified by column chromatography (silica gel 3 g, 2:1 n-hexane/AcOEt) to give alcohol 43 (11.2 mg, 95%) as a pale yellow solid. Rf 0.57 (1:1 n-hexane/AcOEt); mp 195−196 °C (colorless plates from n-hexane/Et2O); [α +29.9 (c 0.249, CHCl3); IR (neat) 3306, 3019, 2955, 2934, 2872, 1757, 1458, 1427, 1393, 1136, 1096, 1045, 1003 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.99 (d, J = 6.3 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.25 (dt, J = 13.0, 9.0 Hz, 1H, one of C1-H2), 1.30 (s, 3H, C4-CH3), 1.46 (dt, J = 14.7, 4.2 Hz, 1H, one of C3-H2), 1.53 (m, 1H, one of C2-H2), 1.69 (ddd, J = 6.3, 13.3, 17.7 Hz, 1H, one of C7-H2), 1.76 (m, 1H, one of C2-H2), 1.85 (dd, J = 9.6, 11.6 Hz, 1H, one of C1-H2), 2.09−2.18 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.19 (t, J = 2.7 Hz, 1H, C13-H), 2.26 (d, J = 4.5 Hz, 1H, C5-H), 2.36 (dd, J = 2.7, 17.1 Hz, 1H, one of C11-H2), 2.59 (dd, J = 2.7, 17.1 Hz, 1H, one of C11-H2), 4.73 (dd, J = 4.5, 6.3 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.3 (CH3), 18.1 (CH2), 22.3 (CH3), 22.6 (CH3), 25.3 (CH2), 28.21 (CH2), 28.22 (CH2), 31.5 (CH2), 32.3 (CH), 39.2 (C), 43.9 (C), 44.8 (CH), 73.77 (C), 73.79 (CH), 76.1 (CH), 80.7 (C), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C17H24O3Na 299.1618; found 299.1623.
Cyllenine C (12). To an ice-cooled (0 °C) mixture of alcohol 43 (6.8 mg, 24.6 μmol) and m-CPBA (ca. 65%, 9.8 mg, 36.9 μmol) in CH2Cl2 (0.3 mL) was added a 0.123 M solution of methanesulfonic acid in CH2Cl2 (0.20 mL, 24.6 μmol) followed by Ph3PAuNTf2 (1.0 mg, 1.3 μmol). After 1 h of stirring at room temperature, the reaction was quenched with saturated aqueous NaHCO3 (1 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (13.7 mg), which was purified by column chromatography (silica gel 2 g, 2:1 n-hexane/AcOEt) to give cyllenine C (12, 6.9 mg, 96%) as a white solid. Rf 0.32 (1:2 n-hexane/AcOEt); mp 164−165 °C (colorless needles from n-hexane/Et2O); [α +22.1 (c 0.69, CH2Cl2) [lit. [11], [α +11.82 (c 0.33, CH2Cl2)]; IR (neat) 3019, 2934, 2870, 1763, 1474, 1458, 1420, 1391, 1273, 1120, 1067, 1042, 993 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.93 (d, J = 6.5 Hz, 3H, C8-CH3), 1.09 (s, 3H, C10-CH3), 1.30 (dt, J = 12.2, 9.8 Hz, 1H, one of C1-H2), 1.31 (s, 3H, C4-CH3), 1.46 (m, 1H, one of C1-H2), 1.49 (m, 1H, one of C3-H2), 1.52 (m, 1H, one of C2-H2), 1.72 (ddd, J = 6.3, 11.9, 16.4 Hz, 1H, one of C7-H2), 1.78 (m, 1H, one of C2-H2), 1.95 (ddd, J = 4.4, 11.4, 13.8 Hz, 1H, one of C11-H2), 2.12 (m, 1H, one of C3-H2), 2.18 (ddq, J = 6.3, 11.9, 6.5 Hz, 1H, C8-H), 2.19 (d, J = 4.5 Hz, 1H, C5-H), 2.22 (ddd, J = 8.9, 11.5, 13.8 Hz, 1H, one of C11-H2), 2.28 (dd, J = 6.3, 16.4 Hz, 1H, one of C7-H2), 2.54 (ddd, J = 4.4, 11.5, 18.8 Hz, 1H, one of C12-H2), 2.62 (ddd, J = 8.9, 11.4, 18.8 Hz, 1H, one of C12-H2), 4.75 (dd, J = 4.5, 6.3 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.4 (CH3), 17.7 (CH2), 22.2 (CH3), 22.4 (CH3), 24.5 (CH2), 27.7 (CH2), 28.2 (CH2), 29.3 (CH2), 31.2 (CH2), 32.3 (CH), 38.6 (C), 44.0 (C), 45.3 (CH), 75.6 (CH), 91.2 (C), 176.9 (C), 183.3 (C); HRMS (ESI) m/z [M + Na]+ calcd for C17H24O4Na 315.1567; found 315.1566.
