2.1. Synthesis and Preliminary Characterization
In a previous publication [
39], some of us explored the effect of selecting different non-conventional synthetic methods for the reaction between
bpa and benzenedicarboxylate (
m-bdc). In the course of these studies, microwave-assisted synthesis was found to be the most efficient method for the prepration of frameworks with higher dimensionality. Therefore, the same synthetic method employed for [Cu
4(m-bdc)
4(bpa)
2dmf]·dmf was used, but with
btec as the polycarboxylic ligand instead. In this case, microwave-assisted synthesis provides a mixture of both blue crystals of the predominant phase
1 and a few green crystals of
1.ah. The same result was obtained for different crystal batches. Crystals were separated manually under a polarized light microscope.
Compound
1 and
1.ah were firstly identified by infrared spectroscopy (FT-IR). The FT-IR spectra, together with the list of the most intense bands and their assignations, are shown in
Figure S1 and Table S1. Both spectra display the characteristic bands of both the
bpa and
btec ligands, as exemplified by the presence of the N=C stretching vibration band at 1300 cm
−1 and a set of different overlapped signals around 1630 cm
−1 related to asymmetrical and symmetrical stretching of C=O bonds from carboxylate groups, which could indicate the presence of
btec ligands in different coordination modes (monodentate, chelating, bridging) [
41]. However, the wide band centered at ca. 3082 cm
−1, which corresponds to the stretching O–H vibrations from water molecules, is narrowed significantly when going from
1 to
1.ah. Although the O–H band can still be observed in the spectrum of
1.ah because of the humidity absorbed by the KBr matrix, it could suggest a lower water content in this compound in comparison to
1.
2.2. Crystal Structures
Compound
1 crystallizes in the triclinic space group
P-1 and its asymmetric unit contains one Cu(II) ion, one half of the
bpa and one half of the
btec ligand and two coordination water molecules (
Figure S2). Both
bpa and
btec are located at an inversion center. The coordination environment of the metal center in a distorted octahedral geometry is CuNO
5, with three oxygen atoms of carboxylate groups belonging to two different ligands, one in a monodentate mode and the other one in a chelating mode, one nitrogen atom from the pyridinic ligand and two coordination water molecules. Bond lengths and angles, together with the values for continuous shape measurements (CSM) [
