
Combined Ensemble Docking and Machine Learning in Identification of Therapeutic Agents with Potential Inhibitory Effect on Human CES1

 , ,
, ,
Abstract
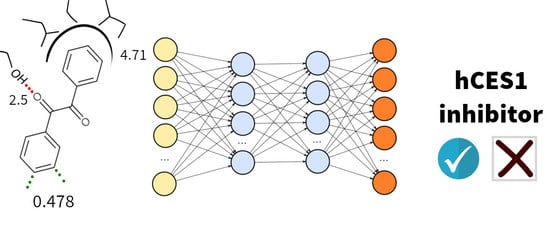
:
1. Introduction
2. Results
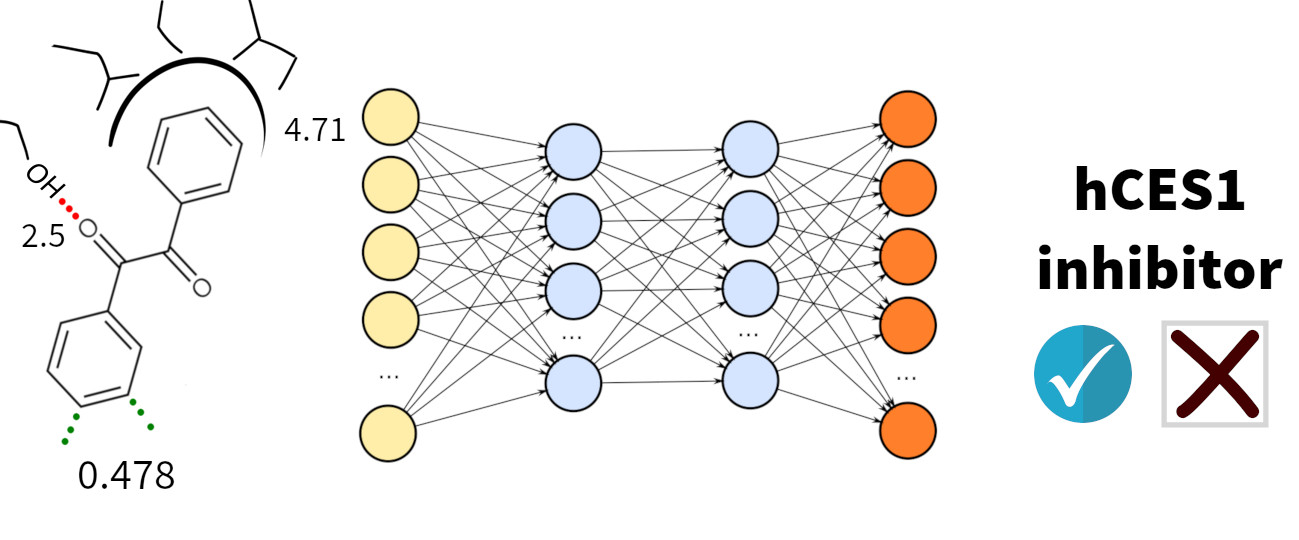
