Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate †


Chemistry Department and Centre for Biotechnology, Brock University, 1812 Sir Isaac Brock Way St. Catharines, ON L2S 3A1, Canada
*
Author to whom correspondence should be addressed.
†
Dedicated to Professor Dieter Shinzer on the occasion of his 65th birthday and in recognition of his contributions to the art and craft of organic synthesis.
Molecules 2019, 24(19), 3477; https://doi.org/10.3390/molecules24193477
Submission received: 6 September 2019
/
Revised: 20 September 2019
/
Accepted: 22 September 2019
/
Published: 25 September 2019
(This article belongs to the Special Issue Total Synthesis of Natural Products: A Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65th Birthday)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The total synthesis of (+)-10-keto-oxycodone was attained from phenethyl acetate in a stereoselective manner. Absolute stereochemistry was established via enzymatic dihydroxylation of phenethyl acetate with the recombinant strain JM109 (pDTG601A) that furnished the corresponding cis-cyclohexadienediol whose configuration corresponds to the absolute stereochemistry of the ring C of (+)-10-keto-oxycodone. Intramolecular Heck reaction was utilized to establish the quaternary carbon at C-13, along with the dibenzodihydrofuran functionality. The C-14 hydroxyl and C-10 ketone were installed via SmI2-mediated radical cyclization, and oxidation of a benzylic alcohol (obtained from an intermediate nitrate azide), respectively. The synthesis of (+)-10-keto-oxycodone was completed in a total of 14 operations (21 steps) and an overall yield of ~2%. Experimental and spectral data are provided for key intermediates and new compounds.
1. Introduction
Interest in the preparation of 10-keto opiates and related derivatives such as the 10-hydroxy-morphinans was aimed at pursuing κ-selective analgesics [1]. Of the three types of opioid receptors (µ, δ, κ), the κ-opioid receptor has been especially interesting because its activation produces analgesia with minimal physical dependence [2]. A highly selective κ-opioid agonist may provide a useful analgesic free from abusive potential [3]. Among the efforts directed toward the preparation of the afore-mentioned 10-keto opiates are those aimed at the preparation of 10-keto, 10α-, 10β-hydroxy-TRK-820 [4], 10-keto-naltrexone, 10-keto-oxymorphone, 10-keto-oxycodone (Figure 1) [5], 10-keto-naloxone, and 10-keto-naloxone 3-methyl ether [6] from the corresponding opiates. These preparations are all conducted from natural morphinans by semi-syntheses. Furthermore, the above 10-keto opiates were also identified as purported intermediates in the decomposition of the parent opiates upon storage [6]. As part of our continuing program in the preparation of morphine alkaloids and related compounds [7,8], we recently had the opportunity to access a suitable intermediate that could potentially be converted to ent-10-keto-oxycodone.
The published methods for the preparation of 10-keto-oxycodone and 10-hydroxy-oxycodone involve the direct oxidation of oxycodone with chromium [5,9,10], or selenium dioxide [11] reagents. Recently, the use of ceric ammonium nitrate (CAN) for such an oxidation led exclusively to the 10-hydroxy product, further oxidized to the C-10-ketone with the use of stronger oxidants such as Dess–Martin periodinane [9,10]. Oxycodone, on the other hand, is prepared in two-steps from thebaine by the oxidation of the diene moiety with a peroxyacid to an enone and subsequent hydrogenation [12,13,14,15,16]. The amount of available thebaine, itself a minor constituent of opium, limits the production of oxycodone. Recently, thebaine and oripavine have become available from genetically modified poppies that produce much higher percentages of these alkaloids [17,18,19,20], now supplied by Tasmanian Alkaloids, Inc. [21]. Since the milestone synthesis of morphine by Gates [22] in 1952, there have been more than 30 total syntheses of morphine and related alkaloids and the academic pursuit continues unabated [23,24,25,26,27]. None of the reported syntheses are practical, however, for an industrial scale production. Even the most efficient academic synthesis, published by Rice [28], may not be amenable for scale-up in the industrial preparation of morphinans. Although the development of a truly practical total synthesis of any morphinan or an opiate-derived agent on a commercial scale seems to be not feasible in the near future, we have attempted to develop methods for the synthesis of oxycodone and derivatives from readily available starting materials. A de novo preparation of oxycodone and related medicinal opiate-derived agents for medicinal use could alleviate the negative impacts of any future unfavorable events that may limit the supply of natural sources—such as climate or political changes in the opium-producing areas.
2. Results and Discussion
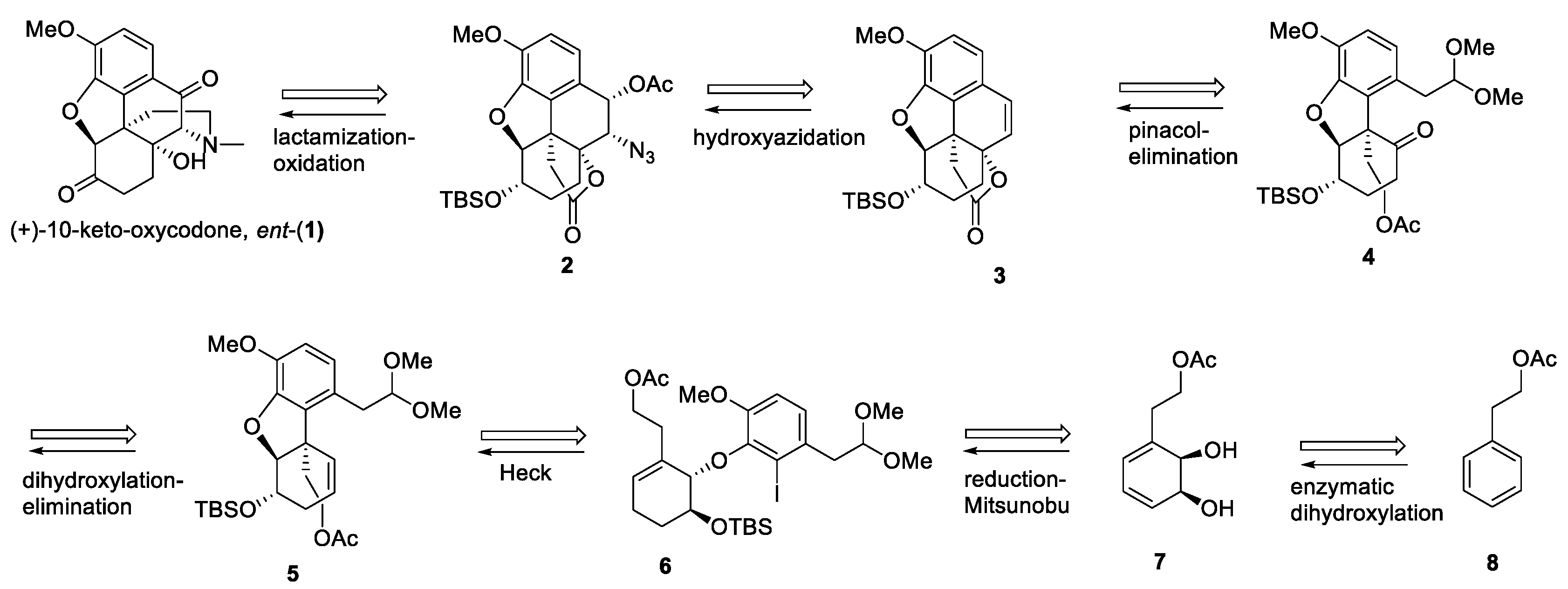
Scheme 1 outlines our retrosynthesis of (+)-10-keto-oxycodone. Disconnection of ring D leads to highly functionalized lactone 2. In the forward sense, Fukuyama’s lactamization approach to oxycodone [29] can be utilized to construct the C-9 stereogenic center. The amino group required for the cyclization reaction is derived from hydroxyazidation of alkene lactone 3 [30,31], which is envisioned to be prepared from the keto acetal 4 following a deprotection of acetal and a SmI2-mediated pinacol-type coupling reaction. The key intermediate 4 can be obtained from alkene 5 via dihydroxylation followed by selective mesylation of the less hindered hydroxyl group and the elimination of the mesylate to reveal the ketone functionality in 4. Alkene 5 is available in two steps from alcohol 7 via a sequence of steps that involves a Mitsunobu coupling with an iodo phenol acetal to furnish aryl ether 6, followed by an intramolecular Heck reaction. The absolute stereochemistry in 7 is incorporated via microbial dihydroxylation of phenethyl acetate 8 in the whole-cell fermentation with toluene dioxygenase, overexpressed in E. coli JM109(pDTG601A) [32]. The enzymatically derived arene cis-dihydrodiols such as 7 have found widespread use in enantioselective synthesis of natural products [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51].