(1S,4R,6R,7R,8S,12R)-7-[4-(tert-Butyldimethylsilyl)oxybutyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (45). To an ice-cooled (0 °C) solution of 3-(tert-butyldimethylsilyl)oxypropylmagnesium bromide (44) [prepared from (3-bromopropoxy)(tert-butyl)dimethylsilane (2.00 g, 7.90 mmol) and magnesium tuning (241 mg, 9.93 mmol)] in THF (14 mL) was added CuBr·SMe2 (129 mg, 0.627 mmol), followed by addition of a solution of epoxide 22 (39.1 mg, 0.156 mmol) in THF (0.20 mL plus 2 × 0.20 mL rinse). After 5 h of stirring at room temperature, the reaction was quenched with saturated aqueous NH4Cl (15 mL), and the resulting mixture was extracted with AcOEt (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow oil (869 mg), which was purified by column chromatography (silica gel 20 g, 10:1 → 6:1 → 3:1 n-hexane/AcOEt) to give TBS ether 45 (42.1 mg, 48%) as a pale yellow amorphous. Rf 0.46 (3:1 n-hexane/AcOEt); [α +23.2 (c 1.00, CHCl3); IR (neat) 3534, 2953, 2927, 2859, 1755, 1471, 1462, 1336, 1253, 1197, 1149, 1099, 994 cm−1; 1H-NMR (500 MHz, C6D6) δ 0.08 (s, 6H, Si(CH3)2), 0.65 (d, J = 6.6 Hz, 3H, C8-CH3), 0.78 (br s, 1H, OH), 0.96 (m, 1H, one of C1-H2), 0.97 (s, 6H, C4-CH3, C10-CH3), 1.00 (s, 9H, SiC(CH3)3), 1.14−1.30 (m, 6H, one of C2-H2, one of C3-H2, C11-H2, C12-H2), 1.35−1.45 (m, 4H, one of C2-H2, one of C7-H2, C13-H2), 1.47 (m, 1H, one of C1-H2), 1.77 (ddq, J = 6.2, 11.2, 6.6 Hz, 1H, C8-H), 1.92 (dd, J = 6.2, 15.8 Hz, 1H, one of C7-H2), 2.00 (d, J = 4.6 Hz, 1H, C5-H), 2.18 (dt, J = 4.3, 13.7 Hz, 1H, one of C3-H2), 3.52 (t, J = 6.1 Hz, 2H, C14-H2), 4.36 (dd, J = 4.6, 6.4 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, C6D6) δ −5.1 (CH3), 16.6 (CH3), 18.51 (C), 18.55 (CH2), 21.9 (CH2), 22.6 (CH3), 22.9 (CH3), 26.1 (CH3), 28.8 (CH2), 28.9 (CH2), 32.0 (CH2), 32.3 (CH), 34.0 (CH2), 34.8 (CH2), 39.8 (C), 43.7 (C), 44.9 (CH), 62.8 (CH2), 75.48 (C), 75.52 (C), 182.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H44O4SiNa 447.2907; found 447.2891.
(1S,4R,6R,7R,8S,12R)-7-Hydroxy-7-(4-hydroxybutyl)-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (46). Bu4NF in THF (1.0 M, 0.38 mL, 0.38 mmol) was added to an ice-cooled (0 °C) solution of TBS ether 45 (32.1 mg, 0.76 mmol) in THF (1.0 mL). After 1 h of stirring at room temperature, the mixture was partitioned between AcOEt (2 mL) and H2O (2 mL), and the aqueous layer was extracted with AcOEt (3 × 2 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow oil (144 mg), which was purified by column chromatography (silica gel 2 g, 1:4 n-hexane/AcOEt) to give diol 46 (19.3 mg, 82%) as a colorless amorphous. Rf 0.33 (1:2 n-hexane/AcOEt); [α +24.4 (c 0.90, CHCl3); IR (neat) 3478, 2951, 2930, 2870, 1747, 1462, 1454, 1350, 1263, 1199, 1149, 1099, 1072, 1041, 991 cm−1; 1H-NMR (500 MHz, C6D6) δ 0.54 (s, 1H, OH), 0.61 (d, J = 6.7 Hz, 3H, C8-CH3), 0.86 (s, 1H, OH), 0.91 (m, 1H, one of C1-H2), 0.95 (s, 3H, C10-CH3), 0.97 (s, 3H, C4-CH3), 1.07−1.30 (m, 8H, one of C1-H2, one of C2-H2, C11-H2, C12-H2, C13-H2), 1.36−1.48 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.73 (ddq, J = 6.2, 11.3, 6.7 Hz, 1H, C8-H), 1.91 (ddd, J = 1.3, 6.2, 15.9 Hz, 1H, one of C7-H2), 2.00 (d, J = 4.8 Hz, 1H, C5-H), 2.18 (dt, J = 4.2, 13.8 Hz, 1H, one of C3-H2), 3.30 (t, J = 6.1 Hz, 2H, C14-H2), 4.35 (ddd, J = 1.3, 4.8, 6.2 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, C6D6) δ 16.6 (CH3), 18.6 (CH2), 21.8 (CH2), 22.5 (CH3), 22.9 (CH3), 28.7 (CH2), 28.9 (CH2), 32.0 (CH2), 32.3 (CH), 33.7 (CH2), 34.7 (CH2), 39.8 (C), 43.7 (C), 44.9 (CH), 62.2 (CH2), 75.4 (C), 75.6 (CH), 182.7 (C); HRMS (ESI) m/z [M + Na]+ calcd for C18H30O4Na 333.2042; found 333.2052.