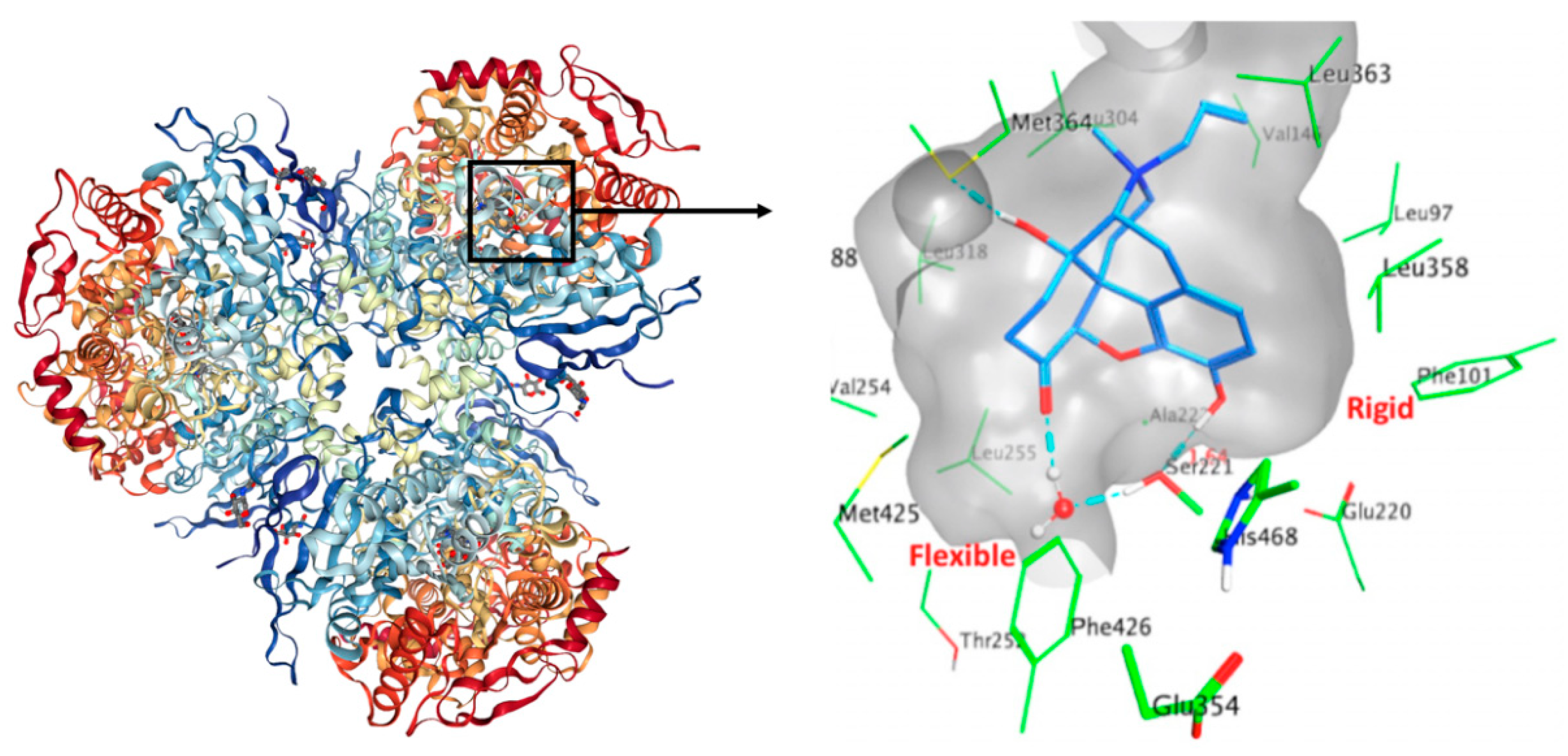
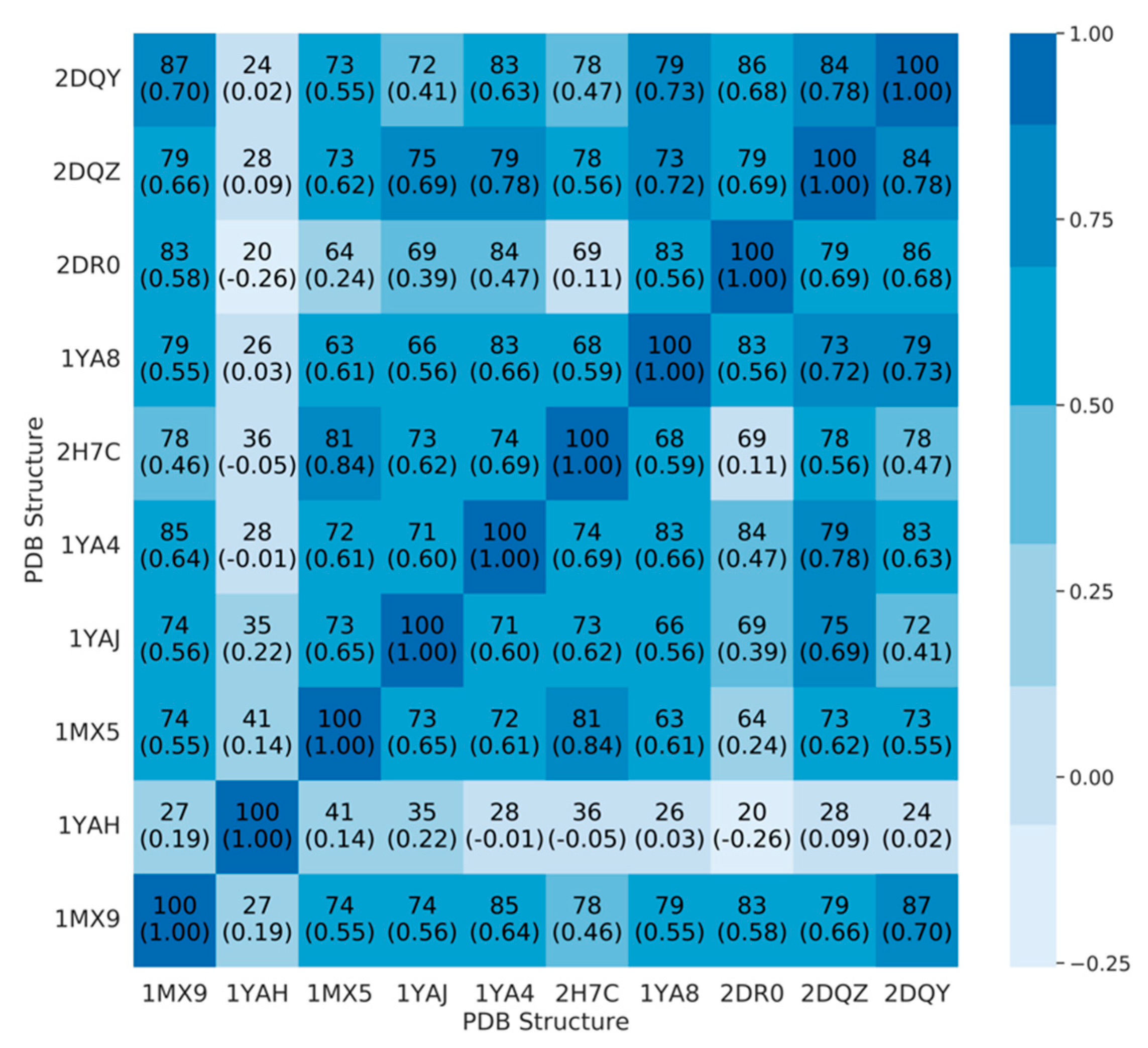
2.1. Binding Site Description
2.1.1. Evaluation and Validation of Docking Protocol
2.1.2. Analysis of the Docking Protocol with Known CES1 Ligands
2.2. Expert System Based on Energy Terms
