Design and Synthesis of a Series of Truncated Neplanocin Fleximers


Abstract
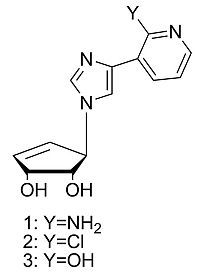
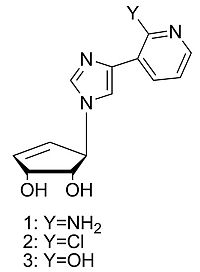
:
1. Introduction

2. Results and Discussion
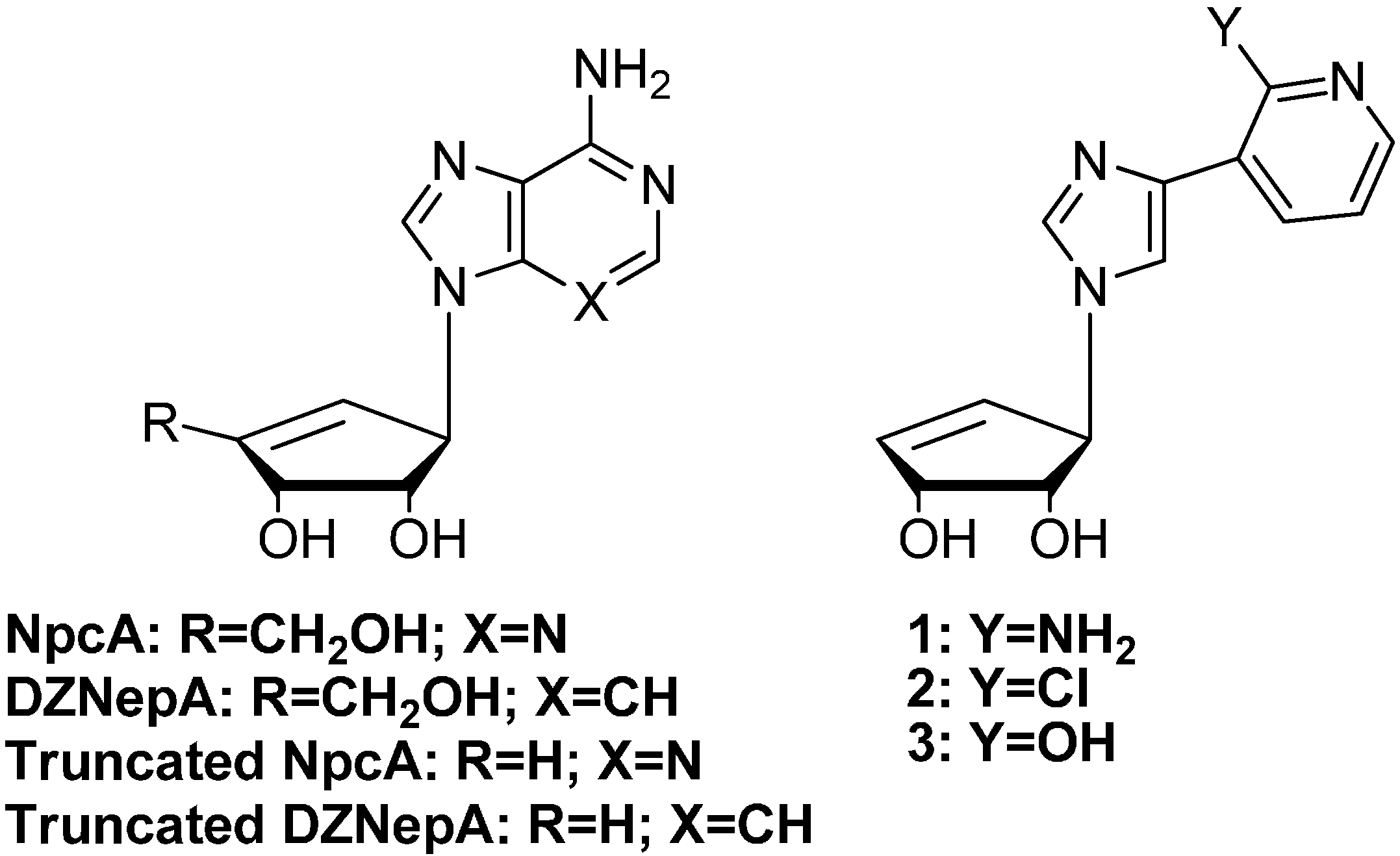
