
Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction


1
Institute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
2
Institute of General and Ecological Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(14), 2565; https://doi.org/10.3390/molecules24142565
Submission received: 5 June 2019
/
Revised: 8 July 2019
/
Accepted: 11 July 2019
/
Published: 15 July 2019
(This article belongs to the Collection Recent Advances in Organocatalysis)
Abstract
:In this manuscript, a novel, decarboxylative Michael reaction between α-substituted azlactones and chromone-3-carboxylic acids is described. The reaction proceeds in a sequence Michael addition followed by decarboxylative deprotonation, and it results in the formation of chromanones bearing an azlactone structural unit. The possibility of transforming an azlactone moiety into a protected α,α-disubstituted α-amino acid derivative is also demonstrated.

1. Introduction
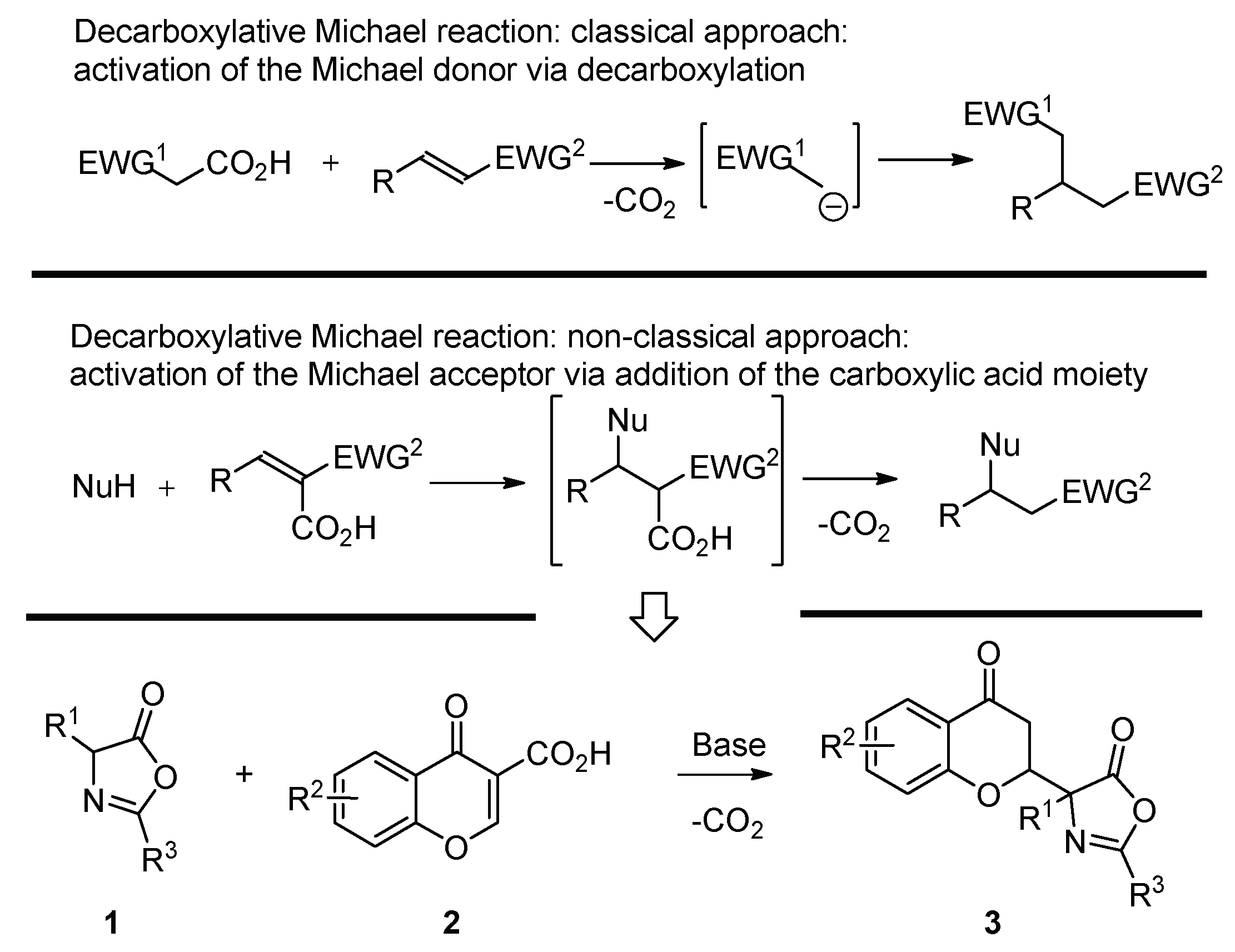
The Michael reaction constitutes one of the most fundamental C–C and C–X bond-forming reactions, allowing access to various useful building blocks for organic synthesis [1,2,3,4,5,6,7,8,9,10]. Its decarboxylative [11] variant has also been described in literature [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. The most common strategy utilized to realize the decarboxylative Michael reaction relies on the activation of Michael donors via decarboxylation (Scheme 1, top) [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30] with malonic acid half-thioesters (MAHT) and related systems [16,17,18,19,20,21,22,23]. The decarboxylation of these molecules leads to the generation of stabilized carbanions readily participating in the subsequent Michael addition. An alternative strategy relies on the activation of the Michael acceptor via the addition of the carboxylic acid group in its α-position (Scheme 1, middle) [31,32,33,34,35,36,37,38]. In such a manner, the electrophilic property of the Michael acceptor is enhanced with the carboxylic acid moiety being readily removed via the decarboxylation of the originally formed Michael adduct. Surprisingly, such decarboxylative Michael reactions [31,32,33,34,35,36,37,38] are very unique in literature, with their enantioselective variant reported, to the best of our knowledge, only in the case of doubly decarboxylative reactions involving MAHT as a Michael donor [23,38].
2. Results
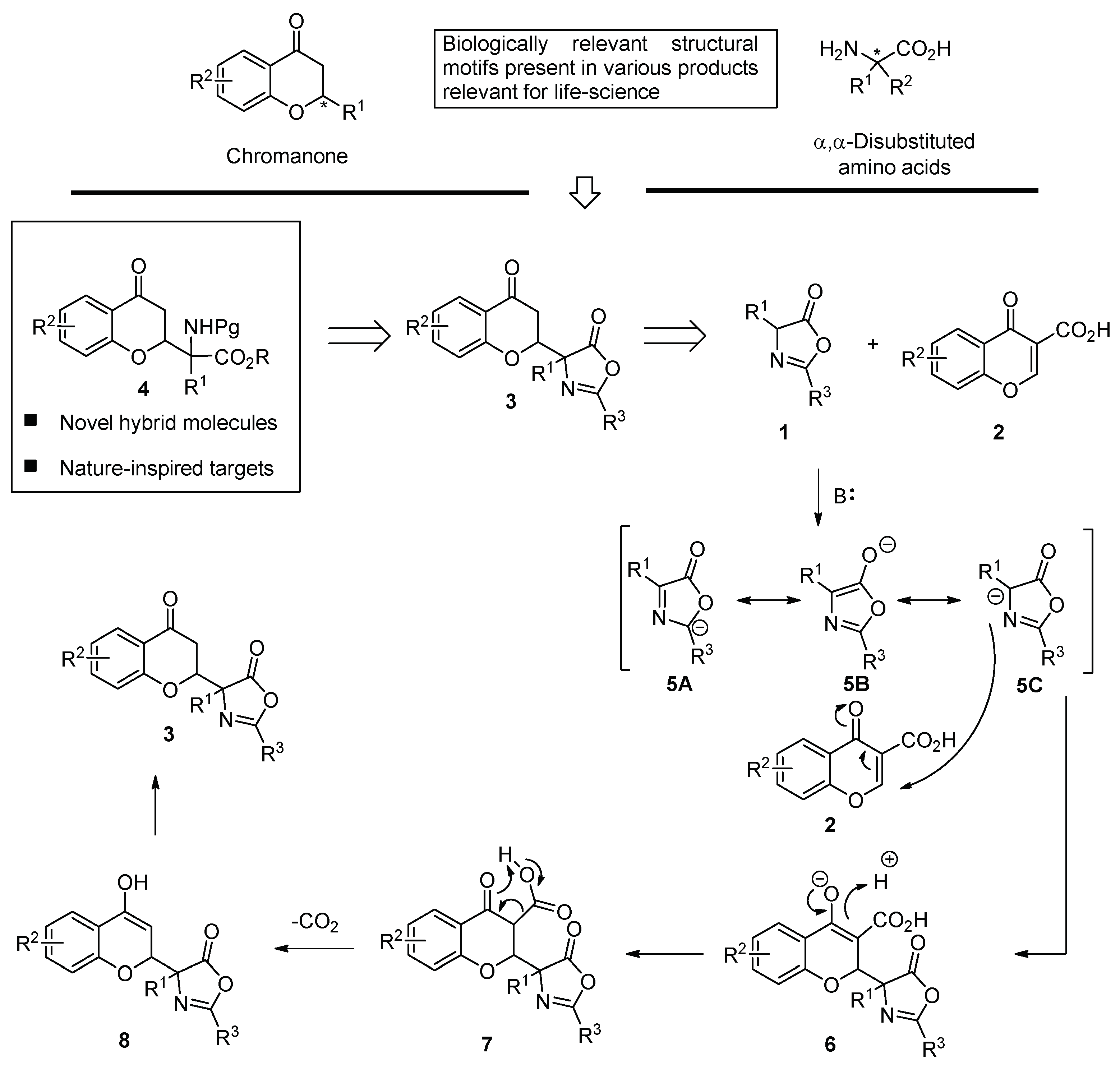
Chromanones and their related heterocyclic ring system (Scheme 2, top to the left) are widely distributed in natural products and biologically active molecules [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58]. Similarly, compounds with an incorporated α,α-disubstituted amino acid moiety exhibit diverse biological activity [59,60,61,62,63,64]. Therefore, given the importance of chromanones and α,α-disubstituted amino acids, the incorporation of both structural motifs into one hybrid molecule seemed like a very attractive synthetic task (Scheme 2, top). It was envisioned that 3 bearing an azlactone moiety will serve as a direct precursor of 4 due to the well-established ability of the oxazol-5-(4H)-one ring to be transformed into the corresponding α,α-disubstituted amino acid moiety [65,66,67,68,69,70,71,72]. It was anticipated that the products 3 should be accessible from α-substituted azlactone 1 and chromone-3-carboxylic acid 2 via a decarboxylative Michael reaction. The mechanism of this transformation is shown in the bottom of Scheme 2. It is initiated through the deprotonation of 1 to give an aromatic anion that is stabilized through a mesomeric effect. The subsequent addition of 5 to 2, acting as a Michael acceptor yields 6, which undergoes protonation to afford 7. The decarboxylation of 7 is the key step of the reaction, allowing for the removal of the activating group. The protonation of the enolate 8 thus obtained yields of the desired chromanone 3 bearing an α,α-disubstituted azlactone moiety. It was anticipated that the use of a chiral Brønsted base 9 as a catalyst of such decarboxylative Michael reaction should afford access to enantio- and diastereomerically enriched products [73,74,75,76,77].