42] are summarized in
Table 1. Each carboxylate ligand acts as a four-connected node between four metal centers forming metal-carboxylate chains running along the [100] direction, whereas the
bpa ligand connects these chains to form covalent layers in the (01–2) plane.
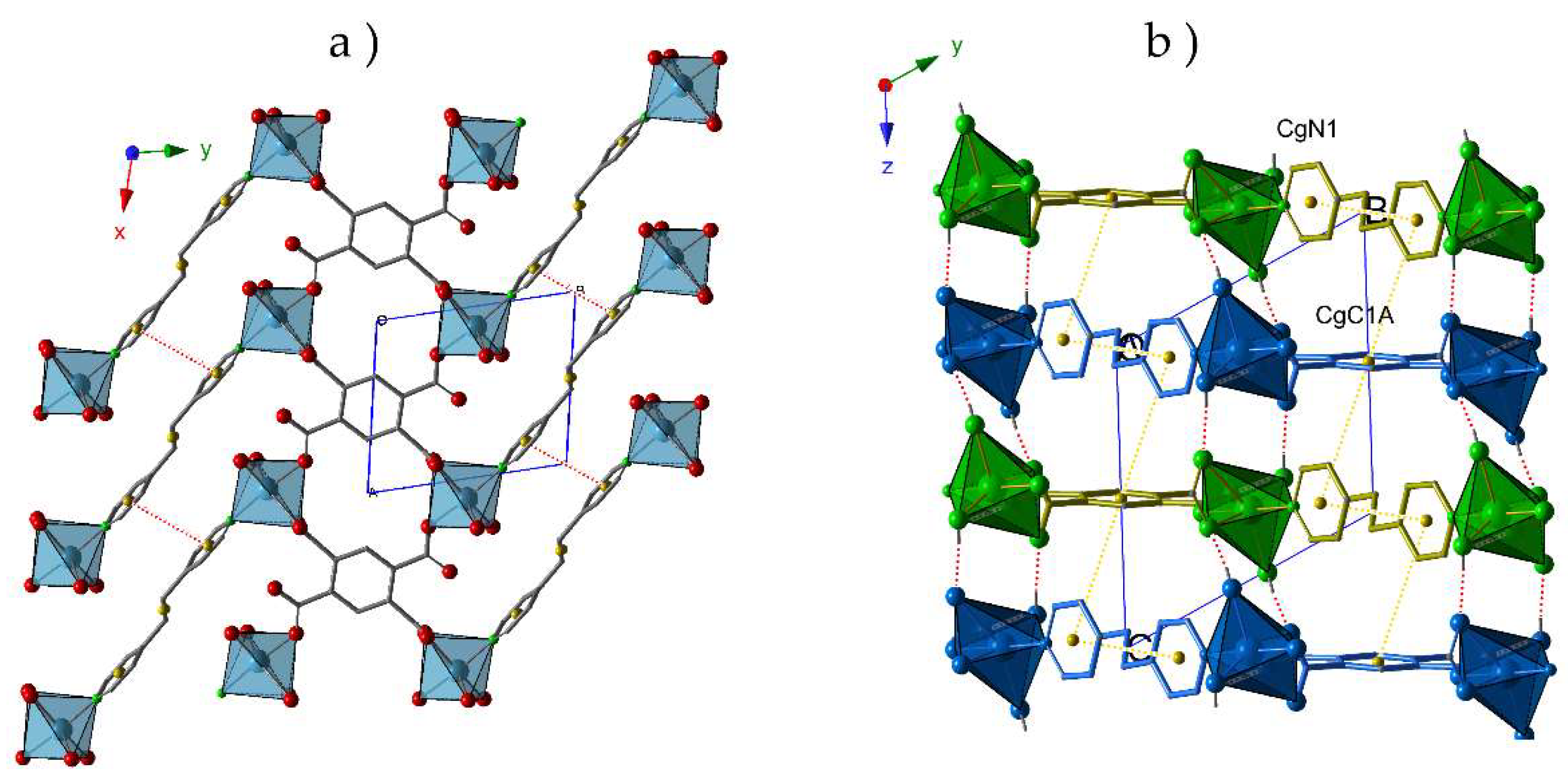
Thus, the crystal packing of compound
1 exhibits a bidimensional character (
Figure 1a) where the π-π interactions between the aromatic rings of different pyridinic ligands (N1···N1) contribute to reinforce these layers. As shown in
Figure 1b, the staking of these layers is antiparallel along the
z-axes, and proceeds through i) T-type π-π interactions between C1A and N1 rings from
btec and
bpa ligands, respectively; and ii) hydrogen bonds established between the coordination water molecules and O atoms from carboxylate groups (O1W-H1WA···O1A, O1W-H1WB···O4A and O2W-H2WA···O2A). Distances and angles of these supramolecular interactions are summarized in
Table 2.
The anhydrous
1.ah shows great differences with respect to the coordination environment of the metal centers in
1 and the arrangement of the ligands. Compound
1.ah crystallizes in the triclinic space group
P−1 and its asymmetric unit contains one Cu(II) ion, and half a ligand of each type (
Figure S3). The loss of coordination water molecules promotes the formation of a centrosymmetric copper (II) dimer, where each metal center displays a CuNO
4 environment with two bridging carboxylate ligands in a μ
2-η
1:η
1 coordination mode, a third bridging carboxylate ligand in a μ
2-η
2 fashion and N atoms from
bpa ligands completing the distorted pentacoordinated geometry. Bond lengths and angles together with the values for CSM are summarized in
Table 3.
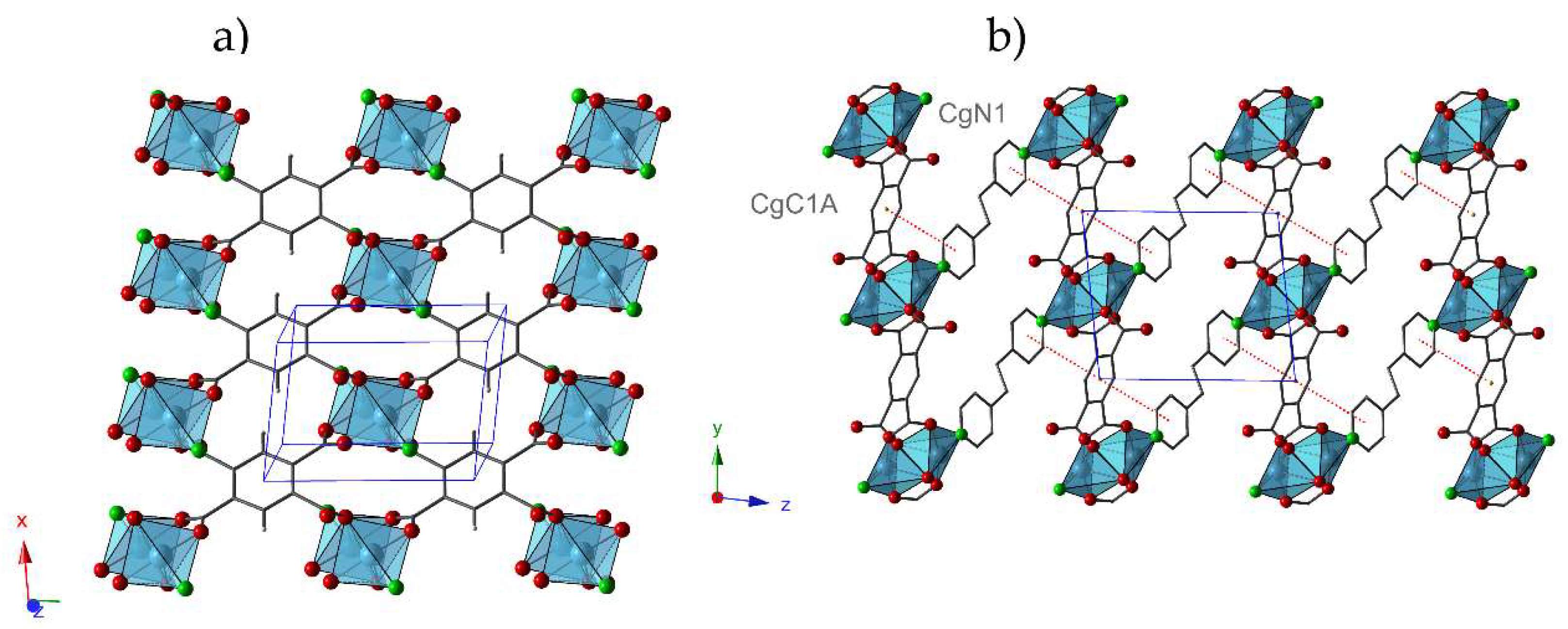
In addition, this structure results in an increase of the dimensionality of the framework in comparison with
1, because Cu(II) dimers in
1.ah can be visualized as being constituted by linking different metal centers located in adjacent layers in
1. In
1.ah, the
btec ligand acts as a tetra-connector node forming layers parallel to the (001) plane (
Figure 2a). These layers are stacked along the z-axis, where
bpa ligands play the role of covalent bridge between dimers resulting in a three-dimensional structure (
Figure 2b). The interactions between the aromatic rings of different types of ligands (N1···C1A) contribute to reinforce the network (
Table 4).