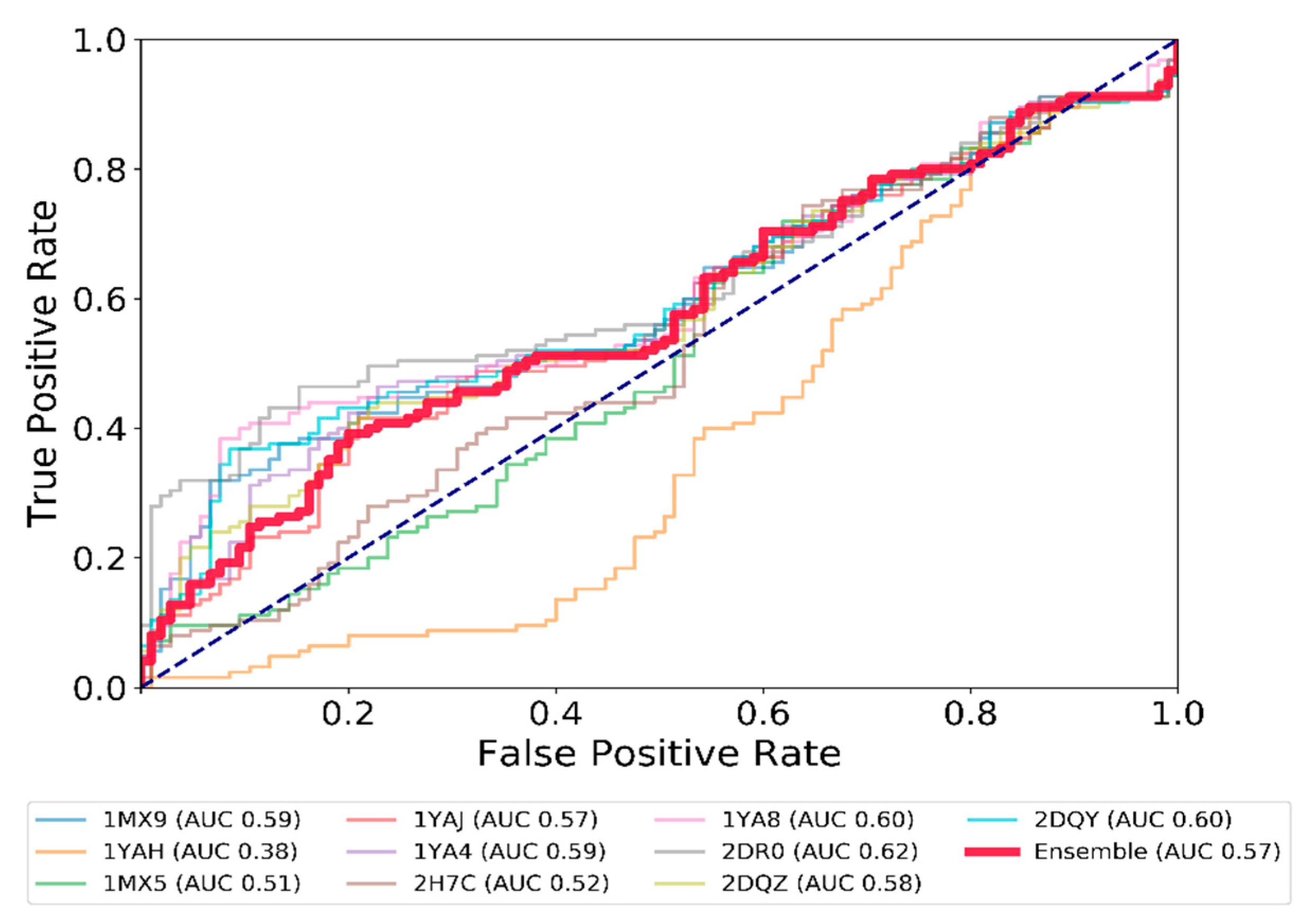
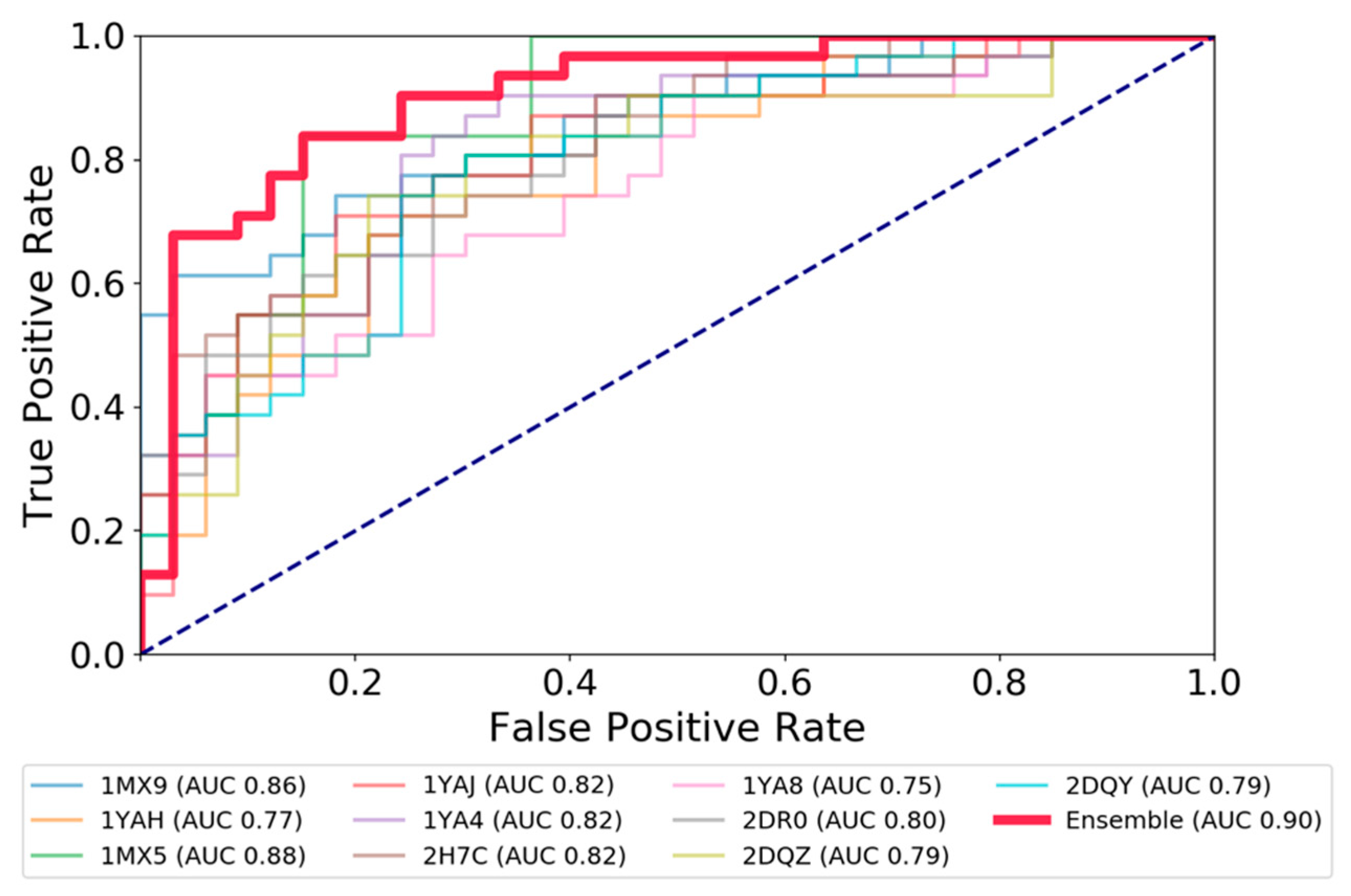
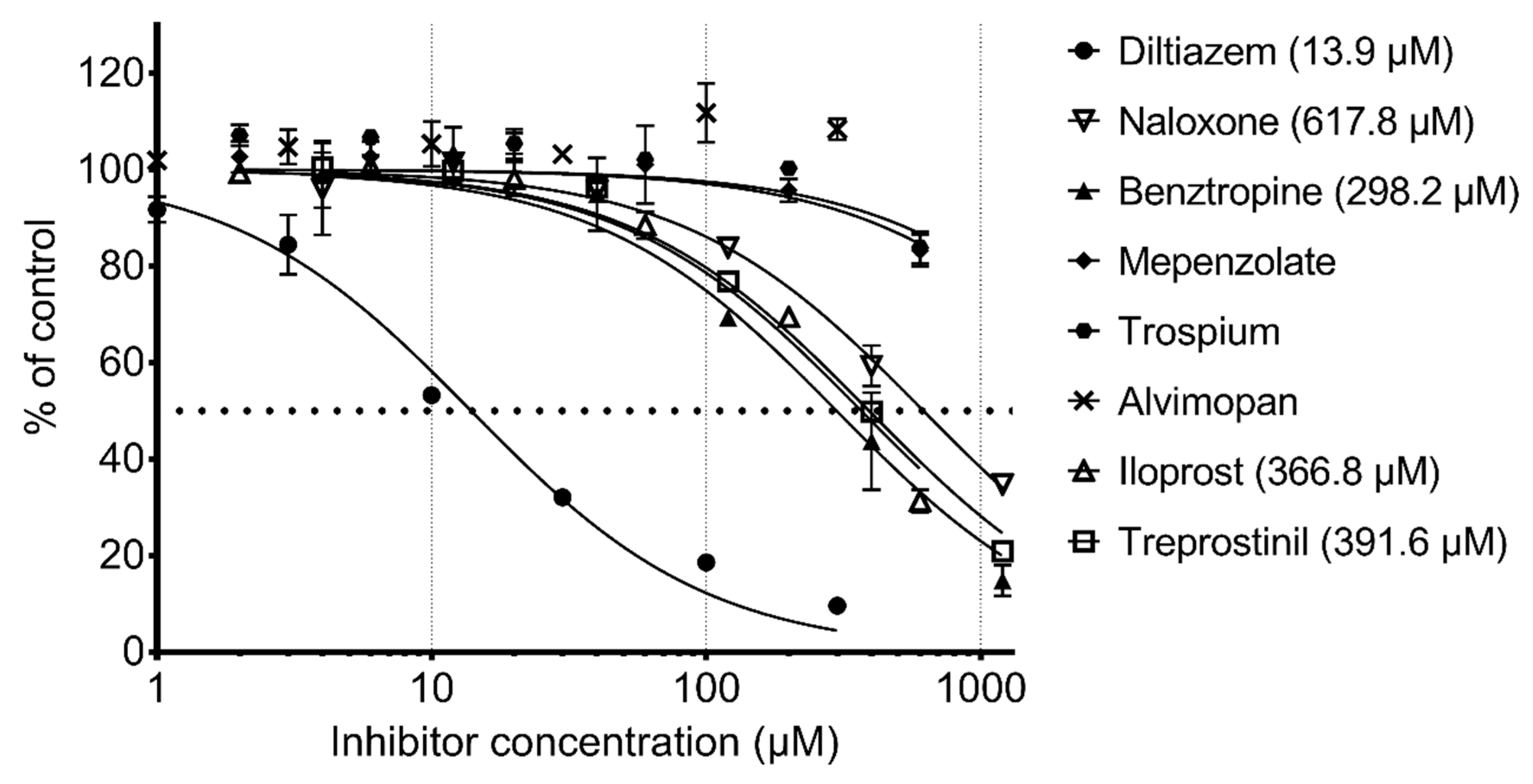
2.3. Test and Validation of the Protocol to Predict CES1 Inhibitors
3. Discussion
4. Materials and Methods
4.1. Collection and Curation of the Chemical Library
4.2. Protein Preparation and Docking Protocol
4.3. Docking Protocol
4.4. Machine Learning Models
4.5. In Vitro Assessment of CES1 Inhibition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
- Henrik Berg Rasmussen, Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark.