Herein, we present our studies on the application of the decarboxylative Michael reaction for the enantioselective synthesis of biologically relevant chromanones bearing an α,α-disubstituted azlactone moiety. The possibility to transform the azlactone ring into a protected α,α-disubstituted amino acid has also been demonstrated.
Initially the Michael reaction between azlactone 1a and 4-chromone 2a was performed (Table 1, entry 1). However, no reaction was observed. To our delight, the incorporation of a carboxylic acid moiety into the structure of the Michael acceptor 2b resulted in the formation of the desired product 3a when quinine 9a was employed as a catalyst (Table 1, Entry 2). The reaction proceeded in a cascade manner, and the initial Michael addition was accompanied by the decarboxylative protonation. Disappointingly, while the diastereoselectivity of the process was good, its enantioselectivity was low. Therefore, a catalyst screening was performed using chromone-3-carboxylic acid 2b as a model Michael acceptor (Table 1, Entries 2–7). Interestingly, the introduction of double H-bonding units into the structure of the cinchona alkaloid (catalysts 9b–f) led to the improvement of reaction stereoselectivity. The best results were obtained when catalyst 9e was used (Table 1, Entry 6) [78]. With the best catalyst identified, the solvent screening was initiated (Table 1, Entries 8–13). However, inferior results were obtained. Subsequently, the effect of concentration (Table 1, Entries 14,15), the relative ratio of reactants (Table 1, Entries 16,17) and temperature (Table 1, Entry 18) on the reaction outcome was evaluated. Disappointingly, no further improvement of the results was observed. Notably, the reaction proved readily scalable with comperable results obtained when 1 g of 2b was used (Table 1, Entry 19).
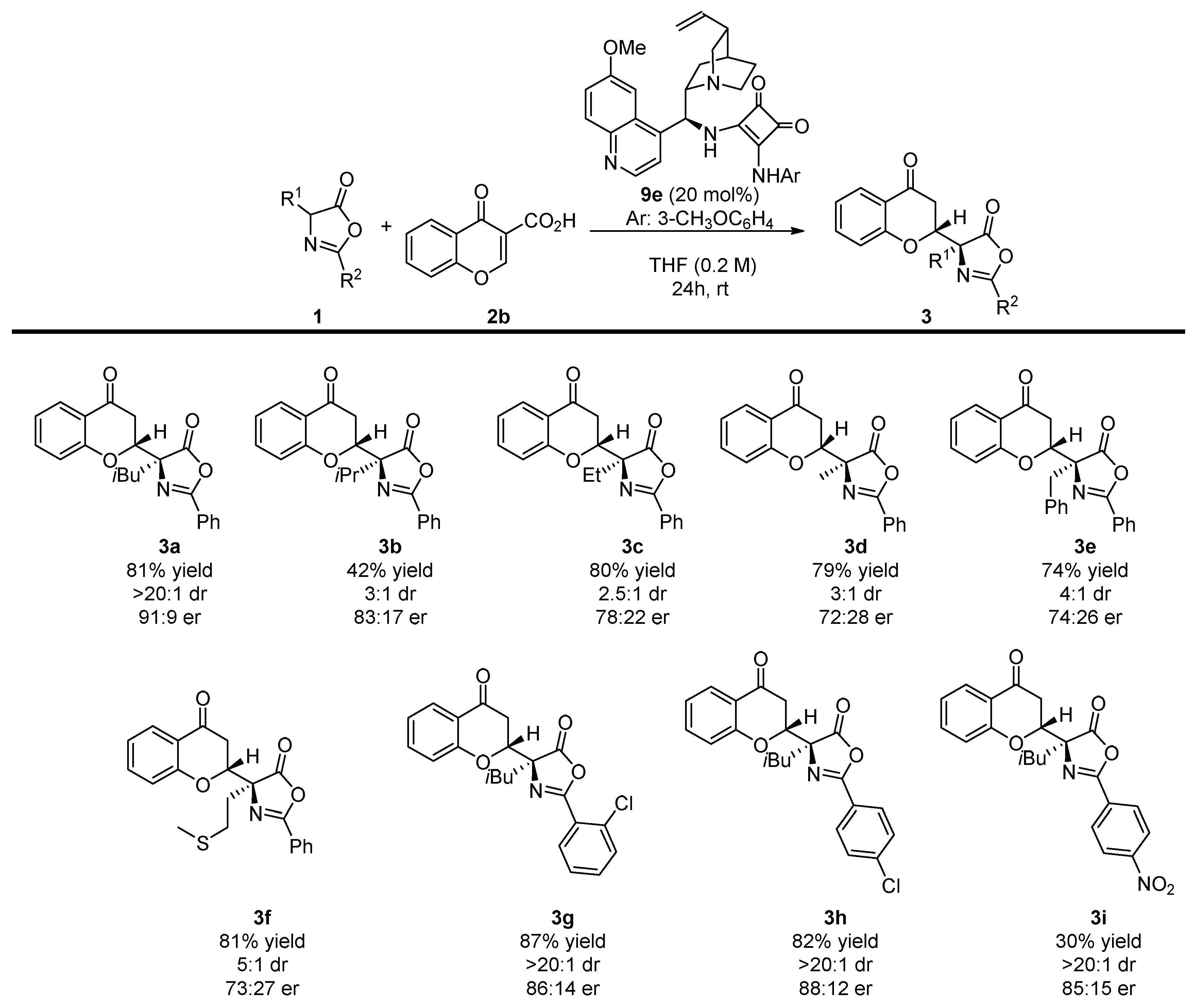
With the optimal reaction conditions established, the scope of the methodology was tested. Initially, various α-substituted azlactones 1 were employed in the developed decarboxylative Michael reaction (Scheme 3). In all of cases, the reaction proceeded with moderate-to-high yields. Disappointingly, regardless the size of the substituent in the α-position of the azlactone ring, lower diastereoselectivities were observed. Similarly, products 3b–f were obtained with a deteriorated enantiomeric enrichment when compared with the model product 3a. Interestingly, the reaction was successfully realized for azlactones 1g–i bearing various R2 groups.
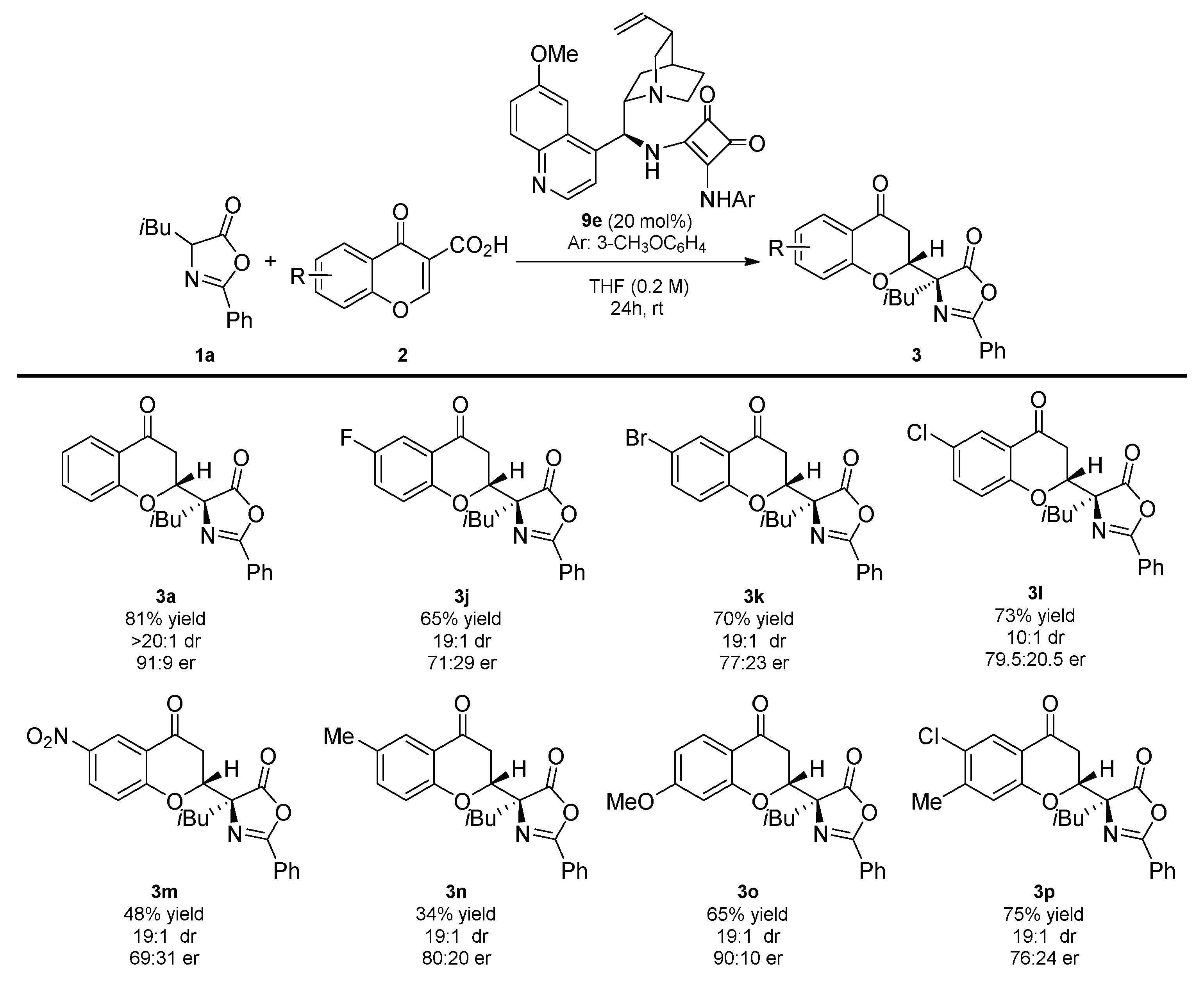
In the second part of the scope studies, the usefulness of various chromone-3-carboxylic acids 2 in the developed reaction was evaluated (Scheme 4). It was found that the diastereoselectivity of the decarboxylative Michael reaction was unbiased towards the electron properties and the position of the substituents on the aromatic ring in the corresponding chromone-3-carboxylic acid 2. Both electron-poor and electron-rich substituents were possibly present in 2, providing products 3 in good-to-high yields and excellent diastereoselectivity. Furthermore, the disubstitution pattern in 2h was also well-tolerated, as shown in the decarboxylative Michael reaction leading to 3p. In all of cases, the enantioselectivity of the process was lower than for the model reaction.