2.3. Thermal Behavior
The thermal stability of
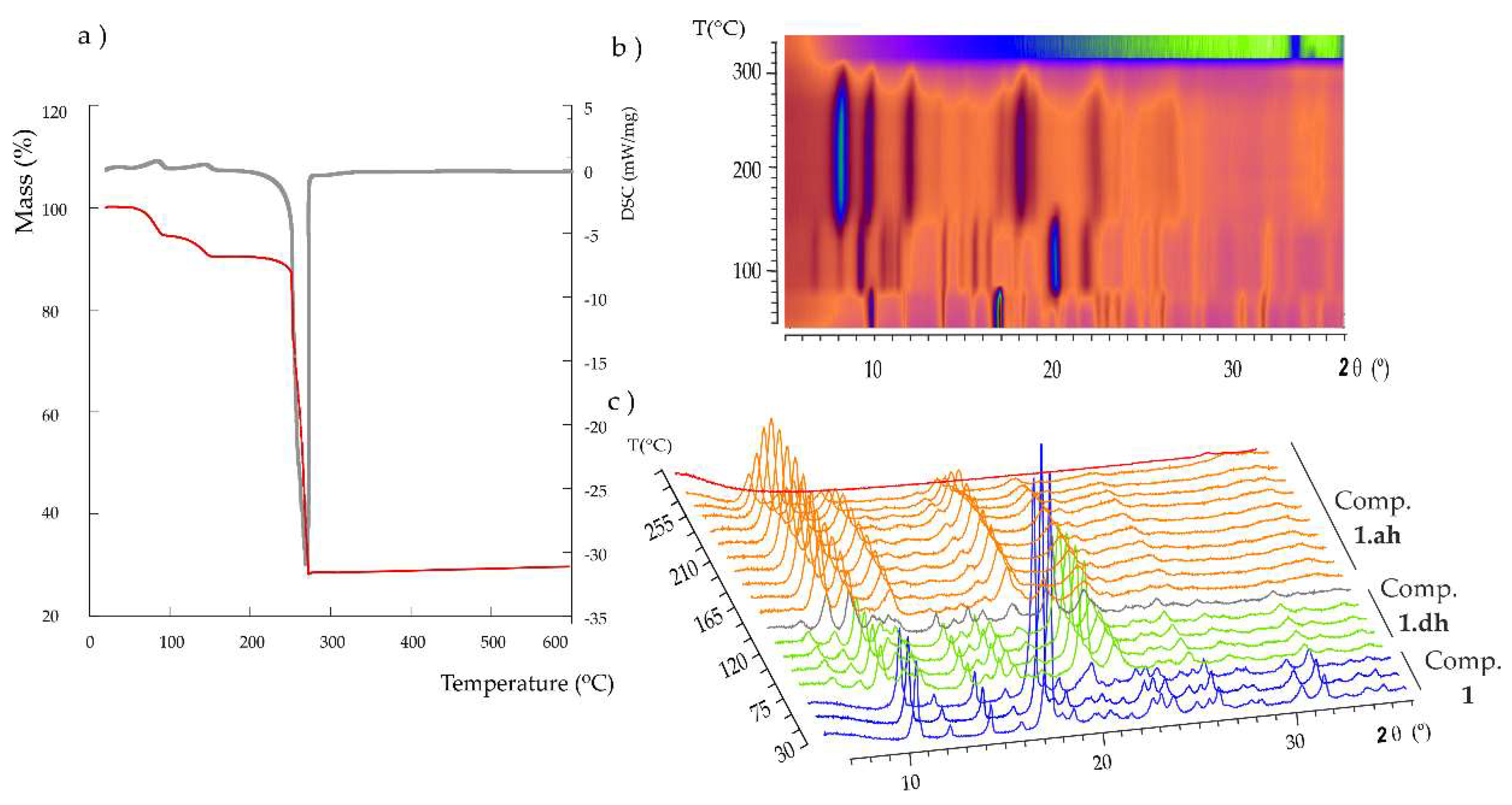
1 was studied by thermogravimetric analyses (TGA/DTA) carried out under synthetic air atmosphere in the temperature range of 25–500 °C at a heating rate of 5 °C/min. As observed in
Figure 3a, thermal decomposition proceeds via three different stages. The first mass loss below 100 °C corresponds to a dehydration process and it implies the release of one water molecule [%mass, calc. (found): 5.6 (5.3)]. The loss of the second coordination water molecule at ca. 160 °C [%mass, calc. (found): 5.6 (4.8)] results in the anhydrous phase, which is stable up to 220 °C. Above this temperature, an exothermic process takes place, which is associated with the combustion of the organic ligands and leads to the final residue at ca. 300 °C [% mass calc. (found) for CuO: 25.0 (28.0)]. The fact that the release of the two coordination water molecules takes place in two well-differentiated stages could be easily explained based on the crystal structure of
1. The first step implies the release of the axial water molecule (OW2), because it exhibits a much longer Cu–O bond length (2.304 Å) in comparison with that shown by the second water molecule (Cu–OW1: 1.975 Å). Furthermore, the latter species establishes two O–H···O type hydrogen bonds, as well as an additional C–H···O type interaction with adjacent carboxylate groups, whereas the former only forms one O–H···O bond (
Table 2).