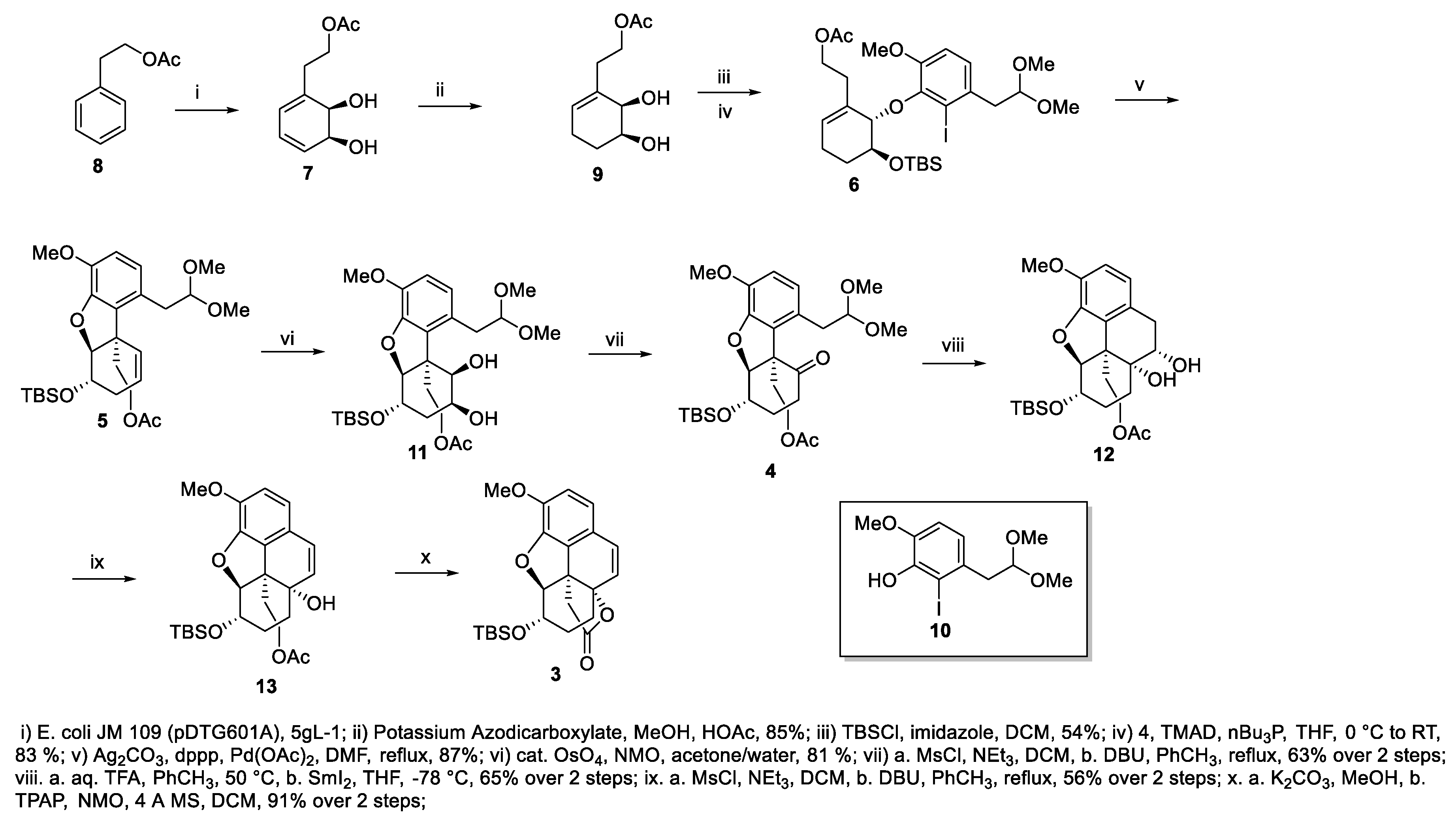
As previously described in our earlier publications on the preparation of ent-oxycodone [30,31], the synthesis began with the microbial dihydroxylation of phenethyl acetate 8 (Scheme 2) via a whole cell fermentation with E. coli JM109 (pDTG601A) to afford the known intermediate cyclohexadiene diol 7 (obtained in 5gL−1 yield) [52,53], which was subjected to a selective reduction of the more accessible alkene to afford the known diol 9 (85% yield) [54,55]. The distal, less hindered, hydroxyl in diol 9 was protected with tert-butyl dimethylsilyl chloride and the free allylic alcohol was then subjected to a Mitsunobu reaction with iodo phenol 10 [56], derived from isovanillin, to furnish the coupled product ether 6 (45% yield over two steps). A subsequent intramolecular Heck reaction of 6 resulted in the formation of olefin 5 (87% yield) whose osmium tetroxide-catalyzed dihydroxylation led to diol 11 (81% yield). This compound possesses the features of the three (ACE) rings of 10-keto-oxycodone. The diol functionality was converted to ketone 4 via mesylation of the less hindered hydroxyl group followed by elimination with excess 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The mechanism may also be viewed formally as a 1,2-hydride shift. [57] of the resulting mesylate (63% yield over two steps). With the attainment of 4, deprotection of the acetal with aqueous TFA followed by a pinacol-type coupling of the intermediate keto aldehyde using SmI2 afforded diol 12, tentatively assigned as the cis isomer (65% yield over two steps) [58]. Mesylation of the less hindered hydroxyl group of diol 12 followed by treatment with excess DBU resulted in the formation of styrene alcohol 13 (56% yield over two steps). Hydrolysis of the acetate followed by oxidation with tetrapropylammonium perruthenate (TPAP) in the presence of N-methylmorpholine N-oxide (NMO) resulted in the formation of the key intermediate lactone 3 (91% over two steps).