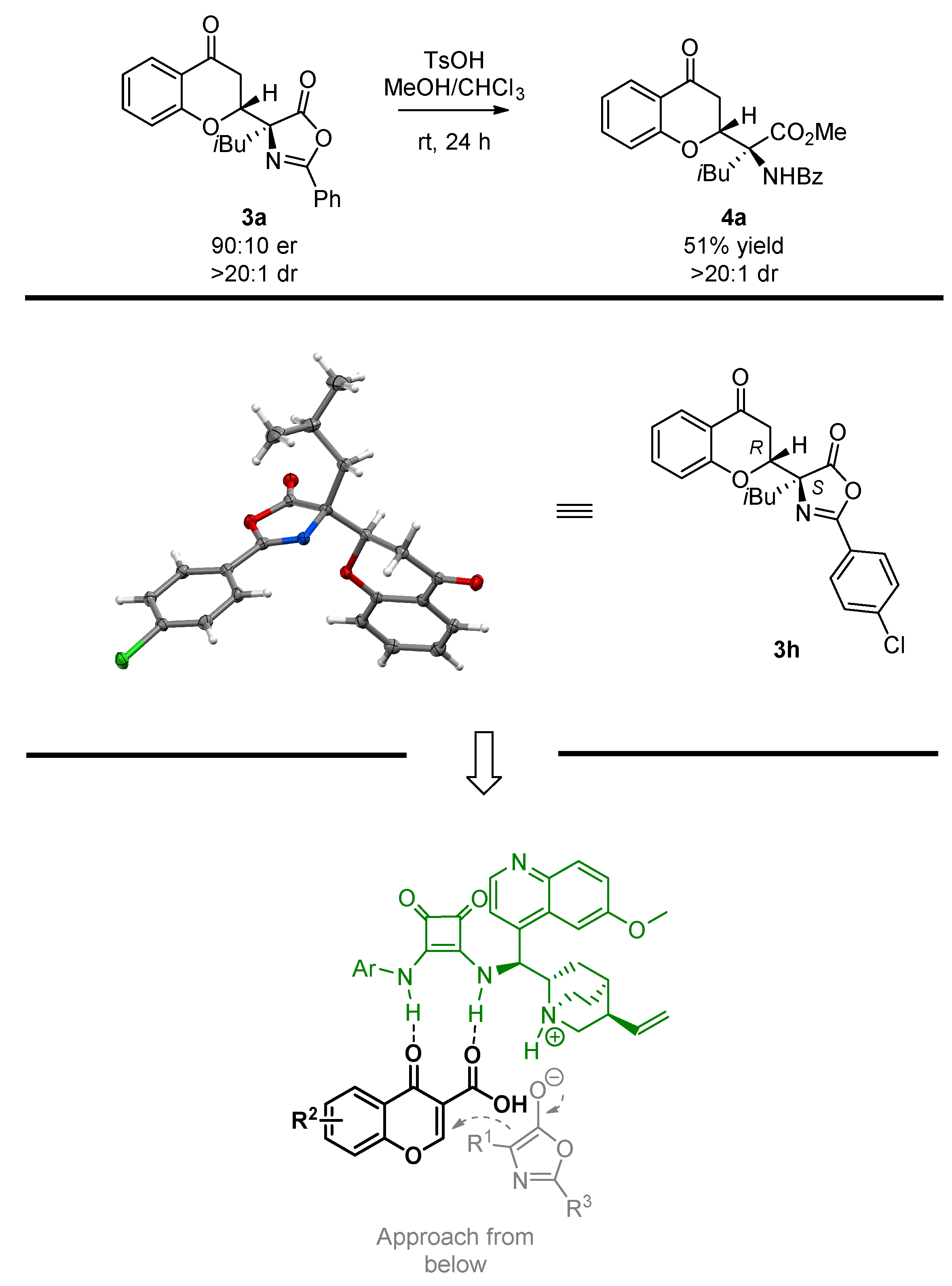
To demonstrate the usefulness of the Michael adduct 3a obtained for the synthesis of α,α-disubstituted amino acids, the azlactone-ring-opening was attempted (Scheme 5, top). It was found that under acidic conditions, product 4a bearing an α,α-disubstituted amino acid moiety was obtained in a 51% yield. Notably, the reaction proceeded with the full preservation of the stereochemical information introduced at the decarboxylative Michael addition step, as 4a was obtained as single diastereoisomer. In the course of further studies, the absolute configuration of the Michael adduct 3h was unambiguously established through single crystal X-ray analysis (Scheme 5, middle) [79]. Notably, the absolute configuration of the remaining products was established by analogy. Given the assignment performed, a transition state model rationalizing the observed stereochemistry was proposed (Scheme 5, bottom). It is postulated that the corresponding chromone-3-carboxylic acid 2 was recognized by the catalyst 9e through the H-bonding interaction with its squaramide moiety. At the same time, azlactone 1 was deprotonated by the tertiary amine moiety present in the quinuclidine ring of 9e. As the result of the ion pair formation between the protonated catalyst and the enolate obtained, the Michael addition occurred in a stereoselective fashion.
3. Conclusion
In conclusion, we have developed a novel, decarboxylative reaction between α-substituted azlactones and chromone-3-carboxylic acids leading to biologically relevant chromanones bearing an azlactone moiety. Its ring-opening realized under acidic conditions constitutes a facile route to protected α,α-disubstituted α-amino acid derivatives. The activation of the Michael acceptor through the introduction of a carboxylic acid moiety proved both necessary and a very convenient means to achieve the desired reactivity pathway.
4. Materials and Methods
4.1. General Methods
NMR spectra were acquired on a Bruker Ultra Shield 700 instrument (Bruker Corporation, Billerica, MA, USA), running at 700 MHz for 1H and 176 MHz for 13C, respectively. Chemical shifts (δ) were reported in ppm relative to residual solvent signals (CDCl3: 7.26 ppm for 1H NMR, 77.16 ppm for 13C NMR). Mass spectra were recorded on a Bruker Maxis Impact spectrometer using electrospray (ES+) ionization (referenced to the mass of the charged species). Analytical thin layer chromatography (TLC) was performed using pre-coated aluminum-backed plates (Merck Kieselgel 60 F254) and visualized by the ultraviolet irradiation or I2 stain. Unless otherwise noted, analytical grade solvents and commercially available reagents were used without further purification. For flash chromatography (FC), silica gel (Silica gel 60, 230–400 mesh, Merck, Darmstadt, Germany) was used. The enantiomeric ratio (er) of the products were determined either by ultra performance convergence chromatography (UPC2) using Daicel Chiralpak IA and IG columns as chiral stationary phases or by chiral stationary phase HPLC (Daicel Chiralpak IF column). Azlactones 1 were synthetized according to the literature procedure [80]. Chromone-3-carboxylic acids 2 were prepared from the corresponding 2-hydroxyacetophenones following the literature procedure [81].
4.2. General Procedure
An ordinary screw-cap vial was charged with a magnetic stirring bar, the corresponding chromone-3-carboxylic acid 2 (0.1 mmol, 1 equivalent), THF (0.2 mL), catalyst 9e (0.02 mmol, 0.2 equivalent), and the corresponding azlactone 1 (0.1 mmol, 1 equivalent). The reaction mixture was stirred at room temperature and monitored by 1H NMR spectroscopy. After the complete consumption of the carboxylic acid 2, the mixture was directly subjected to FC on silica gel (hexane:ethyl acetate 15:1 or 10:1) to afford pure product 3.
(S)-4-Isobutyl-((R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3a) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 10:1) as yellow crystals (m.p. 124–126 °C) in an 81% yield (29.8 mg), dr > 20:1. Major diastereoisomer: IR (film): 3072, 1813, 1691, 1652, 1603, 1463, 1307, 1223, 995, 884, 760 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.03 (d, J = 7.8 Hz, 2H), 7.84 (t, J = 10.2 Hz, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.87 (t, J = 8.4 Hz, 1H), 4.74 (dd, J = 13.0, 2.9 Hz, 1H), 3.22 (dd, J = 16.9, 13.0 Hz, 1H), 2.92 (dd, J = 16.9, 2.9 Hz, 1H), 1.93 (dd, J = 13.8, 6.3 Hz, 1H), 1.84 (dd, J = 13.8, 6.5 Hz, 1H), 1.66–1.60 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 191.1, 178.6, 161.7, 160.6, 136.3, 133.2, 129.0 (2C), 128.4 (2C), 126.9, 125.7, 122.0, 121.0, 118.0, 80.7, 75.9, 41.1, 38.1, 24.8, 24.0, 23.5. HRMS: Calculated for [C22H21NO4+H+]: 364.1543, found: 364.154. The er was determined by HPLC using a Chiralpak IF column [hexane/i-PrOH (80:20)]; flow rate 1.0 mL/min; τmajor = 6.3 min; τminor = 10.0 min, (91:9 er).