Variable-temperature powder X-ray diffraction (termodiffractometry, TXRD) experiments were carried out from 30 to 500 °C at steps of 15 °C (
Figure 3b) to determine the potential phase transformations in
1 upon thermal dehydration and the subsequent range of thermal stability. These results revealed a dynamic thermostructural behavior for
1. The parent hydrated compound transforms into a second partially dehydrated phase (
1.dh) upon release of the first coordination water molecule above 75 °C, as indicated by the clear modifications in either the positions and relative intensity of most of the diffraction maxima in the
2θ = 5–30° angle range (
Figure 3c). More specifically, instead of the most intense diffraction maxima at 10.6 and 18.0° for
1, those at 10.0, 21.2 and 23.0° became predominant in the spectrum of the partially dehydrated phase. It is also worth noting the presence of two maxima of mid-to-low intensity at low
2θ values (7.4 and 8.7°) for the latter phase. A totally anhydrous crystalline phase is completely formed at 150 °C and keeps stable up to 250 °C, above which the diffraction maxima lose intensity until full amorphization is reached at 285 °C. This temperature is in good agreement with the results obtained from the TGA analyses, which show that the thermally stable anhydrous phase starts decomposing at 250 °C. The intensity of the diffraction maximum located at 8.8° increases significantly when going from the partially dehydrated phase to the anhydrous form, whereas those at 10.4, 12.9, 19.3 and 23.6° became the most intense. The final residue at 330 °C was identified as monoclinic
C2/c CuO, also known as tenorite [
43].
Taking into account the structural similarities between
1 and
1.ah, we hypothesized whether the latter compound could be prepared by thermal dehydration of the former. Thus, a crystalline sample of
1 was heated in the oven at 200 °C for 1 h, and immediately afterwards it was analyzed by powder XRD. There was a good agreement in the profile fitting procedure carried out between the experimental spectrum and the cell unit parameters obtained in the single crystal XRD studies (
Figure S4), revealing that
1.ah can be directly obtained by total thermal dehydration of
1. Cell parameters, the displacement of the sample, the shape of the diffraction maxima, as well as the angular evolution of the half-height width were refined using FULLPROF [
44]. Agreement factors of the fitting are summarized in
Table S2. Similarly, we tried to elucidate the crystal structure of the partially dehydrated phase by heating single crystals of
1 at 90 °C for 1 h. Unfortunately, despite our efforts, crystals did not preserve their integrity, and we were not able to perform a full single-crystal XRD data acquisition.
Figure S5 shows the experimental powder X-Ray diffraction patterns in comparison to those simulated from single crystal data for both compounds
1 and
1.ah. The good agreement between experimental and simulated patterns indicate the presence of pure crystalline phases in bulk samples.
2.4. Reversibility of Thermally Triggered Phase Transformation and EPR Espectroscopy
The reversibility of thermally triggered structural transformations that
1 undergoes and the associated solvatochromism (thermochromism) was studied by XRD (
Figure 4). A powdered light blue sample of
1 was heated at 90 °C in an oven and a clear color change to dark blue was observed as a result of modifications in the coordination environment of the Cu(II) center promoted by the thermal evacuation of the first coordination water molecule. When the partially dehydrated phase is exposed to ambient moisture, it reverts to the parent phase
1 upon re-hydration, as evidenced by its color change and the registered powder XRD pattern. As mentioned above, a second sample of
1 was heated to 200 °C, and the anhydrous
1.ah was obtained. The color change from blue to green arises not only from the removal of coordination water molecules, but also from modifications in the coordination modes of
btec ligands. In this case, the dehydration is not reversible because no rehydration is observed neither when the solid is exposed to ambient moisture, nor when it is soaked in water.