Marrulactone (14). TEMPO (2.3 mg, 14.7 μmol) was added to an ice-cooled (0 °C) mixture of diol 46 (13.1 mg, 42.2 μmol) and PhI(OAc)2 (40.5 mg, 0.125 mmol) in CH2Cl2 (0.3 mL). After 4 h of stirring at room temperature, the reaction was quenched with 1 M aqueous Na2S2O3 (1 mL), and the resulting mixture was extracted with AcOEt (3 × 1 mL). The combined organic extracts were washed with brine (3 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (64.0 mg), which was purified by column chromatography (silica gel 1 g, 1:1 n-hexane/AcOEt) to give marrulactone (14, 11.8 mg, 91%) as a colorless amorphous. Rf 0.60 (1:2 n-hexane/AcOEt); [α −11.6 (c 0.61, CHCl3) [lit. [12], [α −23.80 (c 0.22, CHCl3)]; IR (neat) 3017, 2961, 2928, 2872, 1769, 1719, 1458, 1391, 1329, 1261, 1244, 1215, 1188, 1117, 1070, 1030, 1003 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.98 (d, J = 6.3 Hz, 3H, C8-CH3), 1.11 (s, 3H, C10-CH3), 1.31 (s, 3H, C4-CH3), 1.34−1.42 (m, 2H, C1-H2), 1.48 (m, 1H, one of C3-H2), 1.55 (m, 1H, one of C2-H2), 1.74−1.80 (m, 2H, one of C2-H2, one of C11-H2), 1.80−1.94 (m, 4H, one of C7-H2, one of C11-H2, C12-H2), 2.09−2.20 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.25 (ddd, J = 6.2, 11.2, 17.3 Hz, 1H, one of C13-H2), 2.32 (d, J = 4.4 Hz, 1H, C5-H), 2.57 (ddt, J = 2.0, 17.3, 4.1 Hz, 1H, one of C13-H2), 4.78 (ddd, J = 0.7, 4.4, 5.5 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.5 (CH3), 17.8 (CH2), 18.9 (CH2), 22.0 (CH3), 22.8 (CH3), 26.1 (CH2), 28.1 (CH2), 28.6 (CH2), 30.4 (CH2), 30.9 (CH2), 34.3 (CH), 40.8 (C), 44.0 (C), 44.3 (CH), 75.8 (CH), 88.3 (C), 172.2 (C), 183.5 (C); HRMS (ESI) m/z [M + Na]+ calcd for C18H26O4Na 329.1723; found 329.1711.
Marrulanic acid (10). LiOH (18.7 mg, 0.78 mmol) was added to a solution of marrulactone (14, 11.8 mg, 38.5 μmol) in THF/H2O (2:1, 1.2 mL). After 31 h of stirring, the mixture was acidified with 1 M aqueous HCl (3 mL), and the resulting mixture was extracted with AcOEt (3 × 3 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (11.7 mg), which was purified by column chromatography (silica gel 1 g, 1:1 n-hexane/AcOEt → AcOEt → 20:1 AcOEt/MeOH) to give marrulanic acid (10, 10.5 mg, 84%) as a colorless amorphous. Rf 0.19 (1:2 n-hexane/AcOEt); [α +25.4 (c 0.53, CHCl3) [lit. [12], [α −10.8 (c 1.2, CHCl3)]; IR (neat) 3536, 3196, 2949, 2928, 2868, 1761, 1732, 1464, 1412, 1391, 1287, 1260, 1196, 1179, 1159, 1126, 1094, 1078, 1042, 1018 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.92 (d, J = 6.5 Hz, 3H, C8-CH3), 1.04 (s, 3H, C10-CH3), 1.26 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.44−1.80 (m, 9H, one of C1-H2, C2-H2, one of C3-H2, one of C7-H2, C11-H2, C12-H2), 2.04−2.17 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.24 (d, J = 4.6 Hz, 1H, C5-H), 2.38 (dt, J = 1.8, 7.0 Hz, 2H, C13-H2), 4.74 (ddd, J = 1.2, 4.6, 5.8 Hz, 1H, C6-H); 13C-NMR (125.0 MHz, CDCl3) δ 16.5 (CH3), 18.2 (CH2), 20.3 (CH2), 22.3 (CH3), 22.9 (CH3), 28.3 (CH2), 28.6 (CH2), 31.5 (CH2), 32.2 (CH), 34.29 (CH2), 34.31 (CH2), 39.7 (C), 43.8 (C), 44.8 (CH), 75.6 (C), 76.2 (CH), 178.5 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C18H28O5Na 347.1834; found 347.1813.