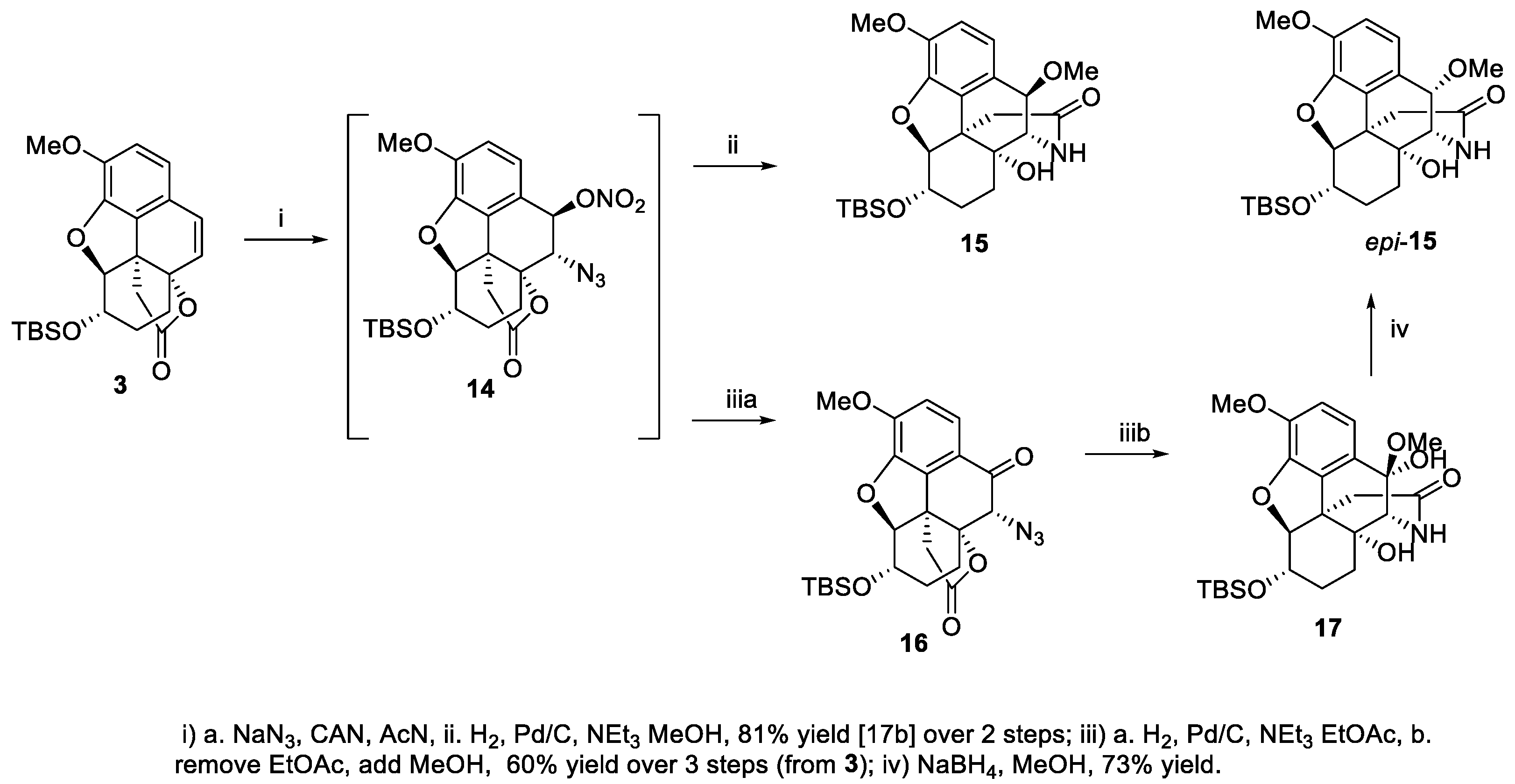
Based on our earlier work on a similar CAN-mediated functionalization of styrene to deliver ent-oxycodone [31], it was envisioned that the lactone 3 could be readily converted to a hydroxy lactam en route to ent-10-keto-oxycodone (Scheme 3). It was previously reported [31] that hydrogenation of azido nitrate 14 intermediate in methanol as solvent resulted in the formation benzylic methoxy ether lactam 15. Therefore, it seemed logical that switching to non-nucleophilic solvent (e.g., EtOAc, THF) would afford the benzylic alcohol instead. To our surprise and disappointment, the reduction of azide functionality did not occur and it appears that the benzylic alcohol derived by hydrogenation of the nitrate was slowly oxidized to the ketone 16 on exposure to air during workup and further manipulation [59]. To achieve the desired reduction of the azide to amine and subsequent lactamization, the solvent had to be changed to methanol. The desired lactam formation did occur, but the product was the hemiketal 17. This route was not followed through as this would necessitate deprotecting the hemiketal to the ketone prior to reduction with borane reagent. The hemiketal, when subjected to reduction with sodium borohydride, resulted in the formation of an ether, tentatively assigned as the C-10 epimeric benzylic methoxy ether (epi-15).
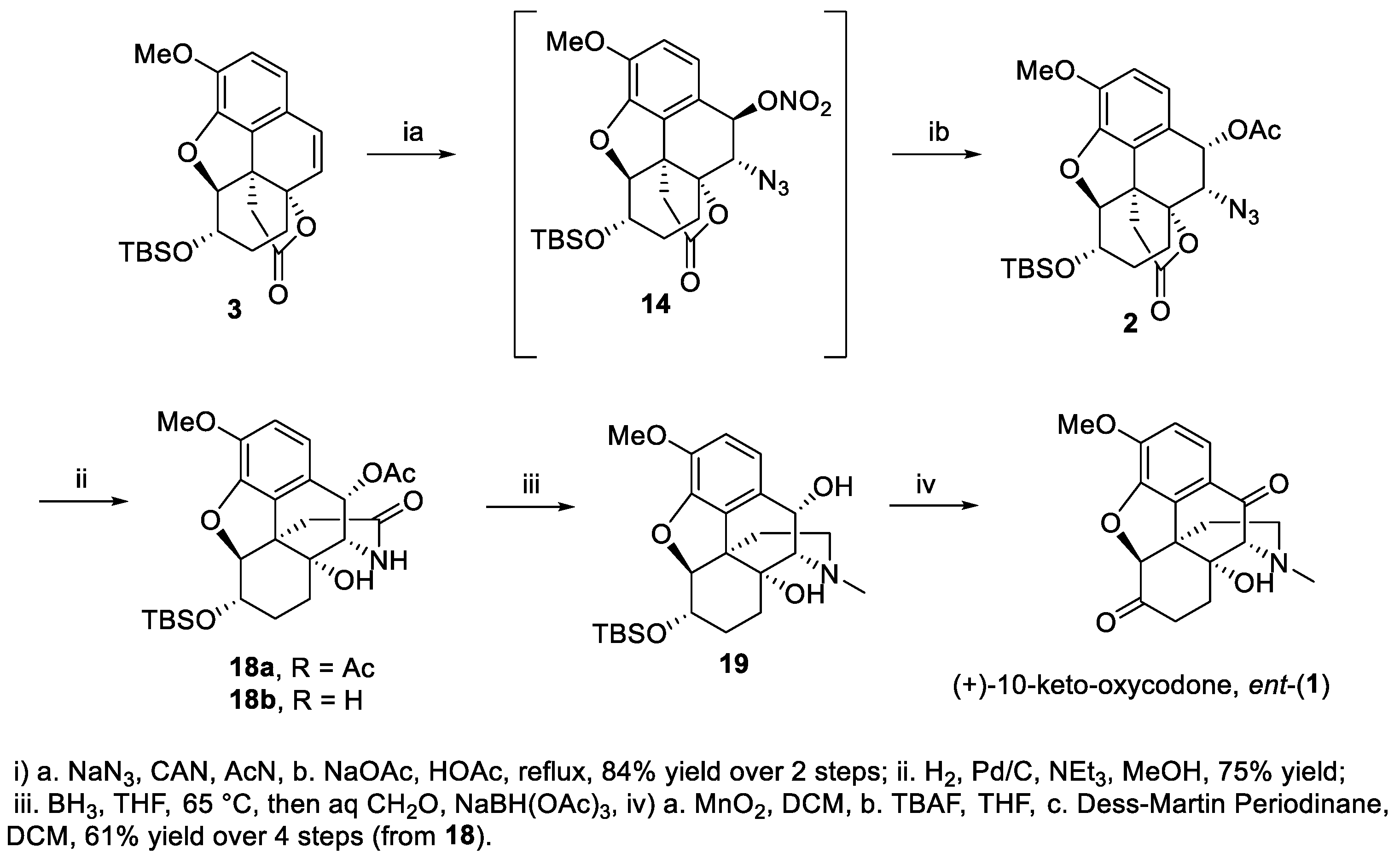
To address the above issue of the premature oxidation of C-10, the intermediate azido nitrate 14 was subjected to nucleophilic substitution with acetate to afford an intermediate acetate azide 2 (Scheme 4), with the acetate serving as a “protecting group” against premature oxidation of C-10. An overall inversion of stereochemistry of benzylic (C-10) carbon was tentatively assigned as in 2, in contrast with the formation of 15 (retention). It is believed that 15 was formed from an intermediate benzylic alcohol upon hydrogenolysis of the nitrate ester followed by lactamization and then nucleophilic displacement of OH by methoxy group. The stereochemical properties of the intermediate benzylic alcohol lactam favored a beta face attack by methanol as nucleophile. The facile displacement of OH in the benzylic position was facilitated by the highly activating methoxy group at the para position. For the formation of epi-15, on the other hand, the hydride served as the nucleophile and thus in the product the methoxy group ended up on the alpha face. The assignment of 15 and epi-15 as beta and alpha epimers was based on the coupling constants (J9-10) between the H-10 (benzylic C-H) and H-9 on similar systems [4,60]. For compounds similar to 15, J values are approximately 5.0 Hz and for epi-15, J values are around 0 Hz. The acetate azide 2 was hydrogenated to the amine, which underwent a spontaneous lactam formation to afford the acetate lactam 18 (63% yield over three steps). Reduction of the lactam 18 with borane followed by reductive amination with formaldehyde converted the lactam to the N-methyl amine 19. Because of the sensitivity (to basic and acidic reagents needed for the deprotection of C-6 hydroxyl) of the benzylic alcohol functionality in 19, it was first oxidized with manganese dioxide to the corresponding ketone to increase stability. It has been suggested by one of the referees that the benzylic alcohol 19 may be subject to a Grob-type fragmentation under basic conditions. Deprotection of the TBS group using TBAF and oxidation of the C-6 hydroxyl afforded ent-10-keto-oxycodone (61% yield over four steps).
In conclusion, a relatively short chemoenzymatic total synthesis of ent-10-keto-oxycodone has been accomplished in 14 operations from phenethyl acetate. To the best of our knowledge this accomplishment constitutes the first synthesis of ent-10-keto-oxycodone.
3. Experimental Section
3.1. General Methods