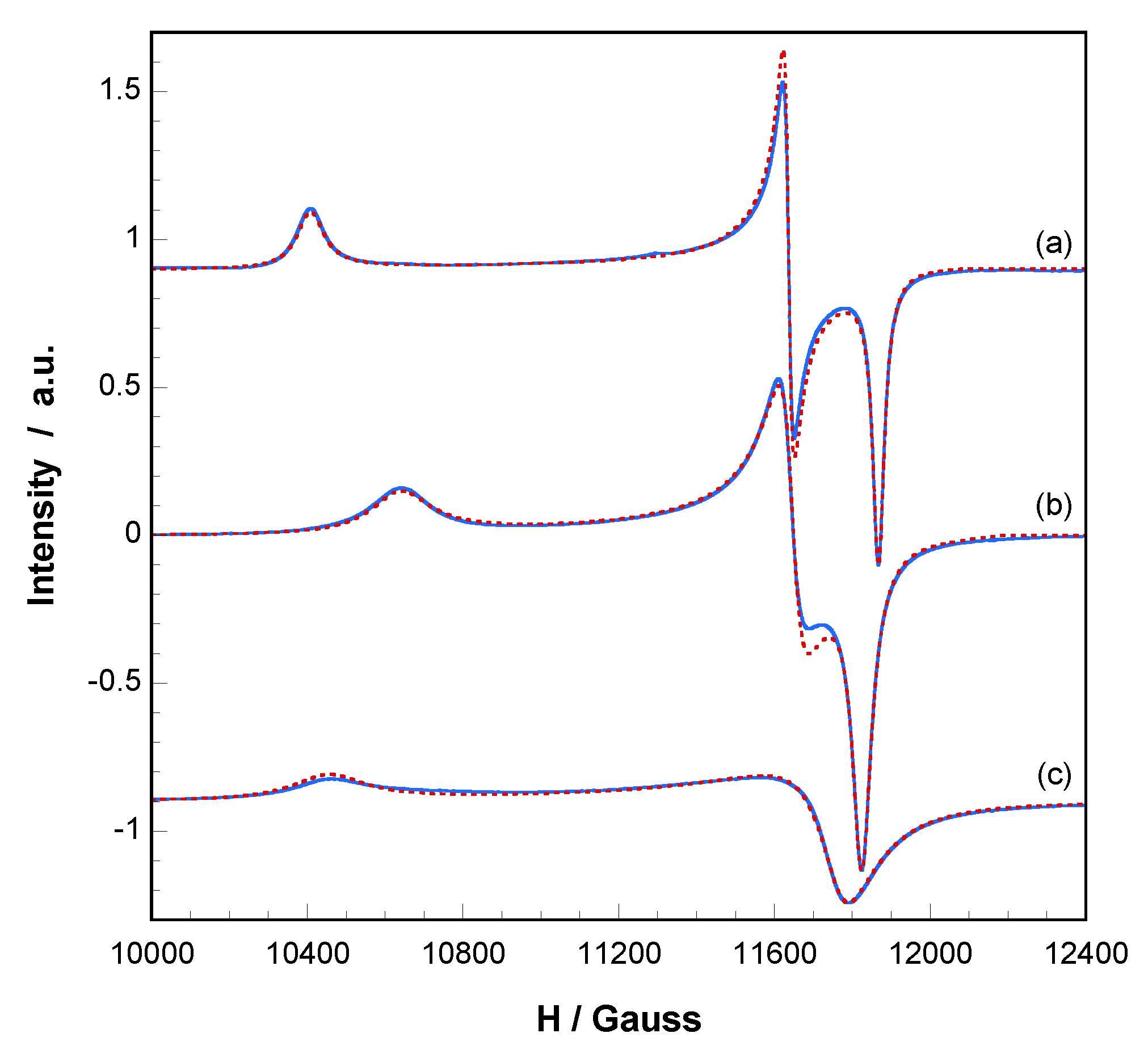
These phase transformations and their reversibility were monitored by electron paramagnetc resonance spectroscopy (EPR). X and Q band EPR measurements were carried out at several temperatures in the 4.2–298 K range. The X-band EPR spectra of
1 and the corresponding dehydrated phases exhibit nearly axial symmetries for the g tensor in the ΔMs = ± 1 region, but an appreciable extent of rhombicity can be detected operating at Q-band (
Figure 5). The spin Hamiltonian parameters were estimated by comparison of the experimental spectra with those obtained by a computer simulation program working at the second order of the perturbation theory. The best-fit results are represented as dashed lines in
Figure 5.
The main components of the g tensor for
1 are g
1 = 2.338, g
2 = 2.091 and g
3 = 2.051 (g
II = 2.338, g
⊥ = 2.071, <g> = 2.160). These values are typical of Cu(II) ions in distorted octahedral environment in good agreement with the structural characteristics of the CuNO
5 chromophore. In addition, the lowest g value deviates appreciably from the free electron value (g
e = 2.0023), indicating a d
x2-
y2 ground state which corresponds to that expected for an axially elongated octahedral Cu(II) ion [
45].
After heating a sample of
1 at 90 °C, the spectrum continues displaying a single and well-defined signal, with rhombic g values of g
1 = 2.287, g
2 = 2.090 and g
3 = 2.058 (g
II = 2.287, g
⊥ = 2.074, <g> = 2.145). The reduction of g
II is in good agreement with the loss of the water molecule in apical position. As the axial field is removed, the copper ion attracts the equatorial ligands more strongly, the d
x2-
y2 orbital becomes more antibonding and the g
II value decreases [
46]. It must be taken into account that, for systems with similar covalence degree, the g values depend mainly on the energy of the d-d transitions, following these Equations:
where λ
0 is the free ion spin-orbit coupling constant (λ
0 = −830 cm
−1 for Cu
2+), K
II and K
⊥ are the covalence factors, and Δ
1 and Δ
2 are the energies of the d
xy→d
x2-
y2 and d
xz,yz→d
x2-
y2 transitions, respectively [
47]. Diffuse reflectance UV-Vis spectra (
Figure S6) showed that the center of gravity of the d-d transitions shift to higher energies when the first water molecule is removed. This effect can be visually observed in its color change (from a lighter to a darker blue) and it is in good agreement with the decrease of the <g> value.