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(3,4-Dihydroxy-2,5-dimethoxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (40). TsOH (0.4 mg, 2.3 μmol) was added to a solution of marrulibacetal A (17, 0.90 mg, 2.3 μmol) in MeOH (1.0 mL), and the mixture was heated at reflux for 5 h. After cooling to 0 °C, the reaction was quenched with half saturated aqueous NaHCO3 (4 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (1.4 mg), which was purified by flash column chromatography (silica gel 0.8 g, 1:1 n-hexane/AcOEt → AcOEt) to give triol 40 (0.24 mg, 25%) as a colorless oil. Rf 0.30 (1:1 n-hexane/AcOEt); [α +16.5 (c 0.46, benzene); IR (neat) 3447, 2930, 1744, 1466, 1449, 1389, 1288, 1198, 1130, 1094, 1042, 991 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.4 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.28 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.45 (m, 1H, one of C3-H2), 1.51 (m, 1H, one of C2-H2), 1.65 (m, 1H, one of C7-H2), 1.74 (m, 1H, one of C2-H2), 1.60−1.98 (m, 5H, one of C1-H2, C11-H2, C12-H2), 2.07 (m, 1H, C8-H), 2.11 (m, 1H, one of C3-H2), 2.14 (m, 1H, one of C7-H2), 2.22 (d, J = 4.5 Hz, 1H, C5-H), 3.40 (s, 3H, C16-OCH3), 3.48 (s, 3H, C15-OCH3), 3.92 (d, J = 3.7 Hz, 1H, C14-H), 4.73 (dd, J = 4.5, 6.5 Hz, 1H, C6-H), 4.77 (s, 1H, C16-H), 4.89 (d, J = 3.7 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 18.1 (CH2), 22.3 (CH3), 22.9 (CH3), 28.0 (CH2), 28.26 (CH2), 28.28 (CH2), 28.4 (CH2), 31.5 (CH2), 32.6 (CH), 40.0 (C), 43.9 (C), 44.9 (CH), 55.1 (CH3), 56.4 (CH3), 75.4 (C), 76.2 (CH), 80.3 (CH), 81.4 (C), 108.3 (CH), 110.7 (CH), 184.0 (C); HRMS (ESI) m/z [M + Na]+ calcd for C22H36O8Na 451.2302; found 451.2308.
Desertine (18). TsOH (1.6 mg, 9.3 μmol) was added to a solution of bisacetal 42 (2.0 mg, 4.7 μmol) in MeOH (0.5 mL), and the mixture was heated at reflux for 3.5 h. After cooling to 0 °C, the reaction was quenched with saturated aqueous NaHCO3 (2 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow oil (3.6 mg), which was purified by flash column chromatography (silica gel 0.8 g, 1:1 n-hexane/AcOEt) to give desertine (18, 0.34 mg, 16%) as a colorless oil. Rf 0.30 (1:1 n-hexane/AcOEt); [α +22.8 (c 0.50, benzene) (lit. [14], [α −56.6); IR (neat) 3464, 2930, 1748, 1463, 1450, 1391, 1256, 1198, 1152, 1099, 1043, 989 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.2 Hz, 3H, C8-CH3), 1.04 (s, 3H, C10-CH3), 1.28 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.45 (m, 1H, one of C3-H2), 1.51 (m, 1H, one of C2-H2), 1.65 (m, 1H, one of C7-H2), 1.74 (m, 1H, one of C2-H2), 1.60−1.98 (m, 5H, one of C1-H2, C11-H2, C12-H2), 2.07 (m, 1H, C8-H), 2.11 (m, 1H, one of C3-H2), 2.14 (m, 1H, one of C7-H2), 2.23 (d, J = 4.0 Hz, 1H, C5-H), 3.41 (s, 3H, C16-OCH3), 3.47 (s, 3H, C15-OCH3), 3.95 (d, J = 3.5 Hz, 1H, C14-H), 4.73 (dd, J = 4.0, 6.9 Hz, 1H, C6-H), 4.76 (s, 1H, C16-H), 4.90 (d, J = 3.5 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.8 (CH3), 18.2 (CH2), 22.2 (CH3), 23.0 (CH3), 27.4 (CH2), 28.3 (CH2), 28.4 (CH2), 28.5 (CH2), 31.6 (CH2), 32.5 (CH), 40.1 (C), 43.9 (C), 45.0 (CH), 55.1 (CH3), 56.4 (CH3), 75.3 (C), 76.2 (CH), 80.2 (CH), 81.2 (C), 108.6 (CH), 110.8 (CH), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C22H36O8Na 451.2302; found 451.2313.