((R)-4-Oxochroman-2-yl)-2-phenyl-(S)-4-isopropan-2-yl-1,3-oxazol-5(4H)-one (3b) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 10:1) as yellow crystals (m.p. 121–122 °C) in a 42% yield (14.7 mg), dr = 3:1. IR (film): 2922, 1813, 1691, 1653, 1605, 1463, 1229, 1180, 993, 881, 763, 700 cm−1. 1H NMR (700 MHz, CDCl3) Major diastereoisomer: δ 8.09 (d, J = 7.3 Hz, 2H), 7.86 (t, J = 7.9 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.41 (t, J = 8.7 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 4.90 (dd, J = 14.0, 2.6 Hz, 1H), 3.31 (dd, J = 16.8, 14.0 Hz, 1H), 2.77 (dd, J = 16.8, 2.6 Hz, 1H), 2.44 (hept, J = 6.9 Hz, 1H), 1.13 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H). Minor diastereoisomer: δ 8.03 (d, J = 7.3 Hz, 2H), 7.86 (t, J = 7.9 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.40 (t, J = 7.6 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 5.03 (dd, J = 13.2, 2.8 Hz, 1H), 3.21 (dd, J = 16.9, 13.3 Hz, 1H), 2.86 (dd, J = 16.9, 2.9 Hz, 1H), 2.38 (hept, J = 6.9 Hz, 1H), 1.18 (d, J = 7.0 Hz, 3H), 0.95 (d, J = 6.5 Hz, 3H). 13C NMR (176 MHz, CDCl3) Major diastereoisomer: δ 191.1, 177.6, 162.2, 160.7, 136.2, 133.2, 129.0 (2C), 128.4 (2C), 127.0, 125.7, 122.2, 121.1, 118.2, 78.6, 78.1, 37.0, 31.6, 17.2, 16.7. Minor diastereoisomer: δ 191.1, 177.0, 161.9, 160.9, 136.3, 133.1, 128.9 (2C), 128.3 (2C), 127.0, 125.6, 122.0, 121.1, 118.1, 78.6, 78.4, 37.9, 31.3, 17.3, 15.7. HRMS: Calculated for [C21H19NO4+H+]: 350.1387, found: 350.1380. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.52 min, τminor = 2.60 min, (83:17 er).
(S)-4-Ethyl-(R)-4-oxochroman-2-yl-2-phenyl-1,3-oxazol-5(4H)-one (3c) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow crystals (m.p. 122–124 °C) in an 80% yield (26.8 mg), dr = 2.5:1. IR (film): 2960, 1816, 1691, 1654, 1604, 1463, 1227, 1152, 994, 882, 761 cm−1. 1H NMR (700 MHz, CDCl3) Major diastereoisomer: δ 8.04 (d, J = 7.7 Hz, 2H), 7.85 (d, J = 8.1 Hz, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.50 (t, J = 7.7 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 4.83 (dd, J = 13.2, 2.8 Hz, 1H), 3.25 (dd, J = 16.8, 13.2 Hz, 1H), 2.90 (dd, J = 16.9, 2.8 Hz, 1H), 1.97 (q, J = 7.3 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). Minor diastereoisomer: 1H NMR (700 MHz, CDCl3) δ 8.07 (d, J = 7.7 Hz, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.7 Hz, 2H), 7.44 (t, J = 7.2 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 4.78 (dd, J = 13.8, 2.6 Hz, 1H), 3.21 (dd, J = 16.7, 13.8 Hz, 1H), 2.75 (dd, J = 16.7, 2.6 Hz, 1H), 2.13 (dq, J = 14.6, 7.4 Hz, 1H), 2.05 (dq, J = 14.4, 7.3 Hz, 1H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (176 MHz, CDCl3) Major diastereoisomer: δ 191.0, 177.9, 162.0, 160.6, 136.2, 133.2, 128.9 (2C), 128.4 (2C), 126.9, 125.6, 122.0, 121.0, 118.0, 80.0, 76.8, 38.0, 26.0, 7.7. Minor diastereoisomer: δ 190.8, 177.3, 162.0, 160.6, 136.2, 133.2, 129.0 (2C), 128.4 (2C), 127.0, 125.5, 122.2, 121.0, 118.2, 79.1, 76.0, 37.5, 26.4, 8.0. HRMS: Calculated for [C20H17NO4+H+]: 336.1230, found: 336.1239. The er was determined by UPC2 using a chiral Chiralpack IG column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.66 min, τminor = 3.40 min, (78:22 er).
(S)-4-Methyl-2-((R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3d) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow crystals (m.p. 112–113 °C) in a 79% yield (25.4 mg) dr = 3:1. IR (film): 3058, 1817, 1692, 1650, 1607, 1464, 1307, 1225, 1154, 993, 880, 762 cm−1. 1H NMR (700 MHz, CDCl3) Major diastereoisomer: δ 8.03 (d, J = 7.8 Hz, 2H), 7.86 (d, J = 7.9 Hz, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 4.80 (dd, J = 13.1, 2.8 Hz, 1H), 3.25 (dd, J = 16.8, 13.2 Hz, 1H), 2.93 (dd, J = 16.8, 2.9 Hz, 1H), 1.56 (s, 3H). Minor diastereoisomer: δ 8.05 (d, J = 7.7 Hz, 2H), 7.86 (d, J = 8.8 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.52 (t, J = 7.7 Hz, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 4.73 (dd, J = 13.9, 2.3 Hz, 1H), 3.21 (dd, J = 16.5, 14.1 Hz, 1H), 2.75 (dd, J = 16.7, 2.4 Hz, 1H), 1.64 (s, 3H). 13C NMR (176 MHz, CDCl3) Major diastereoisomer: δ 190.9, 178.5, 161.7, 160.6, 136.3, 133.2, 128.9 (2C), 128.3 (2C), 126.9, 125.7, 122.1, 121.0, 118.0, 80.2, 72.0, 37.6, 19.5. Minor diastereoisomer: δ 190.8, 177.6, 161.9, 160.5, 136.3, 133.3, 129.0 (2C), 128.3 (2C), 127.0, 125.6, 122.2, 120.9, 118.2, 79.5, 71.3, 37.4, 19.9. HRMS: Calculated for [C19H15NO4+H+]: 322.1074, found: 322.1077. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor =2.58 min, τminor = 2.79 min, (72:28 er).
(S)-4-Benzyl-(R)-4-oxochroman-2-yl-2-phenyl-1,3-oxazol-5(4H)-one (3e) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow crystals (m.p. 124–126 °C) in a 74% yield (29.4 mg), dr = 4:1. Major diastereoisomer: IR (film): 3033, 1817, 1688, 1652, 1603, 1459, 1299, 1225, 1106, 993, 764, 696 cm−1. 1H NMR (700 MHz, CDCl3) δ 7.88–7.86 (m, 3H), 7.54 (t, J = 7.5 Hz, 1H), 7.44–7.40 (m, 3H), 7.20–7.15 (m, 5H), 7.01 (t, J = 7.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 4.96 (dd, J = 13.2, 2.9 Hz, 1H), 3.33 (dd, J = 16.7, 13.2 Hz, 1H), 3.26 (d, J = 13.2 Hz, 1H), 3.16 (d, J = 13.2 Hz, 1H), 3.00 (dd, J = 16.8, 2.9 Hz, 1H). 13C NMR (176 MHz, CDCl3) δ 190.8, 177.0, 161.8, 160.6, 136.3, 133.0, 132.8, 130.4 (2C), 128.8 (2C), 128.5 (2C), 128.2 (2C), 127.8, 127.0, 125.5, 122.1, 121.1, 118.1, 79.9, 77.4, 39.0, 38.2. HRMS: calculated for [C25H19NO4+H+]: 398.1387, found: 398.1381. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 3.29 min, τminor = 4.04 min, (74:26 er).
(S)-4-(2-(Methylthio)ethyl)-2-((R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3f) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as colorless solid (m.p. 146–148 °C) in an 81% yield (30.8 mg), dr = 5:1. Major diastereoisomer: IR (film): 2957, 1818, 32 4 1651, 1603, 1459, 1297, 1225, 993, 893, 762, 696 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.03 (d, J = 7.8 Hz, 2H), 7.86 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.50 (t, J = 7.7 Hz, 2H), 7.40 (t, J = 7.8 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.81 (dd, J = 13.0, 2.9 Hz, 1H), 3.22 (dd, J = 16.8, 13.0 Hz, 1H), 2.91 (dd, J = 16.8, 3.0 Hz, 1H), 2.48 (ddd, J = 13.1, 9.6, 4.8 Hz, 1H), 2.39 (ddd, J = 13.1, 10.0, 6.7 Hz, 1H), 2.30–2.21 (m, 2H), 2.08 (s, 3H). 13C NMR (176 MHz, CDCl3) δ 190.7, 177.8, 162.6, 160.5, 136.3, 133.3, 128.9 (2C), 128.4 (2C), 127.0, 125.5, 122.2, 121.0, 118.0, 80.0, 75.3, 38.1, 32.0, 28.2, 15.4. HRMS: calculated for [C21H19NO4S+H+]: 382.1108, found: 382.1109. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.95 min, τminor = 3.22 min, (73:27 er).
(S)-4-Isobutyl-(2-chlorophenyl)-((R)-4-oxochroman-2-yl)-2-1,3-oxazol-5(4H)-one (3g) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 20:1) as yellow oil in an 87% yield (34.5 mg), dr = >20:1. Major diastereoisomer: IR (film): 3074, 1815, 1690, 1652, 1605, 1579, 1467, 1256, 1228, 995, 884, 762, 735 cm−1. 1H NMR (700 MHz, CDCl3) δ 7.85 (dt, J = 7.8, 1.6 Hz, 1H), 7.79 (dd, J = 7.8, 1.8 Hz, 1H), 7.55–7.49 (m, 1H), 7.47 (tt, J = 8.0, 1.4 Hz, 1H), 7.42 (ddd, J = 8.7, 5.2, 1.8 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.91 (dd, J = 8.5, 2.9 Hz, 1H), 4.75 (dd, J = 13.2, 2.9 Hz, 1H), 3.29–3.15 (m, 1H), 2.93 (dd, J = 16.9, 2.9 Hz, 1H), 1.97 (dd, J = 13.8, 5.9 Hz, 1H), 1.85 (dd, J = 13.8, 6.9 Hz, 1H), 1.72 (dt, J = 13.1, 6.6 Hz, 1H), 0.95 (ddd, J = 19.0, 6.7, 1.7 Hz, 6H). 13C NMR (176 MHz, CDCl3) δ 190.6, 178.0, 160.4, 160.4, 136.2, 134.0, 133.0, 131.4, 131.2, 126.8, 126.8, 125.3, 122.0, 120.8, 117.8, 80.4, 75.8, 40.7, 38.0, 24.6, 23.9, 23.1. HRMS: Calculated for [C22H20ClNO4+H+]: 398, 1154, found: 398.1135. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.70 min, τminor = 3.20 min, (86:14 er).