Inoculum was obtained from viable cells stored −78 °C in cryovials. They were grown in suitable media as previously described [28]. The substrate was fed in 5 mL increments over the course of ~3 h with metabolites being harvested in the usual manner. All non-aqueous reactions were conducted in an argon atmosphere using standard Schlenk techniques for the exclusion of moisture and air. Methylene chloride was distilled from calcium hydride, THF and toluene were dried over sodium/benzophenone. Analytical thin layer chromatography was performed on Silicycle 60 A° 250 mm TLC plates with F-254 indicator. Flash column chromatography was performed using Kieselgel 60 (230–400 mesh). Melting points were recorded on a Hoover Unimelt apparatus (Hoover, VA, USA) and are uncorrected. IR spectra were obtained on a Perkin-Elmer One FT-IR spectrometer (PerkinElmer, Waltham, MA, USA) Optical rotation was measured on a Perkin-Elmer 341 polarimeter (PerkinElmer, Waltham, MA, USA) at a wavelength of 589 nm. 1H and 13C spectra were recorded on a 300 MHz and 400 MHz Bruker spectrometer (Bruker, Billerica, MA, USA). All chemical shifts are referenced to TMS or residual nondeuterated solvent. Data of proton spectra are reported as follows: chemical shift in ppm (multiplicity: singlet (s), doublet (d), triplet (t), quartet(q) and multiplet (m)), coupling constants [Hz], integration). Carbon spectra were recorded with complete proton decoupling and the chemical shifts are reported in ppm (δ) relative to solvent resonance as internal standard. Mass spectra and high resolution mass spectra were performed by the Analytical Division at Brock University. The 1H and 13C NMR spectra of some compounds are in the Supplementary Materials.
3.2. (4bR,5S,6S,8aR,9S)-9-azido-6-((tert-butyldimethylsilyl)oxy)-4,5-epoxy3-methoxy-10-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-12-one (16)
To a stirred solution of lactone 3 (140 mg, 0.34 mmol) in AcN (5 mL) at 0 °C was added NaN3 (140 mg, 2.15 mmol) followed by CAN (616 mg, 1.12 mmol). After 30 min, the reaction mixture was diluted with Et2O/satd. NaHCO3 (30 mL/15 mL). The layers were separated and the aqueous layer was further extracted with Et2O (2 × 20 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated to afford a residue that was used as crude in the next step. To a stirred solution of crude azido nitrate 14 (from previous step) in EtOAc (4.0 mL) was added NEt3 (200 µL, 1.0 mmol) and Pd/C (10 mg, 0.004 mmol). The reaction mixture was evacuated and refilled (3×) with hydrogen gas and was allowed to stir under an atmosphere of hydrogen for 12 h. (A small aliquot was taken, filtered through a plug of Celite and concentrated to afford a solid whose 1H NMR spectrum was obtained.) It was taken in its crude form to the next step.
16: 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 4.45 (d, J = 7.8 Hz, 1H), 4.09 (s, 1H), 4.02 (s, 3H), 3.24–3.35 (m, 1H), 3.16 (d, J = 17.7 Hz, 1H), 3.01 (d, J = 17.7 Hz, 1H), 2.20–2.36 (m, 1H), 1.65–1.85 (m, 3H), 0.89 (s, 9H), 0.09 (s, 3H), 0.02 (s, 3H) ppm.
3.3. (5R,5aR,8S,8aS,13bR,14R)-8-((tert-butyldimethylsilyl)oxy)-5a,14-dihydroxy-10,14-dimethoxy-1,2,5,5a,6,7,8,8a-octahydro-5,13-methanobenzo[2,3]benzofuro[4,3a-c]azepin-3(4H)-one (17)
The solvent (EtOAc) of the reaction from the previous step was exchanged (for MeOH) as follows: EtOAc was removed via rotary evaporation and the resulting residue was diluted with MeOH (4.0 mL). The reaction mixture was evacuated and refilled (3×) with hydrogen gas and was allowed to stir under an atmosphere of hydrogen for another 12 h. The catalyst was separated by filtration through Celite. The Celite was washed with MeOH (2 × 15 mL). The filtrate and washings were combined and evaporated to dryness to afford a crude solid residue that was further purified by chromatography on silica gel using DCM/MeOH (9:1) as eluent to afford the hemiketal lactam product 17 as a white solid (90 mg, 59.8% yield).