4. Conclusions
Total syntheses of eight labdane diterpene lactones, marrubiin (1), marrulanic acid (10), cyllenine C (12), marrulibacetal (13), marrulactone (14), marrulibacetal A (17), desertine (18), and marrubasch F (19), have been accomplished. Since chiral building block 20 could be prepared from (4R,6S)-6-[(tert-butyldimethylsilyl)oxymethyl]-4-methyltetrahydro-2H-pyran-2-one [53] in 37% yield over eight steps, the syntheses proceeded in 19−22 steps involving a stereoselective intramolecular PKR, and these natural products were obtained in yields ranging from 1.1% (marrulibacetal A) to 10.9% (cyllenin C). Stereochemistries of desertine were established by the facts that the compound could be obtained by both osmylation of bisacetal 37 and transacetalization of the diastereomer of marrulibacetal A with MeOH. The syntheses illustrate the synthetic utility of chiral building block 20 for the synthesis of this class of diterpenoids. The synthetic strategy also features elongation of the C9 side chain through an epoxide-opening reaction as well as high convergency and flexibility. The syntheses of other natural products and non-natural analogues for biological and pharmaceutical investigations are in progress in our laboratory and will be reported in due course.
Supplementary Materials
The following are available online. Comparison data for natural products and synthetic materials, and copies of 1H and 13C-NMR spectra for all new compounds.
Author Contributions
Y.S. (Yukari Sakagami), N.K., Y.S. (Yuki Sawayama), and H.Y. performed the experiments. S.N. conceived and designed the project. H.Y. and S.N. directed the investigations and prepared this manuscript. All authors discussed the experimental results. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by the Platform Project for Supporting in Drug Discovery and Life Science Research from Japan Agency for Medical Research and Development (AMED), and Shionogi Award in Synthetic Organic Chemistry, Japan.
Acknowledgments
The authors acknowledge the assistance of the Research Equipment Sharing Center at Nagoya City University. We are grateful to Mr. Kota Suzuki of the Faculty of Pharmaceutical Sciences, Nagoya City University, for experimental assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Meyre-Silva, C.; Cechinel-Filho, V. A Review of the Chemical and Pharmacological Aspects of the Genus Marrubium. Curr. Pharm. Des. 2010, 16, 3503–3518. [Google Scholar] [CrossRef] [PubMed]
- Popoola, O.K.; Elbagory, A.M.; Ameer, F.; Hussein, A.A. Marrubiin. Molecules 2013, 18, 9049–9060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardai, S.E.; Morel, N.; Wibo, M.; Fabre, N.; Llabres, G.; Lyoussi, B.; Quetin-Leclercq, J. The Vasorelaxant Activity of Marrubenol and Marrubiin from Marrubium vulgare. Planta Med. 2003, 69, 75–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piozzi, F.; Bruno, M.; Rosselli, S.; Maggio, A. The Diterpenoids of the Genus Marrubium (Lamiaceae). Nat. Prod. Commun. 2006, 1, 585–592. [Google Scholar] [CrossRef]
- Salei, L.A.; Popa, D.P.; Lazur’evskii, G.V. Diterpenoids from Marrubium peregrinum. Khim. Prir. Soedin. 1966, 2, 249–251. [Google Scholar]
- Iida, A.; Tanaka, Y.; Mihara, T.; Tabata, M.; Honda, G.; Shingu, T.; Takeda, Y.; Takaishi, Y.; Yesilada, E.; Sezik, E.; et al. Marrubinones A and B, New Labdane Diterpenoids from Marrubium astracanicum (Labiatae). Chem. Pharm. Bull. 1995, 43, 1454–1457. [Google Scholar] [CrossRef] [Green Version]
- Hatam, N.A.R.; Porzel, A.; Seifert, K. Polyodonine, a prefuranic labdane diterpene from Marrubium polydon. Phytochemistry 1995, 40, 1575–1576. [Google Scholar] [CrossRef]
- Karioti, A.; Heilmann, J.; Skaltsa, H. Labdane Diterpenes from Marrubium velutinum and Marrubium cylleneum. Phytochemistry 2005, 66, 1060–1066. [Google Scholar] [CrossRef]
- Rigano, D.; Grassia, A.; Borrelli, F.; Aviello, G.; Piozzi, F.; Bruno, M.; Apostolides Arnold, N.; Capasso, R.; Senatore, F. Phytochemical and Pharmacological Studies on the Acetonic Extract of Marrubium globosum ssp. libanoticum. Planta Med. 2006, 72, 575–578. [Google Scholar] [CrossRef]
- Rigano, D.; Grassia, A.; Bruno, M.; Rosselli, S.; Piozzi, F.; Formisano, C.; Apostolides Arnold, N.; Senatore, F. Labdane Diterpenoids from Marrubium globosum ssp. libanoticum. J. Nat. Prod. 2006, 69, 836–838. [Google Scholar] [CrossRef]
- Karioti, A.; Skopeliti, M.; Tsitsilonis, O.; Heilmann, J.; Skaltsa, H. Cytotoxicity and Immunomodulating Characteristics of Labdane Diterpenes from Marrubium cylleneum and Marrubium velutinum. Phytochemistry 2007, 68, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Rigano, D.; Aviello, G.; Bruno, M.; Formisano, C.; Rosselli, S.; Capasso, R.; Senatore, F.; Izzo, A.A.; Borrelli, F. Antispasmodic Effects and Structure−Activity Relationships of Labdane Diterpenoids from Marrubium globosum ssp. libanoticum. J. Nat. Prod. 2009, 72, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Perveen, S.; Malik, A.; Khan, A.N.; Tareen, R.B. Marrusidins A and B, New Epimeric Labdane Diterpenes from Marrubium anisodon. Helv. Chim. Acta 2010, 93, 1101–1104. [Google Scholar] [CrossRef]
- Zaabat, N.; Hay, A.-E.; Michalet, S.; Darbour, N.; Bayet, C.; Skandrani, I.; Chekir-Ghedira, L.; Akkal, S.; Dijoux-Franca, M.-G. Antioxidant and Antigenotoxic Properties of Compounds Isolated from Marrubium deserti de Noé. Food Chem. Toxicol. 2011, 49, 3328–3335. [Google Scholar] [CrossRef]
- Zhang, J.-S.; Zou, Y.-H.; Zhao, J.-J.; Chen, Y.; Bao, J.-M.; Tang, G.-H. Three New Diterpenoids from Marrubium aschersonii. Phytochem. Lett. 2016, 16, 241–244. [Google Scholar] [CrossRef]
- In addition to these named compounds, a large number of unnamed analogues have been isolated and characterized.