(S)-4-Isobutyl-(4-chlorophenyl)-((R)-4-oxochroman-2-yl)-2-1,3-oxazol-5(4H)-one (3h) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 20:1) as colorless crystals (m.p. 188–190 °C) in an 82% yield (32.6 mg), dr = >20:1. Major diastereoisomer: IR (film): 3076, 1816, 1691, 1652, 1605, 1579, 1463, 1278, 1227, 994, 897, 761, 734 cm−1. 1H NMR (700 MHz, CDCl3) δ 7.95 (d, J = 8.7 Hz, 2H), 7.83 (dd, J = 7.8, 1.8 Hz, 1H), 7.46 (d, J = 8.7 Hz, 2H), 7.38 (ddd, J = 8.7, 7.2, 1.8 Hz, 1H), 6.97 (ddd, J = 8.0, 7.2, 1.0 Hz, 1H), 6.86 (dd, J = 8.4, 0.9 Hz, 1H), 4.75 (dd, J = 12.7, 3.0 Hz, 1H), 3.19 (dd, J = 16.8, 12.7 Hz, 1H), 2.92 (dd, J = 16.9, 3.0 Hz, 1H), 1.87 (ddd, J = 57.0, 13.9, 6.4 Hz, 2H), 1.61 (dt, J = 13.1, 6.6 Hz, 1H), 0.90 (t, J = 6.5 Hz, 6H). 13C NMR (176 MHz, CDCl3) δ 190.6, 178.1, 160.8, 160.3, 139.5, 136.1, 129.5 (2C), 129.2 (2C), 126.8, 124.0, 121.9, 120.9, 117.8, 80.5, 76.0, 40.9, 37.9, 24.7, 23.8, 23.3. HRMS: Calculated for [C22H20ClNO4+H+]: 398.1154, found: 398.1165. The er was determined by UPC2 using a chiral Chiralpack IG column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.57 min, τminor = 3.40 min, (88:12 er).
(S)-4-Isobutyl-2-(4-nitrophenyl)((R)-4-oxochroman-2-yl)-1,3-oxazol-5(4H)-one (3i) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow oil in a 30% yield (12.2 mg), dr = >20:1. Major diastereoisomer: IR (film): 3074, 1815, 1690, 1652, 1605, 1552, 1467, 1256, 1228, 995, 762, 736 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.34 (d, J = 8.9 Hz, 2H), 8.31–8.13 (m, 2H), 7.85 (dd, J = 7.9, 1.7 Hz, 1H), 7.39 (ddd, J = 8.8, 7.2, 1.8 Hz, 1H), 6.99 (ddd, J = 8.0, 7.1, 1.0 Hz, 1H), 6.86 (dd, J = 8.4, 1.0 Hz, 1H), 4.79 (dd, J = 12.5, 3.1 Hz, 1H), 3.21 (dd, J = 16.9, 12.5 Hz, 1H), 2.96 (dd, J = 16.9, 3.1 Hz, 1H), 1.96 (dd, J = 13.9, 6.3 Hz, 1H), 1.87 (dd, J = 14.0, 6.4 Hz, 1H), 1.62 (dt, J = 13.1, 6.5 Hz, 1H), 0.91 (dd, J = 6.6, 0.9 Hz, 6H). 13C NMR (176 MHz, CDCl3) δ 190.32, 177.43, 160.16, 160.07, 150.48, 136.17, 130.96, 129.25, 126.82, 123.96, 122.11, 120.91, 117.71, 80.50, 76.42, 40.90, 37.88, 24.72, 23.78, 23.34. HRMS: Calculated for [C22H20N2O6+H+]: 409.1394, found: 409.1402. The er was determined by UPC2 using a chiral Chiralpack IG column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 3.01 min, τminor = 4.00 min, (85:15 er).
(S)-4-Isobutyl-(6-fluoro-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3j) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow oil in a 65% yield (24.8 mg), dr = 19:1. Major diastereoisomer: IR (film): 3073, 1818, 1702, 1648, 1478, 1218, 878, 773, 699 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.03 (d, J = 7.7 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.52–7.46 (m, 3H), 7.10 (ddd, J = 9.1, 7.7, 3.2 Hz, 1H), 6.86 (dd, J = 9.1, 4.1 Hz, 1H), 4.72 (dt, J = 8.6, 4.3 Hz, 1H), 3.20 (dd, J = 17.0, 12.9 Hz, 1H), 2.93 (dd, J = 17.0, 2.9 Hz, 1H), 1.91 (dd, J = 13.8, 6.2 Hz, 1H), 1.83 (dd, J = 13.8, 6.5 Hz, 1H), 1.66–1.59 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 190.3 (d, J = 1.5 Hz), 178.5, 161.8, 157.6 (d, J = 242.9 Hz), 156.8 (d, J = 1.6 Hz), 133.3, 129.0 (2C), 128.3 (2C), 125.6, 123.7 (d, J = 24.6 Hz), 121.5 (d, J = 6.6 Hz), 119.7 (d, J = 7.4 Hz), 112.0 (d, J = 23.5 Hz), 80.9, 75.9, 41.1, 37.9, 24.8, 24.0, 23.5. HRMS: Calculated for [C22H20FNO4+H+]: 382.1449, found: 382.1449. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.15 min, τminor = 2.51 min, (71:29 er).
(6-Bromo-(R)-4-oxochroman-2-yl)-(S)-4-isobutyl-2-phenyl-1,3-oxazol-5(4H)-one (3k) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow solid (m.p. 142–144 °C) in a 70% yield (30.9 mg), dr = 19:1. Major diastereoisomer: IR (film): 2958, 1817, 1696, 1651, 1598, 1464, 1415, 1270, 1221, 884, 753, 702 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.01 (d, J = 7.7 Hz, 2H), 7.94 (d, J = 2.4 Hz, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.44 (dd, J = 8.8, 2.4 Hz, 1H), 6.77 (d, J = 8.8 Hz, 1H), 4.74 (dd, J = 12.5, 3.0 Hz, 1H), 3.20 (dd, J = 17.0, 12.6 Hz, 1H), 2.94 (dd, J = 17.0, 3.0 Hz, 1H), 1.91 (dd, J = 13.8, 6.2 Hz, 1H), 1.83 (dd, J = 13.8, 6.5 Hz, 1H), 1.63 (hept, J = 6.5 Hz, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 189.7, 178.4, 161.9, 159.4, 138.8, 133.3, 129.4, 129.0 (2C), 128.4 (2C), 125.5, 122.3, 120.0, 114.8, 80.9, 75.9, 41.1, 37.8, 24.8, 24.0, 23.5. HRMS: Calculated for [C22H20BrNO4+H+]: 442.0648, found: 442.0644. The er was determined by UPC2 using a chiral Chiralpack IG column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.76 min, τminor = 3.25 min, (77:23 er).
6-Chloro-((S)-4-isobutyl-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3l) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow solid (m.p. 118–120 °C) in a 73% yield (29.0 mg), dr = 10:1. Major diastereoisomer: IR (film): 3070, 1816, 1702, 1648, 1478, 1212, 878, 773, 699 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.01 (dd, J = 8.4, 1.3 Hz, 2H), 7.94 (d, J = 2.5 Hz, 1H), 7.63–7.56 (m, 1H), 7.54–7.47 (m, 2H), 7.44 (dd, J = 8.8, 2.5 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 4.74 (dd, J = 12.6, 3.1 Hz, 1H), 3.20 (dd, J = 17.0, 12.5 Hz, 1H), 2.94 (dd, J = 17.0, 3.1 Hz, 1H), 1.91 (dd, J = 13.8, 6.3 Hz, 1H), 1.83 (dd, J = 13.8, 6.5 Hz, 1H), 1.63 (dt, J = 13.1, 6.6 Hz, 1H), 0.90 (dd, J = 9.5, 6.7 Hz, 6H). 13C NMR (176 MHz, CDCl3) δ 189.5, 178.2, 161.7, 159.3, 138.7, 133.2, 129.2, 128.8 (2C), 128.2 (2C), 125.4, 122.2, 119.9, 114.7, 80.8, 75.8, 41.0, 37.6, 24.7, 23.8, 23.4. HRMS: Calculated for [C22H20ClNO4+H+]: 398.1154, found: 398.1163. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.81 min, τminor = 3.26 min, (79.5:20.5 er).
(S)-4-Isobutyl-(6-nitro-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3m) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow solid (m.p. 188–190 °C) in a 48% yield (19.6 mg), dr = 19:1. Major diastereoisomer: IR (film): 2922, 1819, 1710, 1605, 1585, 1469, 1275, 1233, 1183,1043, 906, 778, 665 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.72 (d, J = 2.8 Hz, 1H), 8.20 (dd, J = 9.2, 2.8 Hz, 1H), 8.09–7.94 (m, 2H), 7.59 (d, J = 7.5 Hz, 1H), 7.51–7.42 (m, 2H), 7.00 (d, J = 9.1 Hz, 1H), 4.89 (dd, J = 11.5, 3.5 Hz, 1H), 3.28 (dd, J = 17.1, 11.5 Hz, 1H), 3.07 (dd, J = 17.1, 3.5 Hz, 1H), 1.97–1.78 (m, 2H), 1.67–1.57 (m, 1H), 0.91 (dd, J = 7.8, 6.6 Hz, 6H). 13C NMR (176 MHz, CDCl3) δ 188.4, 177.9, 164.0, 162.0, 133.4, 130.3, 128.9, 128.7, 128.4, 128.2, 125.1, 123.1, 120.6, 119.1, 81.4, 75.8, 41.0, 37.4, 24.7, 23.8, 23.33, 22.4. HRMS: Calculated for [C22H20N2O6+H+]: 409.1394, found: 409.1382. The er was determined by UPC2 using a chiral Chiralpack IG column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 3.07 min, τminor = 3.34 min, (69:31 er).