When the sample is heated at 200 °C,
1.ah is obtained, and thus, a new EPR signal is registered showing different g values: g
1 = 2.327, g
2 = 2.075 and g
3 = 2.069 (g
II = 2.327, g
⊥ = 2.072, <g> = 2.157). The relative increase of the g
II component implies that the loss of the second water molecule is accompanied by changes in the coordination of the Cu(II) ions, which results in a slight increase of the equatorial distances [
46]. This is in good agreement with the crystallographic data. In addition, it is worth noting that the Cu(II) hyperfine lines could not be resolved in any of these compounds and therefore extensive magnetic exchange is present in all of them. However, the G parameters, which are in the range 3.8–4.8, indicate that the local tetragonal axes of the molecules are aligned parallel or only slightly misaligned [
45]. Therefore, it can be assumed that the calculated g values correspond to the molecular tensors and adequately reflect the characteristics of the environment of the Cu
2+ ions in each compound. Moreover, the EPR spectra of compound
1 and the dehydrated phase at 90 °C remain practically unchanged over the temperature range 4.2–298 K, so the magnetic interactions should be of small magnitude in both compounds. On the contrary, the intensity of the EPR signal of compound
1.ah decrease below 90 K, indicating that moderate antiferromagnetic exchange is operative between the copper(II) ions of the dimeric entity. Thus, temperature dependent magnetic measurements were carried out.
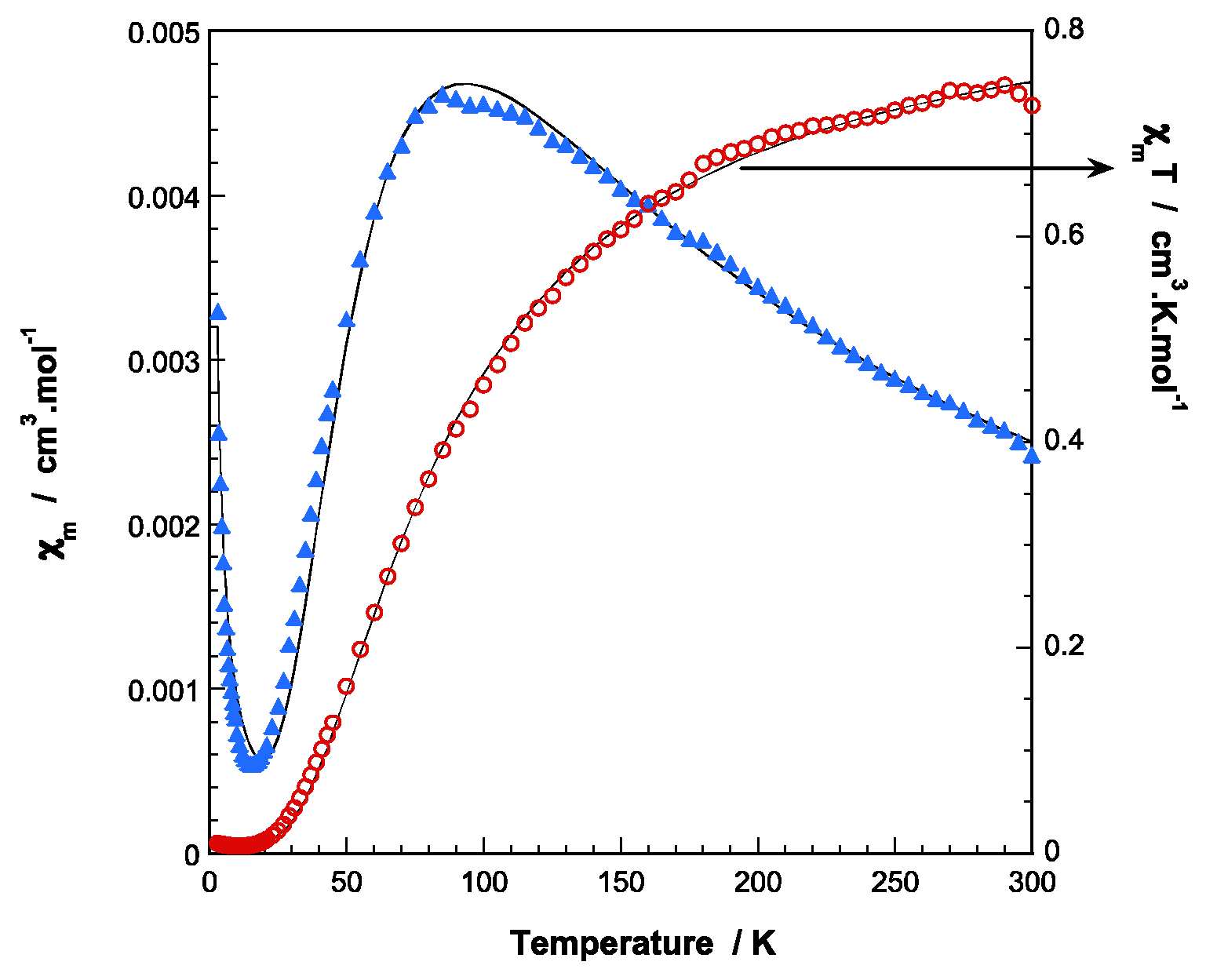
The magnetic susceptibility (
χm) of
1 reveals a Curie-Weiss behavior for the whole temperature range analyzed and the fit of the data to the corresponding expression leads to
Cm and θ values of 0.44 cm
3K/mol and −0.2 K, respectively (
Figure S7). Conversely, temperature-dependent susceptibility data registered for
1.ah displays a maximum at ca. 90 K and decreases to 15 K upon cooling. Below this temperature, there is a drastic increase in magnetic susceptibility as the temperature decreases. The room temperature magnetic moment (
χmT = 0.73 cm
3K/mol,
μeff = 3.42 BM) is considerably lower than that expected for two magnetically isolated copper (II) ions with g=2.16 (
χmT = 0.87 cm
3K/mol,
μeff = 3.74 MB). The
χmT curve continuously decreases when cooling, until it almost vanishes at very low temperatures. These features are consistent with moderately strong antiferromagnetic exchange between the Cu(II) ions within the dimer. The behavior below 15 K could be explained by the presence of a small amount of monomeric Cu(II) species. According to these observations, the experimental data can be well fitted to the Bleaney and Bowers equation for a dinuclear Cu(II) complex [
48] modified with an additional term to take into account the presence of non-coupled Cu(II) impurities following a simple Curie law with the same g factor:
where the singlet-triplet energy gap (2
J) is defined by the Hamiltonian
H = −2
J·S1·S2 (
S1 =
S2 = 1/2),
ρ is the percent of non-coupled component and other symbols have their usual meanings. A good fit to the data (solid lines in
Figure 6) was obtained when g = 2.16,
J = −52.4 cm
−1, and
ρ = 0.013, with an error R =2.4 × 10
−4. According to the topology of the Cu···Cu dimeric entities in
1.ah, different exchange pathways must be taken into account: i) two axial-equatorial monoatomic carboxylate oxygen bridges, ii) two carboxylate bridges in
syn-syn mode. It is well known that axial–equatorial bridging modes between Cu
2+ ions provide weak spin communication between magnetic d
x2−y2 orbitals, thus the observed
J value can be mainly assigned to the
syn-syn bridges [
49].