17: 1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 8.4 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.73 (s, 1H), 4.55 (d, J = 6.3 Hz, 1H), 3.99 (s, 3H), 3.84 (s, 3H), 3.35–3.45 (m, 1H), 3.17 (d, J = 17.7 Hz, 1H), 2.80–2.90 (m, 1H), 2.59 (d, J = 17.7 Hz, 1H), 1.85–1.98 (m, 1H), 1.48–1.56 (m, 1H), 1.40–1.45 (m, 2H), 0.91 (s, 9H), 0.12 (s, 3H), 0.02 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 189.6, 170.2, 150.8, 144.3, 137.3, 121.6, 120.2, 115.6, 95.7, 89.0, 73.7, 72.2, 56.9, 53.6, 48.5, 38.7, 27.3, 26.2, 25.7, 18.0, −4.8, −5.1 ppm; HRMS (CI+) calcd for C24H34O7NSi [M − H] 476.2099 found 476.2090.
3.4. (5S,5aR,8S,8aS,13bR,14S)-8-((tert-butyldimethylsilyl)oxy)-5a-hydroxy-10,14-dimethoxy-1,2,5,5a,6,7,8,8a-octahydro-5,13-methanobenzo[2,3]benzofuro[4,3a-c]azepin-3(4H)-one (epi-15)
To as stirred solution of hemiketal 17 (10 mg, 0.02 mmol) in MeOH (1.0 mL) at 0 °C was added NaBH4 (10 mg, 0.26 mmol). The reaction mixture was stirred at this temperature of 15 min, then it was diluted with acetone (1.0 mL), water (1.0 mL), and Et2O (5 mL). The layers were separated and the aqueous phase was further extracted with Et2O (2 × 5 mL). The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated to afford a crude solid product (7 mg, 72.5% yield). Comparison of 1HNMR data for 15 and epi-15 clearly indicated their relationship as epimeric compounds.
epi-15: 1H NMR (300 MHz, CDCl3) δ 6.95 (d, J = 8.4 Hz, 1H), 6.88(d, J = 8.4 Hz, 1H), 6.62 (br s, 1H), 4.88 (s, 1H), 4.43 (d, J = 6.6 Hz, 1H), 3.89 (s, 3H), 3.43–3.49 (m, 1H), 2.97 (d, J = 17.4 Hz, 1H), 2.43 (d, J = 17.4 Hz, 1H), 1.74–2.04 (m, 1H), 1.52–1.65 (m, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.00 (s, 3H) ppm.
3.5. (4bR,5S,6S,8aR,9S,10R)-9-azido-6-((tert-butyldimethylsilyl)oxy)-4,5-epoxy3-methoxy-12-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-10-yl nitrate (14)
To a stirred solution of lactone 3 [17b] (50 mg, 0.12 mmol) in AcN (5 mL) at 0 °C was added NaN3 (50 mg, 0.77 mmol) followed by CAN (220 mg, 0.40 mmol). After 30 min, the reaction mixture was diluted with Et2O/satd. NaHCO3 (10 mL/5 mL). The layers were separated and the aqueous layer was further extracted with Et2O (2 × 10 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated to afford a residue that was used as crude in the next step. A small sample of azido nitrate 14 was subjected to purification by preparative TLC (SiO2 gel, 2:1 hex/EtOAc as eluent) for characterization purposes. It was found to have limited stability.
14: = −35.8 (c = 0.6, CH2Cl2); Rf = 0.44 (4:1; hexanes/EtOAc); IR (film) v 3434, 2955, 2929, 2855, 2116, 1796, 1645, 1508, 1440, 1274, 837 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.99 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.30 (d, J = 1.8 Hz, 1H), 4.40 (d, J = 7.8 Hz, 1H), 4.10 (d, J = 1.8 Hz, 1H), 3.95 (s, 3H), 3.25–3.29 (m, 1H), 3.04 (d, J = 17.7 Hz, 1H), 2.94 (d, J = 17.7 Hz, 1H), 2.13–2.17 (m, 1H), 1.68–1.77 (m, 3H), 0.89 (s, 9H), 0.10 (s, 3H), 0.00 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 173.6, 151.8, 146.2, 128.9, 122.6, 116.5, 115.3, 99.4, 82.9, 80.5, 69.8, 67.5, 56.8, 49.6, 43.4, 29.7, 28.7, 26.7, 25.7, 18.0, −4.8, −5.2 ppm; MS (EI) m/z (%) 416(15), 386(60), 358(100), 328(30), 288(75), 272(45); HRMS (EI) calcd for C19H21O8N4Si [M − C4H9-H2O], 461.1123 found 461.1118.
3.6. (4bR,5S,6S,8aR,9S,10S)-9-azido-6-((tert-butyldimethylsilyl)oxy)4,5-epoxy-3-methoxy-12-oxo-5,6,7,8,9,10-hexahydro-8a,4b-(epoxyethano)phenanthren-10-yl acetate (2)
To the crude azido nitrate 14 (obtained from the previous step) dissolved in HOAc (1.5 mL) was added NaOAc (150 mg, 1.83 mmol) and the reaction mixture was heated to 100 °C for 2 h. After cooling to room temperature, the mixture was carefully diluted with satd NaHCO3/EtOAc (5 mL/5 mL). The layers were separated and the aqueous phase was further extracted with EtOAc (2 × 5 mL). The combined organic extracts were washed with brine, dried over anhydrous MgSO4, filtered and concentrated to afford a residue that was subjected to chromatography on silica gel using hexanes/EtOAc (2:1) as eluent to afford the product 2 as an oil (52 mg, 84% yield over two steps).
2: = −2.4 (c = 1.0, CH2Cl2); Rf = 0.50 (2:1; hexanes/EtOAc); IR (film) v 2954, 2928, 2855, 2113, 1796, 1747, 1632, 1507, 1441, 1264, 1125, 838, 738 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.92 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.20 (d, J = 2.7 Hz, 1H), 4.42 (d, J = 7.8 Hz, 1H), 3.94 (s, 3H), 3.90 (d, J = 2.7 Hz, 1H), 3.28–3.36 (m, 1H), 3.13 (d, J = 17.7 Hz, 1H), 3.04 (d, J = 17.7 Hz, 1H), 2.19–2.25 (m, 1H), 2.12 (s, 3H), 1.66–1.79 (m, 3H), 0.90 (s, 9H), 0.12 (s, 3H), 0.02 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 173.9, 169.9, 145.8, 145.7, 127.8, 121.4, 120.4, 116.3, 99.3, 83.5, 71.3, 70.3, 69.1, 56.8, 49.8, 43.7, 29.7, 29.3, 26.8, 25.7, 21.1, 18.0, −4.7, −5.2 ppm; MS (EI) m/z (%) 458(80), 430(20), 398(40), 370(100), 343(40), 328(80), 300(45), 254(45), 117(90); HRMS (EI) calcd for C21H24O7N3Si [M − C4H9], 458.1378 found 458.1373.
3.7. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-4a-hydroxy-9-methoxy-2-oxo-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-13-yl acetate (18a)
To a stirred solution of azido acetate 2 (20 mg, 0.04 mmol) in MeOH (0.5 mL) was added NEt3 (30 µL, 0.15 mmol) and Pd/C (2.5 mg, 0.001 mmol). The reaction mixture was evacuated and refilled (3×) with hydrogen gas and was allowed to stir under an atmosphere of hydrogen for 12 h. The catalyst was separated by filtration through Celite. The Celite was washed with MeOH (2 × 5 mL). The filtrate and washings were combined and evaporated to dryness to afford a crude solid residue that was further purified by chromatography on silica gel using DCM/MeOH (9:1) as eluent to afford the desired acetate lactam product 18a and the corresponding alcohol lactam 18b in a ratio of 8:1 as white solids (13 mg, 75% yield). A small sample of acetate lactam was hydrolyzed to the alcohol lactam for characterization purposes.