(S)-4-Isobutyl-(6-methyl-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3n) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow solid (m.p. 102–106 °C) in a 34% yield (12.8 mg), dr = 19:1. Major diastereoisomer: IR (film): 3067, 1819, 1725, 1688, 1651,1558, 1450, 1076, 955, 778, 753 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.03 (d, J = 7.6 Hz, 2H), 7.63 (d, J = 6.0 Hz, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.20 (dd, J = 8.5, 1.9 Hz, 1H), 6.77 (t, J = 8.4 Hz, 1H), 4.70 (dd, J = 13.0, 2.8 Hz, 1H), 3.19 (dd, J = 16.8, 13.0 Hz, 1H), 2.89 (dd, J = 16.9, 2.8 Hz, 1H), 2.25 (s, 3H), 1.92 (dd, J = 13.9, 6.2 Hz, 1H), 1.83 (dd, J = 13.9, 6.5 Hz, 1H), 1.63 (hept, J = 6.5 Hz, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 191.4, 178.7, 161.6, 158.7, 137.3, 133.2, 131.6, 128.9 (2C), 128.4 (2C), 126.5, 125.7, 120.6, 117.8, 80.7, 76.0, 41.1, 38.2, 24.8, 24.0, 23.5, 20.5. HRMS: Calculated for [C23H23NO4+H+]: 378.1700, found: 378.1698. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.43 min, τminor = 3.11 min, (80:20 er).
(S)-4-Isobutyl-(7-methoxy-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3o) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow solid (m.p. 160–161 °C) in a 65% yield (25.5 mg), dr = 19:1. Major diastereoisomer: IR (film): 3071, 1819, 1684, 1651,1582, 1486, 1281, 1214, 1099, 883, 700, 561 cm−1. 1H NMR (700 MHz, CDCl3) δ 8.04 (d, J = 7.3 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.26 (d, J = 3.3 Hz, 1H), 6.99 (dd, J = 9.1, 3.2 Hz, 1H), 6.81 (d, J = 9.1 Hz, 1H), 4.69 (dd, J = 13.1, 2.8 Hz, 1H), 3.75 (s, 3H), 3.19 (dd, J = 16.9, 13.1 Hz, 1H), 2.90 (dd, J = 16.9, 2.9 Hz, 1H), 1.92 (dd, J = 13.9, 6.2 Hz, 1H), 1.83 (dd, J = 13.9, 6.5 Hz, 1H), 1.66–1.60 (m, 1H), 0.91 (d, J = 6.7 Hz, 2H), 0.89 (d, J = 6.6 Hz, 2H). 13C NMR (176 MHz, CDCl3) δ 191.2, 178.6, 161.6, 155.3, 154.6, 133.1, 128.9 (2C), 128.4 (2C), 125.8, 125.3, 120.9, 119.3, 107.4, 80.8, 75.9, 55.9, 41.2, 38.1, 24.8, 24.0, 23.5. HRMS: Calculated for [C23H23NO5+H+]: 394.1649, found: 394.1645. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.58 min, τminor = 3.11 min, (90:10 er).
(S)-4-Isobutyl-(6-chloro-7-methyl-(R)-4-oxochroman-2-yl)-2-phenyl-1,3-oxazol-5(4H)-one (3p) pure product was isolated by flash chromatography on silica gel (hexane:ethyl acetate 15:1) as yellow oil in a 75% yield (30.8 mg), dr = 19:1. Major diastereoisomer: IR (film): 3065, 1819, 1691, 1652, 1611, 1408, 1319, 1154, 873, 703 cm–1. 1H NMR (700 MHz, CDCl3) δ 8.02 (d, J = 7.6 Hz, 2H), 7.78 (s, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 6.78 (s, 1H), 4.71 (dd, J = 12.7, 3.0 Hz, 1H), 3.17 (dd, J = 17.0, 12.7 Hz, 1H), 2.90 (dd, J = 17.0, 3.0 Hz, 1H), 2.27 (s, 2H), 1.90 (dd, J = 13.9, 6.2 Hz, 1H), 1.82 (dd, J = 13.9, 6.5 Hz, 1H), 1.63 (hept, J = 6.6 Hz, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 189.7, 178.5, 161.8, 158.8, 145.4, 133.2, 129.0 (2C), 128.4 (2C), 128.3, 126.6, 125.6, 120.1, 120.0, 81.0, 75.9, 41.1, 37.8, 24.8, 24.0, 23.5, 20.8. HRMS: Calculated for [C23H22ClNO4+H+]: 412.1310, found: 412.1319. The er was determined by UPC2 using a chiral Chiralpack IA column gradient from 100% CO2 up to 40%; i-PrOH, 2.5 mL/min; detection wavelength = 245 nm; τmajor = 2.65 min, τminor = 2.87 min, (76:24 er).
4.3. Synthesis of Methyl 2-Benzamido-4-Methyl-2-(4-Oxochroman-2-yl)Pentanoate (4a)
An ordinary screw-cap vial was charged with a magnetic stirring bar, the chromone 3a (0.05 mmol, 17 mg), MeOH (200 μL), and CHCl3 (100 μL). Then toluenesulphonic acid monohydrate (0.1 mmol, 19 mg) was added, and the reaction mixture was stirred for 1.5 h at 40 °C. The product was isolated using flash chromatography in an eluent gradient (starting from hexane:ethyl acetate—10:1 to hexane:ethyl acetate—5:1), giving 4a as a yellow oil in a 51% yield (10.0 mg), dr = >20:1 dr. Major diastereoisomer: IR (film): 3405, 3064, 1819, 1738, 1691, 1669, 1579, 1464, 1442, 1304, 1224, 1030, 765, 710 cm−1. 1H NMR (700 MHz, CDCl3) δ 7.85 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.53 (t, J = 7.5 Hz, 1H), 7.49 (bs, 1H), 7.46 (t, J = 7.8 Hz, 3H), 7.01 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.9 Hz, 1H), 5.03 (dd, J = 14.0, 2.4 Hz, 1H), 3.87 (s, 3H), 3.05 (dd, J = 14.1, 5.0 Hz, 1H), 3.02 (dd, J = 16.9, 2.5 Hz, 1H), 2.85 (dd, J = 16.9, 14.1 Hz, 1H), 1.95 (dd, J = 14.6, 7.3 Hz, 1H), 1.71–1.63 (m, 1H), 0.96 (d, J = 7.3 Hz, 3H), 0.86 (d, J = 7.2 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 191.5, 173.2, 166.8, 161.0, 135.9, 134.8, 131.8, 128.7 (2C), 127.0 (2C), 127.0, 121.8, 121.0, 117.7, 81.4, 67.2, 53.3, 39.3, 37.6, 24.7, 23.7, 22.3. HRMS: Calculated for [C23H25NO5+H+]: 396.1805, found: 396.1812.
Supplementary Materials
The following are available online. Copies of 1H and 13C spectra of all obtained compounds. Copies of HPLC and UPC data. Crystal structure details.
Author Contributions
A.A., conceptualization; A.A., methodology; A.A. and J.B., investigation; A.A. and J.B., analysis; L.S., crystal structure details; A.A., writing—original draft preparation; A.A., writing—review and editing; A.A., funding acquisition.
Funding
This work was financially supported by the National Science Centre, Poland within the “Sonata” programme realized in the period 2017–2020, project number: UMO-2016/21/D/ST5/01668.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Sibi, M.P.; Manyem, S. Enantioselective Conjugate Additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Röder, A. Recent Advances in Catalytic Enantioselective Michael Additions. Synthesis 2001, 2, 171–196. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael Additions to Nitroalkenes. Eur. J. Org. Chem. 2002, 12, 1877–1894. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Construction of Quaternary Stereocenters: New Perspectives through Enantioselective Michael Reactions. Angew. Chem. Int. Ed. 2003, 42, 1688–1690. [Google Scholar] [CrossRef] [PubMed]
- Christoffers, J.; Koripelly, G.; Rosiak, A.; Rössle, M. Recent Advances in Metal-Catalyzed Asymmetric Conjugate Additions. Synthesis 2007, 9, 1279–1300. [Google Scholar] [CrossRef]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic Enantioselective Michael and Hetero-Michael Reactions. Synthesis 2007, 14, 2065–2092. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 11, 1701–1716. [Google Scholar] [CrossRef]
- Almasi, D.; Alonso, D.A.; Nájera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Schwarz, J.; König, B. Decarboxylative reactions with and without light—A comparison. Green Chem. 2018, 20, 323–361. [Google Scholar] [CrossRef]
- Pan, Y.; Tan, C.H. Catalytic Decarboxylative Reactions: Biomimetic Approaches Inspired by Polyketide Biosynthesis. Synthesis 2011, 13, 2044–2053. [Google Scholar] [CrossRef]
- Wang, Z.L. Recent Advances in Catalytic Asymmetric Decarboxylative Addition Reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Bojanowski, J.; Albrecht, A. Carboxylic-acid-activated olefins in decarboxylative reactions. Asian J. Org. Chem. 2019, 8, 746–754. [Google Scholar] [CrossRef]
- Lubkoll, J.; Wennemers, H. Mimicry of Polyketide Synthases—Enantioselective 1,4-Addition Reactions of Malonic Acid Half-Thioesters to Nitroolefins. Angew. Chem. Int. Ed. 2007, 46, 6841–6844. [Google Scholar] [CrossRef]
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A Heterobimetallic Ni/La-salan Complex for Catalytic Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half-Thioester. Chem. Asian J. 2010, 15, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to β-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of γ-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202. [Google Scholar] [CrossRef]
- Kang, Y.K.; Lee, H.J.; Moon, H.W.; Kim, D.Y. Organocatalytic enantioselective decarboxylative Michael addition of β-ketoacids to α,β-unsaturated ketones. RSC Adv. 2013, 3, 1332–1335. [Google Scholar] [CrossRef]
- Qiao, B.; Liu, Q.; Liu, H.; Yan, L.; Jiang, Z. Asymmetric Decarboxylative 1,4-Addition of Malonic Acid Half Thioesters to Vinyl Sulfones: Highly Enantioselective Synthesis of 3-Monofluoromethyl-3-Arylpropanoic Esters. Chem. Asian J. 2014, 9, 1252–1256. [Google Scholar] [CrossRef]
- Ren, Q.; Sun, S.; Huang, J.; Li, W.; Wu, M.; Guo, H.; Wang, J. An enantioselective cascade reaction between α,β-unsaturated aldehydes and malonic half-thioesters: A rapid access to chiral δ-lactones. Chem. Commun. 2014, 50, 6137–6140. [Google Scholar] [CrossRef]