2.5. Topological Study of the bpa Ligand
Although it exhibits limited coordination modes (monodentate or ambidentate), the
bpa ligand usually acts as linker of metal centers leading to extended structures with very different topologies. An exhaustive bibliographic search in the Cambridge Crystallographic Database (CSD) [
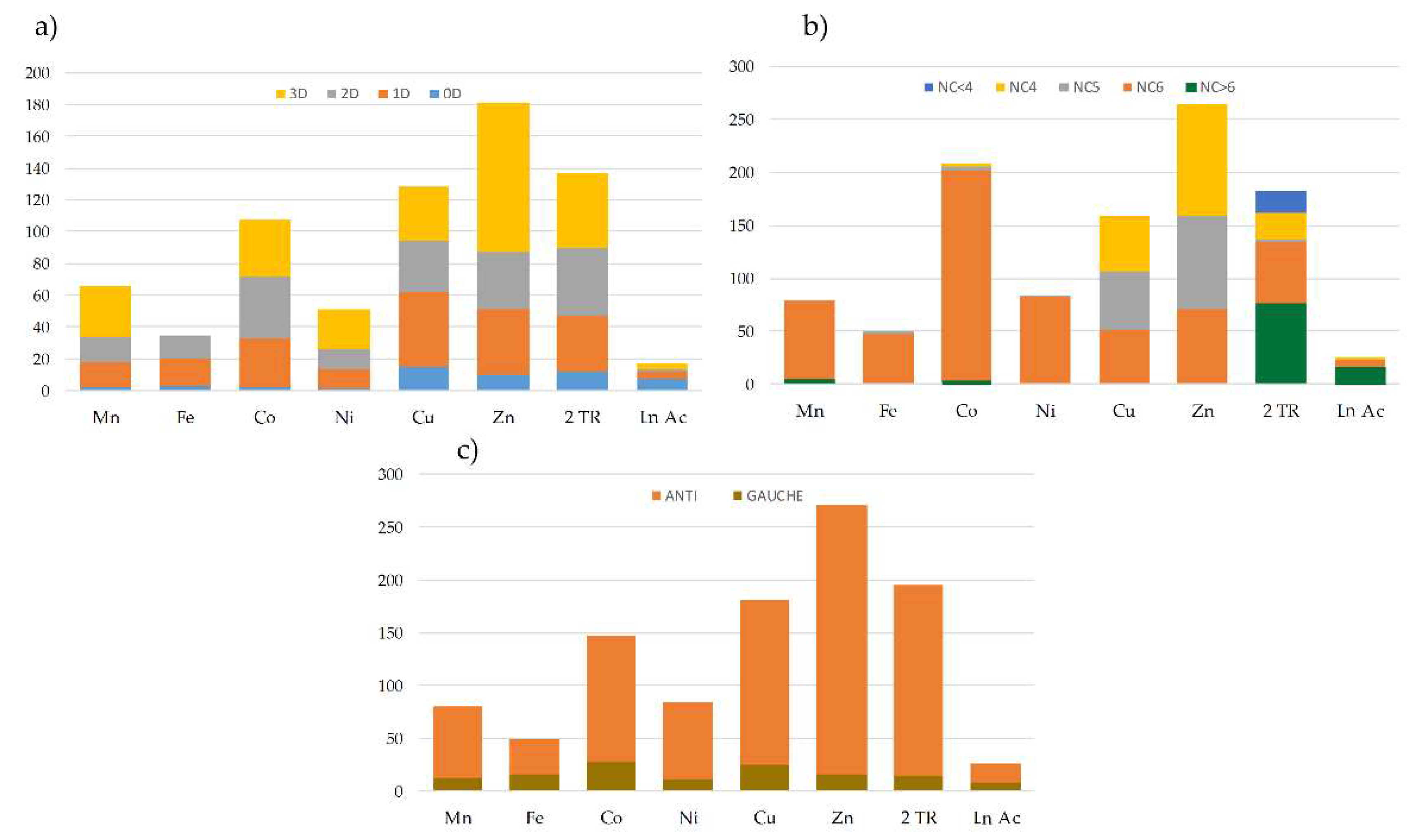
50] of structures containing transition metal ions, lanthanides and actinides provided us with a set of 1197 structures in which the
bpa ligand plays a bridging role between metal centers.