- Zerbe, P.; Chiang, A.; Dullat, H.; O’Neil-Johnson, M.; Starks, C.; Hamberger, B.; Bohlmann, J. Diterpene synthases of the biosynthetic system of medicinally active diterpenoids in Marrubium vulgare. Plant J. 2014, 79, 914–927. [Google Scholar] [CrossRef]
- Karunanithi, P.S.; Dhanota, P.; Addison, J.B.; Tong, S.; Fiehn, O.; Zerbe, P. Functional characterization of the cytochrome P450 monooxygenase CYP71AU87 indicates a role in marrubiin biosynthesis in the medicinal plant Marrubium vulgare. BMC Plant Biol. 2019, 19, 114. [Google Scholar] [CrossRef]
- Mangoni, L.; Adinolfi, M.; Laonigro, G.; Caputo, R. Synthesis of Marrubiin. Tetrahedron 1972, 28, 611–621. [Google Scholar] [CrossRef]
- Laonigro, G.; Lanzetta, R.; Parrilli, M.; Adinolfi, M.; Mangoni, L. The Configuration of the Diterpene Spiroethers from Marrubium vulgare and from Leonotis leonurus. Gazz. Chim. Ital. 1979, 109, 145–150. [Google Scholar]
- Akahori, Y.; Yamakoshi, H.; Sawayama, Y.; Hashimoto, S.; Nakamura, S. Synthesis of Chiral Building Blocks for Oxygenated Terpenoids through a Simultaneous and Stereocontrolled Construction of Contiguous Quaternary Stereocenters by an Ireland−Claisen Rearrangement. J. Org. Chem. 2014, 79, 720–735. [Google Scholar] [CrossRef]
- Saito, S.; Yamakoshi, H.; Nakamura, S. Second-Generation Synthesis of a Chiral Building Block for Oxygenated Terpenoids via a Ring-Contractive Coupling with a Secondary Alcohol. Heterocycles 2019, 99, 1086–1094. [Google Scholar]
- Part of this work has already been communicated: Yamakoshi, H.; Sawayama, Y.; Akahori, Y.; Kato, M.; Nakamura, S. Total Syntheses of (+)-Marrubiin and (−)-Marrulibacetal. Org. Lett. 2016, 18, 3430–3433. [Google Scholar] [CrossRef] [PubMed]
- For a review, see: Jeong, N. The Pauson−Khand Reaction. In Comprehensive Organic Synthesis II, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 5.24, pp. 1106–1178. [Google Scholar]
- For a recent review on the use of Pauson−Khand reaction for synthesis of the natural products, see: Ma, K.; Martin, B.S.; Yin, X.; Dai, M. Natural Product Syntheses via Carbonylative Cyclizations. Nat. Prod. Rep. 2019, 36, 174–219. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.-C.P.; Tsao, W.-C.; Ho, J.-S.; Tai, C.-C.; Chiou, D.-Y.; Tu, L.-H. Rhodium(I)-Catalyzed Intra molecular Cyclohexadienyl Pauson−Khand Reaction: Facile Approach to Tricarbocycles. Organometallics 2004, 23, 792–799. [Google Scholar] [CrossRef]
- Sugihara, T.; Yamada, M.; Ban, H.; Yamaguchi, M.; Kaneko, C. Rate Enhancement of the Pauson−Khand Reaction by Primary Amines. Angew. Chem., Int. Ed. Engl. 1997, 36, 2801–2804. [Google Scholar] [CrossRef]
- Yamanaka, M.; Nakamura, E. Density Functional Studies on the Pauson−Khand Reaction. J. Am. Chem. Soc. 2001, 123, 1703–1708. [Google Scholar] [CrossRef]
- For a recent review, see: Lee, H.-W.; Kwong, F.-Y. A Decade of Advancements in Pauson−Khand-Type Reactions. Eur. J. Org. Chem. 2010, 789–811. [Google Scholar]
- Krafft, M.E.; Boñaga, L.V.R.; Hirosawa, C. Practical Cobalt Carbonyl Catalysis in the Thermal Pauson−Khand Reaction: Efficiency Enhancement Using Lewis Bases. J. Org. Chem. 2001, 66, 3004–3020. [Google Scholar] [CrossRef]
- Morimoto, T.; Fuji, K.; Tsutsumi, K.; Kakiuchi, K. CO-Transfer Carbonylation Reactions. A Catalytic Pauson−Khand-Type Reaction of Enynes with Aldehydes as a Source of Carbon Monoxide. J. Am. Chem. Soc. 2002, 124, 3806–3807. [Google Scholar] [CrossRef]
- Yates, P.; Burke, P.M. Keto Ethers. IV. Products Formed on Reaction of Dihydro-2,2,5,5-tetramethyl- 3(2H)-furanone with Strong Acids. Can. J. Chem. 1987, 65, 1695–1704. [Google Scholar] [CrossRef]
- The same results were obtained by the use of activated carbon supplied by Aldrich (DARCO®), Tokyo Chemical Industry, and Taihei Chemical Industrial, suggesting that the reaction is not effected by contaminants.