- Ren, Q.; Gao, T.; Li, W.; Wan, L.; Hu, Y.; Peng, Y.; Sun, S.; Hu, L.; Wu, M.; Guo, H.; et al. A highly enantioselective Michael reaction between α,β-unsaturated ketones and malonic acid half-thioesters. New. J. Chem. 2015, 39, 5100–5103. [Google Scholar] [CrossRef]
- Nakamura, S.; Toda, A.; Sano, M.; Hatanaka, T.; Funahashi, Y. Organocatalytic Enantioselective Conjugate Addition of Malonic Acid Half Thioesters to Coumarin-3-carboxylic Acids Using N-Heteroarenesulfonyl Cinchona Alkaloid Amides. Adv. Synth. Catal. 2016, 358, 1029–1034. [Google Scholar] [CrossRef]
- Brunner, H.; Baur, M.A. α-Amino Acid Derivatives by Enantioselective Decarboxylation. Eur. J. Org. Chem. 2003, 15, 2854–2862. [Google Scholar] [CrossRef]
- Rogers, L.M.A.; Rouden, J.; Lecomte, L.; Lasne, M.C. Enantioselective decarboxylation–reprotonation of an α-amino malonate derivative as a route to optically enriched cyclic α-amino acid. Tetrahedron Lett. 2003, 44, 3047–3050. [Google Scholar] [CrossRef]
- Seitz, T.; Baudoux, J.; Bekolo, H.; Cahard, D.; Plaquevent, J.C.; Lasne, M.C.; Rouden, J. Organocatalyzed route to enantioenriched pipecolic esters: Decarboxylation of an aminomalonate hemiester. Tetrahedron 2006, 62, 6155–6165. [Google Scholar] [CrossRef]
- Amere, M.; Lasne, M.C.; Rouden, J. Highly Enantioselective Decarboxylative Protonation of α-Aminomalonates Mediated by Thiourea Cinchona Alkaloid Derivatives: Access to Both Enantiomers of Cyclic and Acyclic α-Aminoacids. Org. Lett. 2007, 9, 2621–2624. [Google Scholar] [CrossRef]
- Yuan, H.N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J.A. Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083. Angew. Chem. Int. Ed. 2013, 52, 3869–3873. [Google Scholar] [CrossRef]
- Wei, A.J.; Nie, J.; Zheng, Y.; Ma, J.A. Ni-Catalyzed Highly Chemo-, Regio-, and Enantioselective Decarboxylative Aldol Reaction of β,γ-Unsaturated α-Ketoesters with β-Ketoacids. J. Org. Chem. 2015, 80, 3766–3776. [Google Scholar] [CrossRef]
- Wei, Y.; Guo, R.; Dang, Y.; Nie, J.; Ma, J.A. Organocatalytic Enantioselective Decarboxylative Michael Addition of β-Keto Acids to Dicyanoolefins and Disulfonylolefins. Adv. Synth. Catal. 2016, 358, 2721–2726. [Google Scholar] [CrossRef]
- Wallace, T.W. Conjugate addition to chromones: Synthesis of substituted 4-chromanones. Tetrahedron Lett. 1984, 25, 4299–4302. [Google Scholar] [CrossRef]
- Neo, A.G.; Díaz, J.; Marcaccini, S.; Marcos, C.F. Conjugate addition of isocyanides to chromone 3-carboxylic acid: An efficient one-pot synthesis of chroman-4-one 2-carboxamides. Org. Biomol. Chem. 2012, 10, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Wang, L.; Xu, L.; Zhao, H.; Xiao, J. Facile synthesis of azaarene-2-substituted chromanone derivatives via tandem sp3 C–H functionalization/decarboxylation of azaarenes with 4-oxo-4H-chromene-3-carboxylic acid. RSC Adv. 2014, 4, 53188–53191. [Google Scholar] [CrossRef]
- Xu, L.; Shao, Z.; Wang, L.; Xiao, J. Tandem sp3 C–H Functionlization/Decarboxylation of 2-Alkylazaarenes with Coumarin-3-carboxylic Acids. Org. Lett. 2014, 16, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Xu, L.; Wang, L.; Wie, H.; Xiao, J. Catalyst-free tandem Michael addition/decarboxylation of (thio)coumarin-3-carboxylic acids with indoles: Facile synthesis of indole-3-substituted 3,4-dihydro(thio)coumarins. Org. Biomol. Chem. 2014, 12, 2185–2188. [Google Scholar] [CrossRef] [PubMed]
- Talhi, O.; Brodziak-Jarosz, L.; Panning, J.; Orlikova, B.; Zwergel, C.; Tzanova, T.; Philippot, S.; Pinto, D.C.G.A.; Almeida Paz, F.A.; Gerhäuser, C.; et al. One-Pot Synthesis of Benzopyran-4-ones with Cancer Preventive and Therapeutic Potential. Eur. J. Org. Chem. 2016, 965–975. [Google Scholar] [CrossRef]
- Peng, S.; Wang, L.; Guo, H.; Sun, S.; Wang, J. Facile synthesis of 4-substituted 3,4-dihydrocoumarins via an organocatalytic double decarboxylation process. Org. Biomol. Chem. 2012, 10, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A. Utilization of Chromone-3-Carboxylic Acids as Acceptors in the Michael-Type Decarboxylative Addition. Eur. J. Org. Chem. 2018, 46, 6482–6485. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Applications; Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Saengchantara, S.T.; Wallace, T.W. Chromanols, chromanones, and chromones. Nat. Prod. Rep. 1986, 3, 465–475. [Google Scholar] [CrossRef]
- Kamat, D.P.; Tilve, S.G.; Kamat, V.P.; Kirtany, J.K. Syntheses and Biological Activities of Chroman-2-ones. A Review. Org. Prep. Proc. Int. 2015, 47, 1–79. [Google Scholar] [CrossRef]
- Masters, K.S.; Bräse, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Eggleston, D.S.; Haltiwanger, R.C.; Bean, M.F.; Taylor, P.B.; Caranfa, M.J.; Breen, A.L.; Bartus, H.R.; Johnson, R.K. The inophyllums, novel inhibitors of HIV-1 reverse transcriptase isolated from the Malaysian tree, Calophyllum inophyllum Linn. J. Med. Chem. 1993, 36, 4131–4138. [Google Scholar] [CrossRef] [PubMed]
- Galinis, D.L.; Fuller, R.W.; McKee, T.C.; Cardellina, J.H., II; Gulakowski, R.J.; McMahon, J.B.; Boyd, M.R. Structure−Activity Modifications of the HIV-1 Inhibitors (+)-Calanolide A and (−)-Calanolide B. J. Med. Chem. 1996, 39, 4507–4510. [Google Scholar] [CrossRef] [PubMed]
- Mbwambo, Z.H.; Kapingu, M.C.; Moshi, M.J.; Machumi, F.; Apers, S.; Cos, P.; Ferreira, D.; Marais, J.P.J.; Berghe, D.V.; Maes, L.; et al. Antiparasitic Activity of Some Xanthones and Biflavonoids from the Root Bark of Garcinia livingstonei. J. Nat. Prod. 2006, 69, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Picker, K.; Ritchie, E.; Taylor, W.C. The chemical constituents of Australian Flindersia species. XXI. An examination of the bark and the leaves of F. laevicarpa. Aust. J. Chem. 1976, 29, 2023–2036. [Google Scholar] [CrossRef]
- Zhao, D.L.; Shao, C.L.; Gan, L.S.; Wang, M.; Wang, C.Y. Chromone Derivatives from a Sponge-Derived Strain of the Fungus Corynespora cassiicola. J. Nat. Prod. 2015, 78, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, A.E.; Scheidt, K.A. Asymmetric Methods for the Synthesis of Flavanones, Chromanones, and Azaflavanones. Eur. J. Org. Chem. 2012, 449–462. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.R.; Scheidt, K.A. Pyranone Natural Products as Inspirations for Catalytic Reaction Discovery and Development. Acc. Chem. Res. 2015, 48, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, A.; Bojanowski, J. Decarboxylative Aminocatalytic Cascade for the Synthesis of Dihydroxanthones. Adv. Synth. Catal. 2017, 358, 2907–2911. [Google Scholar] [CrossRef]
- Wen, G.; Su, Y.; Zhang, G.; Lin, Q.; Zhu, Y.; Zhang, Q.; Fang, X. Stereodivergent Synthesis of Chromanones and Flavanones via Intramolecular Benzoin Reaction. Org. Lett. 2016, 18, 3980–3983. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. (−)-β-Pinene-Derived N-Heterocyclic Carbenes: Application to Highly Enantioselective Intramolecular Stetter Reaction. ACS Catal. 2014, 4, 1404–1408. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A. Enantioselective Synthesis of Chromanones Bearing Quaternary Substituted Stereocenters Catalyzed by (1R)-Camphor-Derived N-Heterocyclic Carbenes. J. Org. Chem. 2015, 80, 7468–7476. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.Q.; Li, Y.; Yang, G.Q.; You, S.L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett 2011, 1033–1037. [Google Scholar] [CrossRef]
- Biddle, M.M.; Lin, M.; Scheidt, K.A. Catalytic Enantioselective Synthesis of Flavanones and Chromanones. J. Am. Chem. Soc. 2007, 129, 3830–3831. [Google Scholar] [CrossRef] [PubMed]
- Read de Alaniz, J.; Rovis, T.A. Highly Enantio- and Diastereoselective Catalytic Intramolecular Stetter Reaction. J. Am. Chem. Soc. 2005, 127, 6284–6289. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Runsink, J. The First Asymmetric Intramolecular Stetter Reaction. Preliminary Communication. Helv. Chim. Acta 1996, 79, 1899–1902. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D. Stereoselective synthesis of quaternary α-amino acids. Part 2: Cyclic compounds. Tetrahedron Asymmetry 2000, 11, 645–732. [Google Scholar] [CrossRef]
- Vogt, H.; Bräse, S. Recent approaches towards the asymmetric synthesis of α,α-disubstituted α-amino acids. Org. Biomol. Chem. 2007, 5, 406–430. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D. Recent progress on the stereoselective synthesis of acyclic quaternary α-amino acids. Tetrahedron Asymmetry 2007, 18, 569–623. [Google Scholar] [CrossRef]