Figure 7 shows the general distribution of these structures according to their dimensionality (
Figure 7a), the coordination number of the metal centers (
Figure 7b), and the conformation of the ligand (
Figure 7c). It must be highlighted that most of the structures found in the database are combinations of the
bpa ligand with first raw transition metals in a hexacoordinated environment, and only a few examples belong to the families of 4d and 4f/5f metals. In all cases, the
anti conformation of the
bpa is clearly predominant over the
gauche form (for representations of the extreme conformations see
Figure S8).
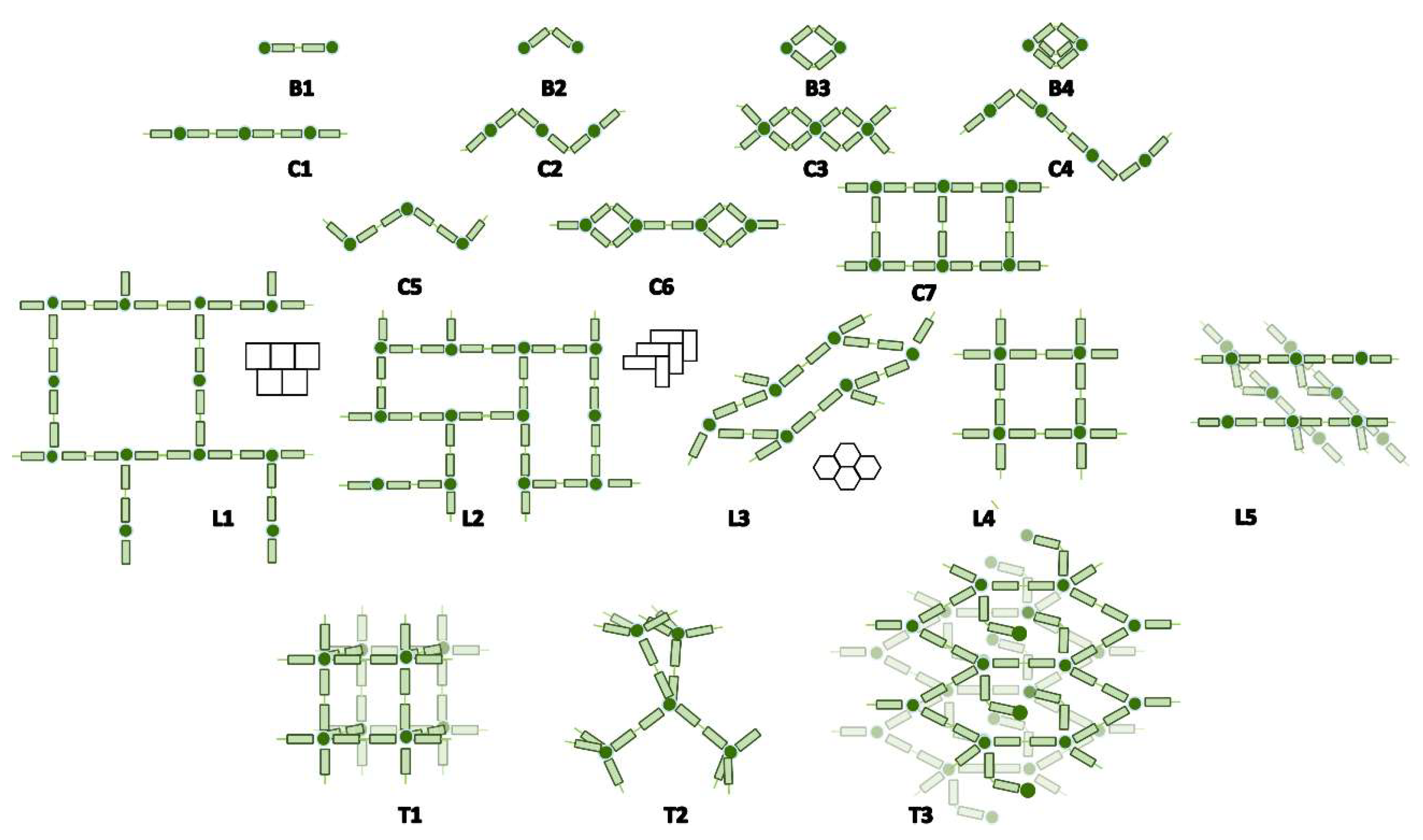
Close inspection of the crystal structures showed four different ways in which the
bpa ligand could contribute to the final dimensionality of the network. Beginning from the most frequent forms, these are: (i) zero connector or bridge (B); (ii) mono-dimensional connector or linear chain (C); (iii) bi-dimensional connector or layers (L); and (iv) three-dimensional network (T). These modes were classified according to their topology as displayed in
Figure 8. Additionally,
Table 5 compiles the number of structures of each possible topology, together with a brief description and a representative example for each case. The
bpa ligand in both compounds
1 and
1.ah reported in this work acts as a B1 linker, which increases the dimensionality of Cu(II)-btec systems from 1D to 2D for
1 and from 2D to 3D for
1.ah.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}