18a: = 48.0 (c = 0.4, CH2Cl2); m.p. 140–141 °C; Rf = 0.42 (9:1; CH2Cl2/MeOH); IR (film) v 3336, 2929, 2855, 1733, 1632, 1608, 1366, 1234, 1021, 837, 755 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.88 (s, 2H), 6.77 (d, J = 4.5 Hz, 1H), 5.95 (d, J = 2.0 Hz, 1H), 4.49 (d, J = 6.0 Hz, 1H), 3.92 (s, 3H), 3.68 (dd, J = 4.5 Hz, 2.0 Hz, 1H), 3.50–3.53 (m, 1H), 2.90 (d, J = 17.7 Hz, 1H), 2.48 (d, J = 17.7 Hz, 1H), 2.10 (s, 3H), 1.8–1.97 (m, 2H), 1.63–1.68 (m, 1H), 1.50–1.53 (m, 1H), 0.92 (s, 9H), 0.15 (s, 3H), 0.05 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 171.1, 170.5, 146.0, 144.3, 133.7, 122.1, 119.5, 115.9, 96.0, 73.1, 72.2, 69.0, 60.9, 56.8, 46.7, 39.9, 29.6, 26.3, 25.7, 21.1, 18.0, −4.7, −5.1 ppm; MS (EI+) m/z (%) 433(25), 432(80), 373(40), 372(100), 354(20), 330(20), 285(65), 274(75); HRMS (EI) calcd for C25H36O7NSi [M + H], 490.2256 found 490.2261.
3.8. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-4a,13-dihydroxy-9-methoxy-4,4a,5,6,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-2(3H)-one (18b)
18b: = 135.8 (c = 0.3, CH2Cl2); m.p. 135–137 °C; Rf = 0.37 (9:1; CH2Cl2/MeOH); IR (film) v 3391, 2954, 2929, 2856, 1644, 1634, 1440, 1258, 838 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.73 (br s, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H), 4.88 (br s, 1H), 4.44 (d, J = 6.0 Hz, 1H), 3.89 (s, 3H), 3.69 (s, 1H), 3.43–3.49 (m, 1H), 2.88 (d, J = 17.7 Hz, 1H), 2.63 (d, J = 17.7 Hz, 1H), 1.98–2.15 (m, 1H), 1.79–1.91 (m, 1H), 1.55–1.59 (m, 1H), 1.41–1.44 (m, 1H), 0.91 (s, 9H), 0.12 (s, 3H), 0.02 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 172.7, 145.5, 144.4, 132.6, 123.2, 121.9, 115.7, 96.5, 72.4, 69.2, 66.6, 63.4, 56.7, 46.6, 39.6, 29.7, 26.5, 25.8, 18.0, −4.8, −5.1 ppm; MS (EI) m/z (%) 390(70), 372(100), 330(25), 285(95), 274(60), 259(25); HRMS (EI) calcd for C19H24O6NSi [M − C4H9], 390.1367 found 390.1370.
3.9. (4S,4aR,7S,7aS,12bR,13S)-7-((tert-butyldimethylsilyl)oxy)-9-methoxy-3-methyl-1,2,3,4,5,6,7,7a-octahydro-4aH-4,12-methanobenzofuro[3,2-e]isoquinoline-4a,13-diol (19)
To a stirred solution of acetate lactam 18a (60 mg, 0.12 mmol) in THF (1.5 mL) at 0 °C was added BH3 in THF (6.8 mL, 6.6 mmol). After stirring at this temperature for 10 min, the mixture was heated to 65 °C for 16 h. The reaction mixture was allowed to cool to room temperature and quenched by the dropwise addition of MeOH (5 mL) and acetic acid (500 µL). To the reaction mixture was added 30% aq. CH2O (1.2 mL, 11.2 mmol) and NaBH(OAc)3 (650 mg, 3.03 mmol) at room temperature. After 1.5 h, the reaction mixture was diluted with DCM/satd. NaHCO3 (30 mL/30 mL). The layers were separated and the aqueous phase was further extracted with DCM (2 × 30 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated to afford a residue that was used as crude in the next step. A small sample was taken whose 1H NMR spectrum was obtained. This compound has limited stability so it was taken as a crude product to the next step.
19: 1H NMR (300 MHz, CDCl3) δ 6.94 (d, J = 8.4 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 5.08 (s, 1H), 4.45 (d, J = 6.3 Hz, 1H), 3.92 (s, 3H), 3.43–3.51 (m, 1H), 2.86 (s, 1H), 2.55 (s, 3H), 2.50–2.60 (m, 1H), 2.17–2.25 (m, 1H), 1.97–2.05 (m, 2H), 1.59–1.67 (m, 2H), 1.4–1.55 (m, 2H), 0.91 (s, 9H), 0.13 (s, 3H), 0.03 (s, 3H) ppm.
3.10. ent-10-keto-oxycodone (ent-(1))
To the crude N-methyl amine alcohol 19 (obtained from the previous step) dissolved in DCM (1.0 mL) was added MnO2 (191 mg, 2.2 mmol). The suspension was stirred vigorously at room temperature for 1 day. The reaction mixture was filtered through a plug of Celite and the residue was washed with DCM (5 mL). The filtrate was concentrated by rotary evaporation and the residue was used as crude in the next step. To a stirred solution of N-methyl ketone (obtained from the previous step) in THF (1 mL) was added TBAF (250 µL, 0.25 mmol). The reaction mixture was stirred at room temperature for 5 h after which it was diluted with EtOAc/H2O (5 mL/5 mL). The layers were separated and the aqueous layer was further extracted with DCM (2 × 5 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated to afford a residue that was used as crude in the next step. To the crude alcohol dissolved in DCM (3 mL) was added Dess-Martin periodinane (93.3 mg, 0.22 mmol). The reaction mixture was stirred at rt for 2 h. It was diluted with satd. Na2S2O3 (1 mL), followed by satd. NaHCO3 solution (1 mL). The layers were separated and the aqueous phase was further extracted with DCM (2 × 10 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated to afford a residue that was chromatographed on silica gel using DCM/MeOH as eluent (9:1) to afford the product ent-(1) as a solid (24 mg, 0.07 mmol, 60.7% yield over four steps).
ent-(1): = 156.8 (c = 0.3, CH2Cl2); m.p. 239–241 °C, lit. [5] m.p. 242–243 °C Rf = 0.57 (9:1; CH2Cl2/MeOH); IR (film) v 3434, 2924, 2851, 1727, 1674, 1619, 1443, 1291, 1115 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 4.84 (br s, 1H), 4.77 (s, 1H), 4.03 (s, 3H), 3.09 (dd, J = 14.4, 4.8 Hz, 3.02 (s, 1H), 2.57–2.72 (m, 2H), 2.49 (s, 3H), 2.20–2.38 (m, 2H), 1.92–1.96 (m, 1H), 1.65–1.77 (m, 1H), 1.45–1.63 (m, 1H) ppm; 13C NMR (75 MHz, CDCl3) δ 207.2, 192.5, 150.2, 144.9, 136.8, 123.8, 119.5, 115.0, 89.9, 73.7, 71.2, 56.9, 52.0, 45.7, 43.3, 35.9, 31.6, 29.6 ppm; MS (EI) m/z (%) 329(100), 273(25), 244(25), 217(30), 112(10); HRMS (EI+) calcd for C18H19O5N, 329.1258 found 329.1258.
Supplementary Materials
Copies of spectral data (1H NMR and 13C NMR) are provided for the following compounds: 2, 18a, 18b, 14, ent-(1).