- Wheeler, D.M.S.; Wheeler, M.M.; Fetizon, M.; Castine, W.H. Synthesis of Diterpenoid Acids—VII: The Stereochemistry of Marrubiin. Tetrahedron 1967, 23, 3909–3921. [Google Scholar] [CrossRef]
- When reductive workup with NaBH4 was employed after ozonolysis, enone 26 could be converted to lactone 23 in two steps without intervening purification, albeit in low yield (18%).
- Welch, S.C.; Rao, A.S.C.P.; Lyon, J.T.; Assercq, J.-M. Synthesis of 2,2-Disubstituted Oxetanes from Ketones with S-Methyl-S-(sodiomethyl)-N-(4-tolylsulfonyl)sulfoximine. J. Am. Chem. Soc. 1983, 105, 252–257. [Google Scholar] [CrossRef]
- Araki, S.; Butsugan, Y. Regio-controlled Prenylation and Geranylation of 3-Furylmethylmagnesium Bromide. Selective Syntheses of 3-Substituted Furanoid and 2-Substituted 3-Methylfuranoid Terpenes. Bull. Chem. Soc. Jpn. 1983, 56, 1446–1449. [Google Scholar] [CrossRef] [Green Version]
- Cocker, W.; Cross, B.E.; Duff, S.R.; Edward, J.T.; Holley, T.F. The Constitution of Marrubiin. Part I. J. Chem. Soc. 1953, 2540–2548. [Google Scholar] [CrossRef]
- Isomerization of Grignard reagent 32 was also observed by Sperry and Wright. Sperry, J.B.; Wright, D.L. Annulated Heterocycles through a Radical-Cation Cyclization: Synthetic and Mechanistic Studies. Tetrahedron 2006, 62, 6551–6557. [Google Scholar] [CrossRef]
- Grignard reagent 34 was prepared from [2-(trimethylsilyl)furan-3-yl]methanol [52] by the following two- step sequence: 1) PBr3, Et2O, 0 °C, 30 min; 2) Mg, Et2O, −10 °C, 1.5 h.
- Bures, E.J.; Keay, B.A. The Regiospecific Formation and Reactions of 4-Lithio-2-(t-butyldimethylsilyl)-3- (hydroxymethyl)furan: An Approach to 3,4-Disubstituted Furans. Tetrahedron Lett. 1988, 29, 1247–1250. [Google Scholar] [CrossRef]
- Harding, W.W.; Schmidt, M.; Tidgewell, K.; Kannan, P.; Holden, K.G.; Gilmour, B.; Navarro, H.; Rothman, R.B.; Prisinzano, T.E. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Semisynthesis of Salvinicins A and B and Other Chemical Transformations of Salvinorin A. J. Nat. Prod. 2006, 69, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Mendoza, E.; Guevara-Salazar, J.A.; Ramírez-Apan, M.T.; Frontana-Uribe, B.A.; Cogordan, J.A.; Cárdenas, J. Electro-Oxidation of Hispanolone and Anti-Inflammatory Properties of the Obtained Deriva tives. J. Org. Chem. 2005, 70, 4538–4541. [Google Scholar] [CrossRef]
- Hönel, M.; Mosher, H.S. Selective Permanganate Oxidation of cis- vs. trans-2,5-Dihydro-2,5-dimethoxy furan. J. Org. Chem. 1985, 50, 4386–4388. [Google Scholar]
- Kuwajima, I.; Urabe, H. Regioselective Synthesis of Δ3-Butenolides via Oxidation of 2-Trimethylsilyl furans. Tetrahedron Lett. 1981, 22, 5191–5194. [Google Scholar] [CrossRef]
- Goldsmith, D.; Liotta, D.; Saindane, M.; Waykole, L.; Bowen, P. 3-Alkylfurans as Useful Synthetic Equiva lents for Substituted Δ2-Butenolides. Tetrahedron Lett. 1983, 24, 5835–5838. [Google Scholar] [CrossRef]
- Shu, C.; Liu, M.-Q.; Sun, Y.-Z.; Ye, L.-W. Efficient Synthesis of γ-Lactones via Gold-Catalyzed Tandem Cycloisomerization/Oxidation. Org. Lett. 2012, 14, 4958–4961. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Fujita, S.; Nagai, M.; Fukumoto, K.; Kametani, T. A Novel Strategy for the Stereoselective Total Synthesis of C-17 Spiro Steroids. Total Synthesis of 19-Norcanrenone, a Formal Total Synthesis of 19-Norspironolactone. J. Am. Chem. Soc. 1988, 110, 2931–2938. [Google Scholar] [CrossRef]