- Ohfune, Y.; Shinada, T. Enantio and Diastereoselective Construction of α,α-Disubstituted α-Amino Acids for the Synthesis of Biologically Active Compounds. Eur. J. Org. Chem. 2005, 5127–5143. [Google Scholar] [CrossRef]
- Nájera, C. From α-Amino Acids to Peptides: All You Need for the Journey. Synlett 2002, 9, 1388–1404. [Google Scholar] [CrossRef]
- Alba, A.N.; Rios, R. Oxazolones in Organocatalysis, New Tricks for an Old Reagent. Chem. Asian. J. 2011, 6, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Scala, A.; Risitano, F.; Grassi, G. Oxazol-5-(4H)-Ones. Part 1. Synthesis and Reactivity as 1,3-dipoles. Curr. Org. Chem. 2014, 18, 2691–2710. [Google Scholar] [CrossRef]
- De Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Reyes, E.; Alemán, J.; Milelli, A.; Kobbelgaard, S.; Jørgensen, K.A. Organocatalytic Asymmetric Synthesis of α,α-Disubstituted α-Amino Acids and Derivatives. J. Am. Chem. Soc. 2008, 130, 12031–12037. [Google Scholar] [CrossRef] [PubMed]
- Balaguer, A.N.; Companyó, X.; Calvet, T.; Font-Bardia, M.; Moyano, A.; Rios, R. Highly Regio and Diastereoselective Oxazol-5-one Addition to Nitrostyrenes. Eur. J. Org. Chem. 2009, 199–203. [Google Scholar] [CrossRef]
- Alba, A.N.R.; Companyó, X.; Valero, G.; Moyano, A.; Rios, R. Enantioselective Organocatalytic Addition of Oxazolones to 1,1-Bis(phenylsulfonyl)ethylene: A Convenient Asymmetric Synthesis of Quaternary α-Amino Acids. Chem. Eur. J. 2010, 16, 5354–5361. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Dzięgielewski, M.; Kowalczyk, D.; Albrecht, L. A Convenient Approach to a Novel Group of Quaternary Amino Acids Containing a Geminal Bisphosphonate Moiety. Synthesis 2014, 46, 3233–3238. [Google Scholar] [CrossRef]
- Jiang, H.; Gschwend, B.; Albrecht, L.; Hansen, S.G.; Jørgensen, K.A. Asymmetric Trienamine Catalysis for the Construction of Structurally Rigid Cyclic α,α-Disubstituted Amino Acid Derivatives. Chem. Eur. J. 2011, 17, 9032–9036. [Google Scholar] [CrossRef]
- Marcelli, T.; van Maarseveen, J.H.; Hiemstra, H. Cupreines and Cupreidines: An Emerging Class of Bifunctional Cinchona Organocatalysts. Angew. Chem. Int. Ed. 2006, 45, 7496–7504. [Google Scholar] [CrossRef] [PubMed]
- Jew, S.S.; Park, H.G. Cinchona-based phase-transfer catalysts for asymmetric synthesis. Chem. Commun. 2009, 46, 7090–7103. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Oiarbide, M.; López, R. Asymmetric organocatalysis by chiral Brønsted bases: Implications and applications. Chem. Soc. Rev. 2009, 38, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.; Rodríguez-Docampo, Z.; Connon, S.J. Highly tunable arylated cinchona alkaloids as bifunctional catalysts. Chem. Commun. 2012, 48, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, H.; Dzięgielewski, M.; Deredas, D.; Albrecht, A.; Albrecht, L. Chiral Iminophosphoranes—An Emerging Class of Superbase Organocatalysts. Chem. Eur. J. 2015, 21, 10268–10277. [Google Scholar] [CrossRef] [PubMed]
- For the synthesis of catalyst 9e see: Bera, K.; Namboothiri, I.N.N. Quinine-Derived Thiourea and Squaramide Catalyzed Conjugate Addition of α-Nitrophosphonates to Enones: Asymmetric Synthesis of Quaternary α-Aminophosphonates. J. Org. Chem. 2015, 80, 1402–1413. [Google Scholar] [CrossRef] Analytical data of 9e was in accordance with the literature. Purity was confirmed by HRMS analysis: Calculated for [C31H33N4O4+H+]: 525.2496, found: 525.2507.
- CCDC 1895323 Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge from The Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/structure (accessed on 1 June 2019).
- Liang, J.; Ruble, J.C.; Fu, G.C. Dynamic Kinetic Resolutions Catalyzed by a Planar-Chiral Derivative of DMAP: Enantioselective Synthesis of Protected α-Amino Acids from Racemic Azlactones. J. Org. Chem. 1998, 63, 3154–3155. [Google Scholar] [CrossRef]
- Ishizuka, N.; Matsunori, K.; Sakai, K.; Fujimoto, M.; Mihara, S.; Yamamori, T. Structure−Activity Relationships of a Novel Class of Endothelin-A Receptor Antagonists and Discovery of Potent and Selective Receptor Antagonist, 2-(Benzo[1,3]dioxol-5-yl)-6-isopropyloxy-4-(4-methoxyphenyl)-2H-chromene-3- carboxylic Acid (S-1255). 1. Study on Structure−Activity Relationships and Basic Structure Crucial for ETA Antagonism. J. Med. Chem. 2002, 45, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3a, 3h are available from the authors. |
Scheme 1.
Decarboxylative Michael reactions.

Scheme 2.
The relevance of a chromanone, α,α-disubstituted amino acid structural motifs, and new hybrid molecules being the objectives of this work.
Scheme 2.
The relevance of a chromanone, α,α-disubstituted amino acid structural motifs, and new hybrid molecules being the objectives of this work.

Scheme 3.
Decarboxylative enantioselective synthesis of chromanones 3 bearing an azlactone unit—α-substituted azlactones 1 scope. Reactions performed on a 0.1 mmol scale using 1 (1 equivalent) and 2b (1 equivalent) in 0.2 mL of the solvent. For details see Supplementary Materials.
Scheme 3.
Decarboxylative enantioselective synthesis of chromanones 3 bearing an azlactone unit—α-substituted azlactones 1 scope. Reactions performed on a 0.1 mmol scale using 1 (1 equivalent) and 2b (1 equivalent) in 0.2 mL of the solvent. For details see Supplementary Materials.

Scheme 4.
Decarboxylative enantioselective synthesis of chromanones 3 bearing an azlactone unit—chromone-3-carboxylic acids 4 scope. Reactions performed on a 0.1 mmol scale using 1a (1 equivalent) and 2 (1 equivalent) in 0.2 mL of the solvent. For details see Supplementary Materials.
Scheme 4.
Decarboxylative enantioselective synthesis of chromanones 3 bearing an azlactone unit—chromone-3-carboxylic acids 4 scope. Reactions performed on a 0.1 mmol scale using 1a (1 equivalent) and 2 (1 equivalent) in 0.2 mL of the solvent. For details see Supplementary Materials.

Scheme 5.
Azlactone-ring opening in 3a, relative configuration assignment, and transition state proposal.
Scheme 5.
Azlactone-ring opening in 3a, relative configuration assignment, and transition state proposal.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Decarboxylative enantioselective synthesis of chromanone 3 bearing an azlactone unit—optimization studies a.
Table 1.
Decarboxylative enantioselective synthesis of chromanone 3 bearing an azlactone unit—optimization studies a.

| Solvent | 2/9 | Conv. (Yield) [%] b | dr c | er d | ee d [%] | |
|---|---|---|---|---|---|---|
| 1 | THF | 2a/9a | <5 | n.d. | n.d. | n.d. |
| 2 | THF | 2b/9a | >95 | 3:1 | 55:45 | 10 |
| 3 | THF | 2b/9b | >95 | 5:1 | 75:25 | 50 |
| 4 | THF | 2b/9c | >95 | 3:1 | 84:16 | 68 |
| 5 | THF | 2b/9d | >95 | 10:1 | 84:16 | 68 |
| 6 | THF | 2b/9e | >95 (81) | >20:1 | 91:9 | 82 |
| 7 | THF | 2b/9f | >95 | >20:1 | 79:21 | 58 |
| 8 | CH2Cl2 | 2b/9e | >95 | 5:1 | 81:19 | 62 |
| 9 | Toluene | 2b/9e | >95 | 5:1 | 91:9 | 82 |
| 10 | 1,4-Dioxane | 2b/9e | >95 | 10:1 | 87:13 | 74 |
| 11 | CPME | 2b/9e | >95 | 4:1 | 84:16 | 68 |
| 12 | Et2O | 2b/9e | >95 | 6:1 | 70:30 | 40 |
| 13 | 2-MeTHF | 2b/9e | >95 | 15:1 | 89:11 | 78 |
| 14 e | THF | 2b/9e | >95 | 20:1 | 90:10 | 80 |
| 15 f | THF | 2b/9e | >95 | 20:1 | 90:10 | 80 |
| 16 g | THF | 2b/9e | >95 | 10:1 | 70:30 | 40 |
| 17 h | THF | 2b/9e | >95 | 20:1 | 84:16 | 68 |
| 18 i | THF | 2b/9e | >95 | 20:1 | 84:16 | 68 |
| 19 j | THF | 2b/9e | >95 (76) | >20:1 | 91:9 | 82 |
a Reactions performed on a 0.1 mmol scale using 1a (1 equivalent) and 2 (1 equivalent) in 0.2 mL of the solvent. b Determined by 1H NMR of a crude reaction mixture. In parenthesis isolated yields are given. c Determined by 1H NMR of a crude reaction mixture. d Determined by a chiral stationary phase UPC2. e Reaction performed in 0.1 mL of THF. f Reaction performed in 1.0 mL of THF. g Reaction performed using 1a (1.5 equivalent). h Reaction performed using 1.5 equivalent of 2b. i Reaction performed at 0 °C. j Reaction performed using 1 gram of 2b.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bojanowski, J.; Sieroń, L.; Albrecht, A. Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules 2019, 24, 2565. https://doi.org/10.3390/molecules24142565
AMA Style
Bojanowski J, Sieroń L, Albrecht A. Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction. Molecules. 2019; 24(14):2565. https://doi.org/10.3390/molecules24142565
Chicago/Turabian StyleBojanowski, Jan, Lesław Sieroń, and Anna Albrecht. 2019. "Enantioselective Synthesis of Chromanones Bearing an α,α-Disubstituted α-Amino Acid Moiety via Decarboxylative Michael Reaction" Molecules 24, no. 14: 2565. https://doi.org/10.3390/molecules24142565