- Ditte Bjerre, Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark
- Majbritt Busk Madsen, Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark.
- Laura Ferrero, Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark.
- Kristian Linnet, Section of Forensic Chemistry, Department of Forensic Medicine, Faculty of Health Sciences, University of Copenhagen, Denmark.
- Ragnar Thomsen, Section of Forensic Chemistry, Department of Forensic Medicine, Faculty of Health Sciences, University of Copenhagen, Denmark.
- Gesche Jürgens, Clinical Pharmacological Unit, Zealand University Hospital Roskilde, Denmark.
- Kim Dalhoff, Department of Clinical Pharmacology, Bispebjerg University Hospital, Copenhagen, Denmark.
- Claus Stage, Department of Clinical Pharmacology, Bispebjerg University Hospital, Copenhagen, Denmark.
- Hreinn Stefansson, CNS Division, deCODE Genetics, Reykjavik, Iceland.
- Thomas Hankemeier, The Leiden/Amsterdam Center for Drug Research LACDR, Leiden University, Gorlaeus laboratories, Leiden, The Netherlands.
- Rima Kaddurah-Daouk, Department of Psychiatry and Behavioral Sciences, Duke University, Durham, NC, USA.
- Søren Brunak, Center for Biological Sequence Analysis, Technical University of Denmark, Kgs. Lyngby, Denmark.
- Olivier Taboureau, Center for Biological Sequence Analysis, Technical University of Denmark, Kgs. Lyngby, Denmark.
- Grace Shema Nzabonimpa, Center for Biological Sequence Analysis, Technical University of Denmark, Kgs. Lyngby, Denmark.
- Tine Houmann, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Pia Jeppesen, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Kristine Kaalund-Jørgensen, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Peter Riis Hansen, Department of Cardiology, Copenhagen University Hospital, Hellerup, Denmark.
- Karl Emil Kristensen, Department of Cardiology, Copenhagen University Hospital, Hellerup, Denmark.
- Anne Katrine Pagsberg, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Kerstin Plessen Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Poul-Erik Hansen, Department of Science, Systems and Models, Roskilde University, Roskilde, Denmark.
- Thomas Werge, Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark.
- Jørgen Dyrborg, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Maj-Britt Lauritzen, Centre for Child and Adolescent Mental Health, Mental Health Services in the Capital Region of Denmark.