Author Contributions
T.H. and M.A.E.-A. conceived and designed the experiments. H.D.P. and M.A.E.-A. performed the experiments. T.H. and M.A.E.-A. analyzed the data and wrote the manuscript.
Funding
The authors are grateful to the following agencies for financial support of this work: Natural Sciences and Engineering Research Council of Canada (NSERC) (Idea to Innovation and Discovery Grants), Canada Research Chair Program, Canada Foundation for Innovation (CFI), TDC Research, Inc., TDC Research Foundation, the Ontario Partnership for Innovation and Commercialization (OPIC), Noramco, Inc., and The Advanced Biomanufacturing Centre (Brock University).
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Nagase, H.; Hayakawa, J.; Kawamura, K.; Kawai, K.; Takezawa, Y.; Matsuura, H.; Tajima, C.; Endo, T. Discovery of a structurally novel opioid κ-agonist derived from 4,5-epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. [Google Scholar] [CrossRef]
- Millan, M.J. Kappa-opioid receptors and analgesia. Trends Pharmacol. Sci. 1990, 11, 70–76. [Google Scholar] [CrossRef]
- Cowan, A.; Gmerek, D.E. In-vivo studies on kappa opioid receptors. Trends Pharmacol. Sci. 1986, 7, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Horikiri, H.; Hirano, N.; Tanaka, Y.; Oishi, J.; Hatakeyama, H.; Kawamura, K.; Nagase, H. Syntheses of 10-oxo, 10-hydroxy, and 10-hydroxy derivatives of a potent κ-opioid receptor agonist, TRK-820. Chem. Pharm. Bull. 2004, 52, 664–669. [Google Scholar] [CrossRef]
- Archer, S.; Seyed-Mozaffari, A.; Ward, S.; Kosterlitz, H.W.; Paterson, S.J.; McKnight, A.T.; Corbett, A.D. 10-Ketonaltrexone and 10-ketooxymorphone. J. Med. Chem. 1985, 28, 974–976. [Google Scholar] [CrossRef]
- Cain, G.A.; Drummond, S., Jr. Sequential benzylic oxidation of naloxone 3-methyl ether. Synth. Commun. 2000, 30, 4513–4521. [Google Scholar] [CrossRef]
- Hudlicky, T. Recent advances in process development for opiate-derived pharmaceutical agents. Can. J. Chem. 2015, 93, 492–501. [Google Scholar] [CrossRef]
- Reed, J.W.; Hudlicky, T. The quest for a practical synthesis of morphine alkaloids and their derivatives by chemoenzymatic methods. Acct. Chem. Res. 2015, 48, 674–687. [Google Scholar] [CrossRef]
- Rapoport, H.; Masamune, S. The stereochemistry of 10-hydroxycodeine derivatives. J. Am. Chem. Soc. 1955, 77, 4330–4335. [Google Scholar] [CrossRef]
- Sun, H.; Johnson, J.E., Jr.; Fenton, R.L.; Dorn, S.M. Preparation of 10-keto morphinans by benzylic oxidation. U.S. Patent 2011071296 (A1), 1 September 2011. [Google Scholar]
- Uyeda, R.T.; Wuonola, M.A.; Read, J.M., Jr. 10-Keto opiates. Tetrahedron Lett. 1989, 30, 5725–5728. [Google Scholar] [CrossRef]
- Freund, M.; Speyer, E. DE 286431, 1914.
- Freund, M.; Speyer, E. DE 296916, 1916.
- Freund, M.; Speyer, E. Transformation of thebaine into hydroxycodeione and its derivatives. J. Prakt. Chem. 1916, 94, 135–178. [Google Scholar] [CrossRef]
- Lutz, R.E.; Small, L. Reduction studies in the morphine series. IX. Hydroxycodeinone. J. Org. Chem. 1939, 4, 220–233. [Google Scholar] [CrossRef]
- Kok, G.B.; Scammells, P.J. Improved synthesis of 14-hydroxy opioid pharmaceuticals and intermediates. RSC Adv. 2012, 2, 11318–11325. [Google Scholar] [CrossRef]
- Millgate, A.G.; Pogson, B.J.; Wilson, I.W.; Kutchan, T.M.; Zenk, M.H.; Gerlach, W.L.; Fist, A.J.; Larkin, P.J. Morphine-pathway block in top1 poppies. Nature 2004, 431, 413–414. [Google Scholar] [CrossRef]
- Fist, A.J.; Byrne, C.J.; Gerlach, W.L. Papaver Somniferum Strain with High Concentration of Thebaine. U.S. Patent 606749, 30 May 2000. [Google Scholar]
- Fist, A.J. Papaver Somniferum Strain with High Concentration of Thebaine and Oripavine. U.S. Patent Application No. 20090227796, 10 September 2009. [Google Scholar]
- Thodey, K.; Galanie, S.; Smolke, C.D. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014, 10, 837–844. [Google Scholar] [CrossRef]
- Tasmanian Alkaloids Pty Ltd.: Westbury, TAS, Australia. Available online: https://tasalk.com.au/ (accessed on 2 September 2019).
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1952, 74, 1109–1110. [Google Scholar] [CrossRef]
- Rinner, U.; Hudlicky, T. Alkaloid synthesis. Top. Curr. Chem. 2012, 309, 33–66. [Google Scholar]
- Zezula, J.; Hudlicky, T. Recent Progress in the synthesis of morphine alkaloids. Synlett 2005, 388–405. [Google Scholar] [CrossRef]
- Taber, D.F.; Neubert, T.D.; Schlecht, M.F. The enantioselective synthesis of morphine. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 5, p. 353. [Google Scholar]
- Novak, B.H.; Hudlicky, T.; Reed, J.W.; Mulzer, J.; Trauner, D. Recent progress in the synthesis of morphine alkaloids. Curr. Org. Chem. 2000, 4, 343. [Google Scholar] [CrossRef]
- Hudlicky, T.; Butora, G.; Fearnley, S.; Gum, A.; Stabile, M. A historical perspective of morphine syntheses. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 18, p. 43. [Google Scholar]
- Rice, K. Synthetic opium alkaloids and derivatives. A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners. J. Org. Chem. 1980, 45, 3135–3137. [Google Scholar] [CrossRef]
- Kimishima, A.; Umihara, J.; Mizoguchi, A.; Yokoshima, S.; Fukuyama, T. Synthesis of (−)-oxycodone. Org. Lett. 2014, 16, 6244–6247. [Google Scholar] [CrossRef]
- Endoma-Arias, M.A.A.; Makarova, M.; Dela Paz, H.E.; Hudlicky, T. Chemoenzymatic total synthesis of (+)-oxycodone from phenethyl acetate. Synthesis 2019, 225–232. [Google Scholar] [CrossRef]
- Makarova, M.; Endoma-Arias, M.A.A.; Dela Paz, H.E.; Simionescu, R.; Hudlicky, T. Chemoenzymatic total synthesis of ent-oxycodone: Second, third and fourth generation strategies. J. Am. Chem. Soc. 2019, 141, 10883–10904. [Google Scholar] [CrossRef]
- Zylstra, G.J.; Gibson, D.T. Toluene degradation by Pseudomonas putida F1. Nucleotide sequence of the todC1C2BADE genes and their expression in Escherichia coli. J. Biol. Chem. 1989, 264, 14940–14946. [Google Scholar]
- Hudlicky, T. Benefits of unconventional methods in the total synthesis of natural products. ACS Omega 2018, 3, 17326–17340. [Google Scholar] [CrossRef]
- Taher, E.S.; Banwell, M.G.; Buckler, J.N.; Yan, Q.; Lan, P. The exploitation of enzymatically derived cis-1,2-dihydrocatechols and related compounds in the synthesis of biologically active natural products. Chem. Rec. 2018, 18, 239–264. [Google Scholar] [CrossRef]