- Hansen, T.M.; Florence, G.J.; Lugo-Mas, P.; Chen, J.; Abrams, J.N.; Forsyth, C.J. Highly Chemoselective Oxidation of 1,5-Diols to δ-Lactones with TEMPO/BAIB. Tetrahedron Lett. 2003, 44, 57–59. [Google Scholar] [CrossRef]
- Byun, H.-S.; Zhong, N.; Bittman, R. 6A-O-p-Toluenesulfonyl-β-cyclodextrin. Org. Synth. 2000, 77, 225–230. [Google Scholar]
- House, H.O.; Chu, C.-Y.; Wilkins, J.M.; Umen, M.J. The Chemistry of Carbanions. XXVII. A Convenient Precursor for the Generation of Lithium Organocuprates. J. Org. Chem. 1975, 40, 1460–1469. [Google Scholar] [CrossRef]
- Dong, J.-Q.; Wong, H.N.C. Biomimetic Total Synthesis of (±)-Pallavicinolide A. Angew. Chem., Int. Ed. 2009, 48, 2351–2354. [Google Scholar] [CrossRef]
- Ahmed, A.; Hoegenauer, E.K.; Enev, V.S.; Hanbauer, M.; Kaehlig, H.; Öhler, E.; Mulzer, J. Total Synthesis of the Microtubule Stabilizing Antitumor Agent Laulimalide and Some Nonnatural Analogues: The Power of Sharpless’ Asymmetric Epoxidation. J. Org. Chem. 2003, 68, 3026–3042. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Structures of marrubiin and related natural products.

Figure 2.
Structure of building block 20.

Scheme 1.
Retrosynthetic analysis.

Scheme 2.
Construction of the tricyclic lactone. KHMDS = potassium bis(trimethylsilyl)amide, THF = tetrahydrofuran.
Scheme 2.
Construction of the tricyclic lactone. KHMDS = potassium bis(trimethylsilyl)amide, THF = tetrahydrofuran.

Scheme 3.
Completion of the synthesis of advanced intermediate 22. Ts = p-toluenesulfonyl.

Scheme 4.
Total syntheses of marrubiin (1), marrulibacetal (13), marrulibacetal A (17), and marrubasch F (19) from epoxide 22.
Scheme 4.
Total syntheses of marrubiin (1), marrulibacetal (13), marrulibacetal A (17), and marrubasch F (19) from epoxide 22.

Scheme 5.
Total syntheses of cyllenine C (12), marrulactone (14), and marrulanic acid (10) from epoxide 22. DMSO = dimethyl sulfoxide, Tf = trifluoromethanesulfonyl, Ms = methanesulfonyl, TBS = tert-butyldimethylsilyl.
Scheme 5.
Total syntheses of cyllenine C (12), marrulactone (14), and marrulanic acid (10) from epoxide 22. DMSO = dimethyl sulfoxide, Tf = trifluoromethanesulfonyl, Ms = methanesulfonyl, TBS = tert-butyldimethylsilyl.

Scheme 6.
Conversion of marrulibacetal A (17) and its isomer 42 to triol 40 and desertine (18).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sakagami, Y.; Kondo, N.; Sawayama, Y.; Yamakoshi, H.; Nakamura, S. Total Syntheses of Marrubiin and Related Labdane Diterpene Lactones. Molecules 2020, 25, 1610. https://doi.org/10.3390/molecules25071610
AMA Style
Sakagami Y, Kondo N, Sawayama Y, Yamakoshi H, Nakamura S. Total Syntheses of Marrubiin and Related Labdane Diterpene Lactones. Molecules. 2020; 25(7):1610. https://doi.org/10.3390/molecules25071610
Chicago/Turabian StyleSakagami, Yukari, Naoki Kondo, Yuki Sawayama, Hiroyuki Yamakoshi, and Seiichi Nakamura. 2020. "Total Syntheses of Marrubiin and Related Labdane Diterpene Lactones" Molecules 25, no. 7: 1610. https://doi.org/10.3390/molecules25071610