- Tim Hughes, Department of Medical Genetics, Oslo; NORMENT, Institute of Clinical Medicine, University of Oslo, Oslo
Conflicts of Interest
References
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandreli, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Tatonetti, N.P.; Liu, T.; Altman, R.B. Predicting drug side-effects by chemical systems biology. Genome Biol. 2009, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Dumbreck, S.; Flynn, A.; Nairn, M.; Wilson, M.; Treweek, S.; Mercer, S.W.; Alderson, P.; Thompson, A.; Payne, K.; Guthrie, B. Drug-disease and drug-drug interactions: Systematic examination of recommendations in 12 UK national clinical guidelines. BMJ 2015, 350, h949. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, M. Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs. Molecules 2008, 13, 412–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, R.S.; Wright, M.W.; Laulederkind, S.J.; Cox, L.A.; Hosokawa, M.; Imai, T.; Ishibashi, S.; Lehner, R.; Miyazaki, M.; Perkins, E.J.; et al. Recommended nomenclature for five mammalian carboxylesterase gene families: Human, mouse, and rat genes and proteins. Mamm. Genome. 2010, 21, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Potter, P.M. Mammalian carboxylesterases: From drug targets to protein therapeutics. Drug Discov. Today 2005, 10, 313–325. [Google Scholar] [CrossRef]
- Sun, Z.; Murry, D.J.; Sanghani, S.P.; Davis, W.I.; Kedishvilli, N.Y.; Zou, Q.; Hurley, T.D.; Bosron, W.F. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J. Pharmacol. Exp. Ther. 2004, 310, 469–476. [Google Scholar] [CrossRef]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef]
- Humerickhouse, R.; Lohrbach, K.; Li, L.; Bosron, W.F.; Dolan, M.E. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000, 60, 1189–1192. [Google Scholar]
- Tabata, T.; Katoh, M.; Tokudome, S.; Nakajima, M.; Yokoi, T. Identification of the cytosolic carboxylesterase catalyzing the 5′-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab. Dispos. 2004, 32, 1103–1110. [Google Scholar] [CrossRef]
- Shi, D.; Yang, J.; Yang, D.; LeCluyse, E.L.; Black, C.; You, L.; Akhlaghi, F.; Yan, B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J. Pharmacol. Exp. Ther. 2006, 319, 1477–1484. [Google Scholar] [CrossRef]
- Pindel, E.V.; Kedishvili, N.Y.; Abraham, T.L.; Brzezinski, M.R.; Zhang, J.; Dean, R.A.; Borson, W.F. Purification and cloning of a broad substrate specificity human liver carboxylesterase that catalyzes the hydrolysis of cocaine and heroin. J. Biol. Chem. 1997, 272, 14769–14775. [Google Scholar] [CrossRef]
- Zhang, J.; Burnell, J.C.; Dumaual, N.; Bosron, W.F. Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J. Pharmacol. Exp. Ther. 1999, 290, 314–318. [Google Scholar]
- Ghosh, S. Cholesteryl ester hydrolase in human monocyte/macrophage: Cloning, sequencing, and expression of full-length cDNA. Physiol.Genomics. 2000, 2, 1–8. [Google Scholar] [CrossRef]
- Streit, T.M.; Borazjani, A.; Lentz, S.E.; Wierdl, M.; Potter, P.M.; Gwaltney, S.R.; Ross, M.K. Evaluation of the “side door” in carboxylesterase-mediated catalysis and inhibition. Biol. Chem. 2008, 389, 149–162. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C.L.; Howard-Williams, E.L.; Danks, M.K.; Potter, P.M.; Redinbo, M.R. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat. Struct. Biol. 2002, 9, 337–342. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C.L.; Xue, Y.; Potter, P.M.; Redinbo, M.R. Structural basis of heroin and cocaine metabolism by a promiscuous human drug processing enzyme. Nat. Struct. Biol. 2003, 10, 349–356. [Google Scholar] [CrossRef]
- Fleming, C.D.; Bencharit, S.; Edwards, C.C.; Hyatt, J.L.; Tsurkan, L.; Bai, F.; Fraga, C.; Morton, C.L.; Howard-Williams, E.L.; Potter, P.M.; et al. Structural insights into drug processing by human carboxylesterase 1: Tamoxifen, mevastatin, and inhibition by benzil. J. Mol. Biol. 2005, 352, 165–177. [Google Scholar] [CrossRef]
- Vistoli, G.; Pedretti, A.; Mazzolari, A.; Testa, B. In silico prediction of human carboxylesterase-1 (hCES1) metabolism combining docking analyses and MD simulations. Bioorg. Med. Chem. 2010, 18, 320–329. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Otto, T.C.; Kirkpatrick, M.G.; Kovaleva, E.; Brown, S.; Buchman, G.; Cerasoli, D.M.; Sussman, J.L. Structure of recombinant human carboxylesterase 1 isolated from whole cabbage looper larvae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Arena de Souza, V.; Scott, D.J.; Nettleship, J.E.; Rahman, N.; Charlton, M.H.; Walsh, M.A.; Owens, R.J. Comparison of the Structure and Activity of Glycosylated and Aglycosylated Human Carboxylesterase 1. PLoS ONE 2015, 10, e0143919. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. The mammalian carboxylesterases: From molecules to functions. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef]