- Banwell, M.G.; Bolte, B.; Buckler, J.N.; Chang, E.L.; Lan, P.; Taher, E.S.; White, L.V.; Willis, A.C. Chemoenzymatic Pathways for the Synthesis of Biologically Active Natural Products; Royal Society of New South Wales: Bowral, NSW, Australia, 2016; Parts 1&2; pp. 34–50. [Google Scholar]
- Lewis, S.E. Applications of biocatalytic arene ipso, ortho cis-dihydroxylation in synthesis. Chem. Commun. 2014, 50, 2821–2830. [Google Scholar] [CrossRef]
- Rinner, U. Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 2, pp. 240–267. [Google Scholar]
- Bon, D.J.-Y.D.; Lee, B.; Banwell, M.G.; Cade, I.A. Biocatalytically-derived cis-1,2-dihydrocatechols—enantiopure and versatile building blocks for chemical synthesis. Chim. Oggi 2012, 30, 22–27. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Celebrating 20 Years of Synlett—Special account on the merits of biocatalysis and the impact of arene cis-dihydrodiols on eantioselective synthesis. Synlett 2009, 685–703. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132. [Google Scholar] [CrossRef]
- Boyd, D.R.; Bugg, T.D.H. Arene cis-dihydrodiol formation: from biology to application. Org. Biomol. Chem. 2006, 4, 181–192. [Google Scholar] [CrossRef]
- Johnson, R.A. Organic Reactions; Overman, L.E., Ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2004; Volume 63, pp. 117–264. [Google Scholar]
- Banwell, M.G.; Edwards, A.J.; Harfoot, G.J.; Jolliffe, K.A.; McLeod, M.D.; McRae, K.J.; Stewart, S.G.; Vogtle, M. Chemoenzymatic methods for the enantioselective preparation of sesquiterpenoid natural products from aromatic precursors. Pure Appl. Chem. 2003, 75, 223–230. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D. Enzymatic and chemoenzymatic synthesis of arene trans-dihydrodiols. J. Mol. Catal. B Enzym. 2002, 19, 31–42. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Allen, C.C.R. Aromatic dioxygenases: Molecular biocatalysis and applications. Curr. Opin. Biotechnol. 2001, 12, 564–573. [Google Scholar] [CrossRef]
- Hudlicky, T.; Gonzalez, D.; Gibson, D.T. Enzymatic dihydroxylation of aromatics in enantioselective synthesis: Expanding asymmetric methodology. Aldrichim. Acta 1999, 32, 35–62. [Google Scholar]
- Boyd, D.R.; Sheldrake, G.N. The dioxygenase-catalysed formation of vicinal cis-diols. Nat. Prod. Rep. 1998, 15, 309–324. [Google Scholar] [CrossRef]
- Reed, J.W.; Hudlicky, T. Advances in Asymmetric Synthesis; Hassner, A., Ed.; JAI Press: Greenwich, CT, USA, 1995; Volume 1, pp. 271–312. [Google Scholar]
- Brown, S.M.; Hudlicky, T. Organic Synthesis: Theory and Applications; Hudlicky, T., Ed.; JAI Press: Greenwich, CT, USA, 1993; Volume 2, pp. 113–176. [Google Scholar]
- Carless, H.A.J. The use of cyclohexa-3,5-diene-1,2-diols in enantiospecific synthesis. Tetrahedron Asymmetry 1992, 3, 795–826. [Google Scholar] [CrossRef]
- Widdowson, D.A.; Ribbons, D.W.; Thomas, D.D. The use of substituted cyclohexadiene diols as versatile chiral synthons. Janssen Chim. Acta 1990, 8, 3–9. [Google Scholar]
- Hudlicky, T.; Endoma, M.A.A.; Butora, G. Nucleophilic substitution of protected β-bromoethyl cyclohexadiene-cis-diol as an alternative to direct microbial oxidation of β-functionalized phenethyl substrates. J. Chem. Soc. Perkin Trans. 1 1996, 17, 2187–2192. [Google Scholar] [CrossRef]
- Endoma, M.A.; Bui, V.P.; Hansen, J.; Hudlicky, T. Medium-scale preparation of useful metabolites of aromatic compounds via whole-cell fermentation with recombinant organisms. Org. Proc. Res. Dev. 2002, 6, 525–532. [Google Scholar] [CrossRef]
- Endoma-Arias, M.A.A.; Hudlicky, J.R.; Simionescu, R.; Hudlicky, T. Chemoenzymatic formal total synthesis of ent-codeine and other morphinans via nitrone cycloadditions and/or radical cyclizations. Comparison of strategies for control of C-9/C-14 stereogenic centers. Adv. Synth. Cat. 2014, 356, 333–339. [Google Scholar] [CrossRef]
- Hudlicky, T.; Endoma-Arias, M.A.A. Chemoenzymatic total synthesis of (+)-galanthamine and (+)-narwedine from phenethyl acetate. Chem. Eur. J. 2016, 22, 14540–14543. [Google Scholar]
- Koizumi, H.; Yokoshima, S.; Fukuyama, T. Total synthesis of (−)-morphine. Chem. Asian J. 2010, 5, 2191–2198. [Google Scholar] [CrossRef]
- Wiemann, J.; Deckelmann, A.M.; Csuk, R. A remarkably simple and convergent partial synthesis of pomolic acid. Tetrahedron Lett. 2016, 57, 3952–3953. [Google Scholar] [CrossRef]
- Oxidation of the diol 12 to afford a keto alcohol followed by NaBH4 reduction resulted in the formation of an epimeric (tentatively assigned as trans) diol. Attempts to form the acetonide of this supposed trans isomer failed. The diol 12 was smoothly converted to the cyclic carbonate [30]. It was also readily converted to the corresponding acetonide and benzylidene acetal. Unpublished observations.
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef]
- Meredith, W.; Nemeth, G.A.; Boucher, R.; Carney, R.; Haas, M.; Sigvardson, K.; Teleha, C.A. Isolation and synthesis of 2-chloro-10-α-hydroxynaltrexone, a new naltrexone degradant. Tetrahedron Lett. 2003, 44, 7381–7384. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from the authors. |
Figure 1.
Structure of (−)-10-keto-oxycodone (natural series enantiomer, with morphine numbering system shown).
Figure 1.
Structure of (−)-10-keto-oxycodone (natural series enantiomer, with morphine numbering system shown).

Scheme 1.
Retrosynthetic analysis of the route to ent-10-keto-oxycodone.

Scheme 2.
Synthesis of lactone 3.

Scheme 3.
Initial studies on hydroxy lactam formation.

Scheme 4.
Completion of the synthesis of ent-10-keto-oxydocone via lactam 18.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Endoma-Arias, M.A.; Dela Paz, H.; Hudlicky, T. Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate. Molecules 2019, 24, 3477. https://doi.org/10.3390/molecules24193477
AMA Style
Endoma-Arias MA, Dela Paz H, Hudlicky T. Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate. Molecules. 2019; 24(19):3477. https://doi.org/10.3390/molecules24193477
Chicago/Turabian StyleEndoma-Arias, Mary Ann, Helen Dela Paz, and Tomas Hudlicky. 2019. "Chemoenzymatic Total Synthesis of (+)-10-Keto-Oxycodone from Phenethyl Acetate" Molecules 24, no. 19: 3477. https://doi.org/10.3390/molecules24193477