- Redinbo, M.R.; Bencharit, S.; Potter, P.M. Human carboxylesterase 1: From drug metabolism to drug discovery. Biochem. Soc. Trans. 2003, 31, 620–624. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. Structure, function and regulation of carboxylesterases. Chem. Biol. Interact. 2006, 162, 195–211. [Google Scholar] [CrossRef]
- Wheelock, C.E.; Severson, T.F.; Hammock, B.D. Synthesis of new carboxylesterase inhibitors and evaluation of potency and water solubility. Chem. Res. Toxicol. 2001, 14, 1563–1572. [Google Scholar] [CrossRef]
- Wheelock, C.E.; Colvin, M.E.; Uemura, I.; Olmstead, M.M.; Sanborn, J.R.; Nakagawa, Y.; Jones, A.D.; Hammock, B.D. Use of ab initio calculations to predict the biological potency of carboxylesterase inhibitors. J. Med. Chem. 2002, 45, 5576–5593. [Google Scholar] [CrossRef]
- Hyatt, J.L.; Stacy, V.; Wadkins, R.M.; Yoon, K.J.; Wierdl, M.; Edwards, C.C.; Zeller, M.; Hunter, A.D.; Danks, M.K.; Crundwell, G.; et al. Inhibition of carboxylesterases by benzil (diphenylethane-1,2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J. Med. Chem. 2005, 48, 5543–5550. [Google Scholar] [CrossRef]
- Hyatt, J.L.; Moak, T.; Hatfield, M.J.; Tsurkan, L.; Edwards, C.C.; Wierdl, M.; Danks, M.K.; Wadkins, R.M.; Potter, P.M. Selective inhibition of carboxylesterases by isatins, indole-2,3-diones. J. Med. Chem. 2007, 50, 1876–1885. [Google Scholar] [CrossRef]
- Casida, J.E.; Quistad, G.B. Serine hydrolase targets of organophosphorus toxicants. Chem. Biol. Interact. 2005, 157, 277–283. [Google Scholar] [CrossRef]
- Sogorb, M.A.; Vilanova, E. Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol Lett. 2002, 128, 215–228. [Google Scholar] [CrossRef]
- Wadkins, R.M.; Hyatt, J.L.; Wei, X.; Yoon, K.J.; Wierdl, M.; Edwards, C.C.; Morton, C.L.; Obenauer, J.C.; Damodaran, K.; Beroza, P.; et al. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J. Med. Chem. 2005, 48, 2906–2915. [Google Scholar] [CrossRef]
- Hicks, L.D.; Hyatt, J.L.; Moak, T.; Edwards, C.C.; Tsurkan, L.; Wierdl, M.; Ferreira, A.M.; Wadkins, R.M.; Potter, P.M. Analysis of the inhibition of mammalian carboxylesterases by novel fluorobenzoins and fluorobenzils. Bioorganic Med. Chem. 2007, 15, 3801–3817. [Google Scholar] [CrossRef] [Green Version]
- Rhoades, J.; Peterson, Y.K.; Zhu, H.J.; Appel, D.I.; Peloquin, C.; Markowitz, J.S. Prediction and in vitro evaluation of selected protease inhibitor antiviral drugs as inhibitors of carboxylesterase 1: A potential source of drug-drug interactions. Pharm. Res. 2012, 29, 972–982. [Google Scholar] [CrossRef]
- Zhu, H.J.; Appel, D.I.; Peterson, Y.K.; Wang, Z.; Markowitz, J.S. Identification of selected therapeutic agents as inhibitors of carboxylesterase 1: Potential sources of metabolic drug interactions. Toxicology 2010, 270, 59–65. [Google Scholar] [CrossRef]
- Vistoli, G.; Pedretti, A.; Mazzolari, A.; Testa, B. Homology modeling and metabolism prediction of human carboxylesterase-2 using docking analyses by GriDock: A parallelized tool based on AutoDock 4.0. J. Comput. Aided Mol. Des. 2010, 24, 771–787. [Google Scholar] [CrossRef]
- Stoddard, S.V.; Yu, X.; Potter, P.M.; Wadkins, R.M. In Silico Design and Evaluation of Carboxylesterase Inhibitors. J. Pest. Sci. 2010, 35, 240–249. [Google Scholar] [CrossRef]
- Zhu, H.J.; Patrick, K.S.; Yuan, H.J.; Wang, J.S.; Donovan, J.L.; DeVane, C.L.; Malcom, R.; Johnson, J.A.; Younblood, G.L.; Sweet, D.H.; et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: Clinical significance and molecular basis. Am. J. Hum. Genet. 2008, 82, 1241–1248. [Google Scholar] [CrossRef]
- Nemoda, Z.; Angyal, N.; Tarnok, Z.; Gadoros, J.; Sasvari-Szekely, M. Carboxylesterase 1 gene polymorphism and methylphenidate response in ADHD. Neuropharmacology 2009, 57, 731–733. [Google Scholar] [CrossRef]
- Nzabonimpa, G.S.; Rasmussen, H.B.; Brunak, S.; Taboureau, O. INDICES Consortium. Investigating the impact of missense mutations in hCES1 by in silico structure-based approaches. Drug Metab. Pers. Ther. 2016, 31, 97–106. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, O.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Tikhonova, I.G.; Sum, C.S.; Neumann, S.; Engel, S.; Raaka, B.M.; Costanzi, S.; Gershengom, M.C. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J. Med. Chem. 2008, 51, 625–633. [Google Scholar] [CrossRef]
- Barreiro, G.; Guimarães, C.R.W.; Tubert-Brohman, I.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L. Search for non-nucleoside inhibitors of HIV-1 reverse transcriptase using chemical similarity, molecular docking, and MM-GB/SA scoring. J. Chem. Inf. Model. 2007, 47, 2416–2428. [Google Scholar] [CrossRef]
- Wei, D.Q.; Zhang, R.; Du, Q.S.; Gao, W.N.; Li, Y.; Gao, H.; Wang, S.Q.; Zhang, X.; Li, A.X.; Sirois, S.; et al. Anti-SARS drug screening by molecular docking. Amino Acids 2006, 31, 73–80. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Hankemeier, T.; Scholl, E.H.; Baillie, R.; Harms, A.; Stage, C.; Dalhoff, K.P.; Jürgens, G.; Taboureau, O.; Nzabonimpa, G.S.; et al. Pharmacometabolomics informs about pharmacokinetic profile of methylphenidate. CPT Pharmacometrics Syst. Pharmacol. 2018, 7, 525–533. [Google Scholar] [CrossRef]
- Bencharit, S.; Edwards, C.C.; Morton, C.L.; Howard-Williams, E.L.; Kuhn, P.; Potter, P.M.; Redinbo, M.R. Multisite promiscuity in the processing of endogenous substrates by carboxylesterase 1. J. Mol. Biol. 2006, 363, 201–214. [Google Scholar] [CrossRef]
- Carugo, O. How large B-factors can be in protein crystal structures. BMC Bioinformatics 2018, 19, 61. [Google Scholar] [CrossRef]
- Deller, M.C.; Rupp, B. Models of protein-ligand crystal structures: Trust, but verify. J. Comput. Aided Mol. Des. 2015, 29, 817–836. [Google Scholar] [CrossRef]
- Alghamedy, F.; Bopaiah, J.; Jones, D.; Zhang, X.; Weiss, H.L.; Ellingson, S.R. Incorporating protein dynamics through ensemble docking in machine learning models to predict drug binding. AMIA Jt Summits Transl. Sci. Proc. 2018, 2017, 26–34. [Google Scholar]
- Quiroga, R.; Villareal, M.A. Vinardo: A scoring function based on autodock vina improves scoring, docking and virtual screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Thomsen, R.; Rasmussen, H.B.; Linnet, K. In vitro drug metabolism by human carboxylesterase 1: Focus on angiotensin-converting enzyme inhibitors. Drug Metab Dispos. 2014, 42, 126–133. [Google Scholar] [CrossRef]
- Fukami, T.; Takahashi, S.; Nakagawa, N.; Maruichi, T.; Nakajima, M.; Yokoi, T. In vitro evaluation of inhibitory effects of antidiabetic and antihyperlipidemic drugs on human carboxylesterase activities. Drug Metab Dispos. 2010, 38, 2173–2178. [Google Scholar] [CrossRef]
- Shimizu, M.; Fukami, T.; Nakajima, M.; Yokoi, T. Screening of specific inhibitors for human carboxylesterases or Arylacetamide. Drug. Metab. Dispo. 2014, 42, 1103–1109. [Google Scholar] [CrossRef]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov. 2015, 10, 1–13. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhaite, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Lovric, M.; Molero, J.M.; Kern, R. PySpark and RDKit: Moving towards big data in cheminformatics. Mol. Inform. 2019, 38, e1800082. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.T.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine learning in python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Threshold | 1 µM | 10 µM | 100 µM |
|---|---|---|---|
| Number of active ligands | |||
| of the 230 in the training set | 76 (33%) | 107 (46%) | 127 (55%) |
| of the 64 in the test set | 19 (30%) | 33 (52%) | 36 (56%) |
| Vina score (no statistical model) | |||
| Ensemble AUC (training set) | 0.54 | 0.57 | 0.63 |
| Best AUC among all individual structures | 0.58 | 0.62 | 0.66 |
| Ensemble MLP model | |||
| AUC (std. dev.) 5CV (training set) | 0.84 (0.05) | 0.81 (0.03) | 0.75 (0.06) |
| AUC (test set) | 0.82 | 0.90 | 0.92 |
| Matthews coeff. (test set) | 0.49 | 0.56 | 0.59 |
| Ensemble LDA model | |||
| AUC (std. dev.) 5CV (training set) | 0.78 (0.10) | 0.77 (0.08) | 0.81 (0.07) |
| AUC (test set) | 0.77 | 0.77 | 0.77 |
| Matthews coeff. (test set) | 0.38 | 0.34 | 0.43 |
| Ensemble QDA model | |||
| AUC (std. dev.) 5CV (training set) | 0.79 (0.12) | 0.77 (0.08) | 0.81 (0.10) |
| AUC (test set) | 0.76 | 0.78 | 0.75 |
| Matthews coeff. (test set) | 0.31 | 0.47 | 0.39 |
| Name | 2D Structure | IC50 (µM) | Pharmacological Action |
|---|---|---|---|
| Diltiazem |  | 13.9 | Cardiovascular diseases. Antihypertensive and vasodilating properties. |
| Naloxone |  | 617.8 | Indicated for the rapid reversal of symptoms of central nervous system depression in opioid overdose. |
| Benztropine |  | 298.2 | Treatment of Parkinson’s disease. |
| Mepenzolate |  | NS | Gastrointestinal disorders. Decrease gastric acid and pepsin secretion. |
| Iloprost |  | 366.8 | Pulmonary arterial hypertension. |
| Trospium |  | NS | Antispasmodic agent. |
| Alvimopan |  | NS | Drug for constipation. Accelerates the gastrointestinal recovery after bowel surgery. |
| Treprostinil |  | 391.6 | Pulmonary arterial hypertension. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Briand, E.; Thomsen, R.; Linnet, K.; Rasmussen, H.B.; Brunak, S.; Taboureau, O. Combined Ensemble Docking and Machine Learning in Identification of Therapeutic Agents with Potential Inhibitory Effect on Human CES1. Molecules 2019, 24, 2747. https://doi.org/10.3390/molecules24152747
Briand E, Thomsen R, Linnet K, Rasmussen HB, Brunak S, Taboureau O. Combined Ensemble Docking and Machine Learning in Identification of Therapeutic Agents with Potential Inhibitory Effect on Human CES1. Molecules. 2019; 24(15):2747. https://doi.org/10.3390/molecules24152747
Chicago/Turabian StyleBriand, Eliane, Ragnar Thomsen, Kristian Linnet, Henrik Berg Rasmussen, Søren Brunak, and Olivier Taboureau. 2019. "Combined Ensemble Docking and Machine Learning in Identification of Therapeutic Agents with Potential Inhibitory Effect on Human CES1" Molecules 24, no. 15: 2747. https://doi.org/10.3390/molecules24152747