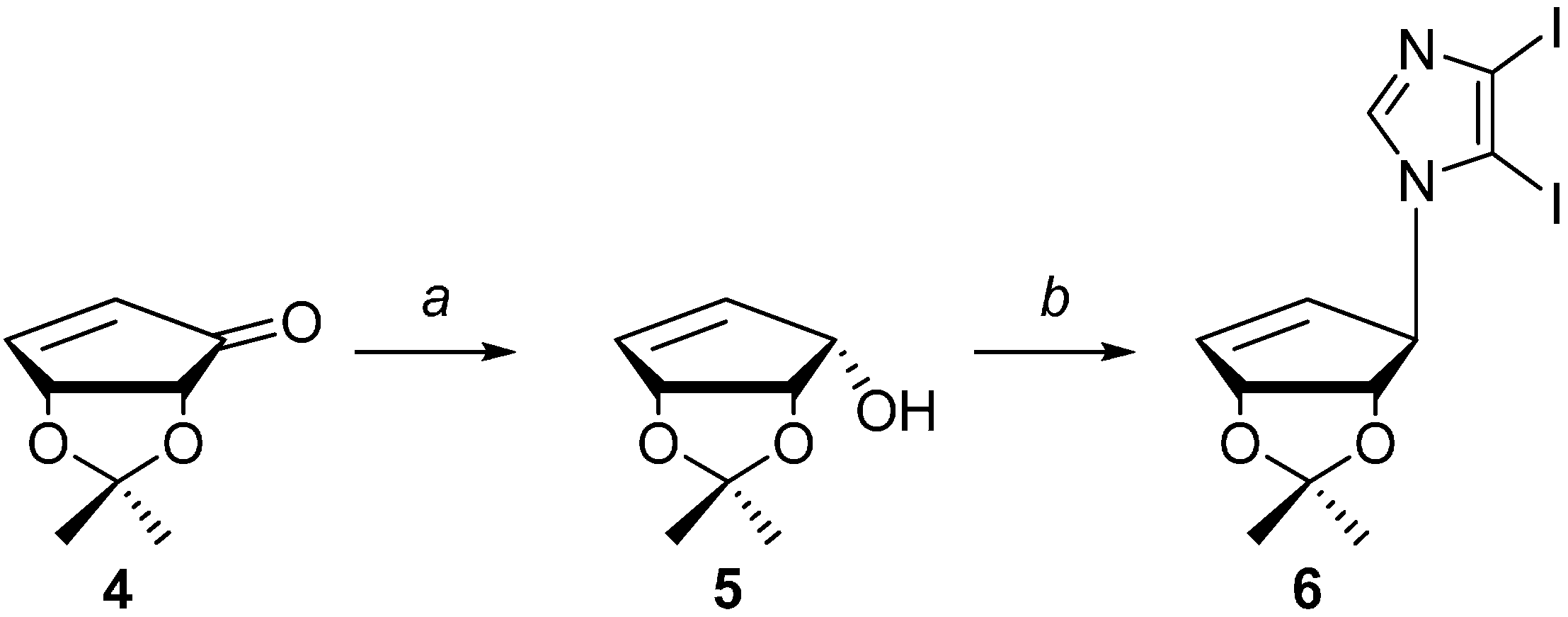
2.1. Chemistry


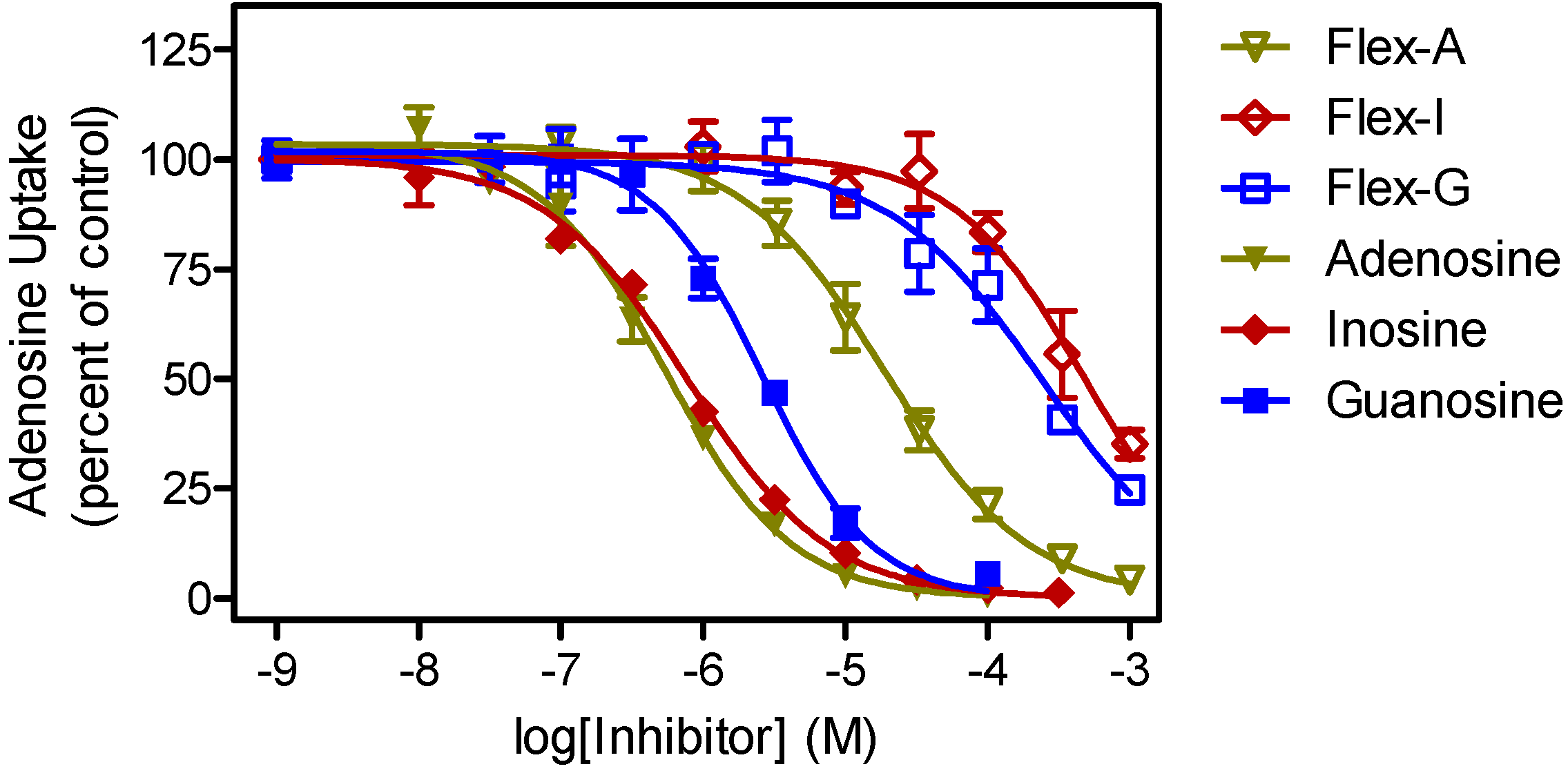
2.2. Trypanosomiasis Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Average EC50 (µM) |
|---|---|
| 1 | 216 ± 21 |
| 2 | 212 ± 31 |
| 3 | 287 ± 24 |
| pentamidine | 0.0044 ± 0.0001 |


| P1 Ki (μM) | P2 Ki (µM) | |||||
|---|---|---|---|---|---|---|
| Nucleoside 1 | Fleximer | δ(ΔG0) | Nucleoside 1 | Fleximer | δ(ΔG0) | |
| Adenosine | 0.36 ± 0.05 | 35 ± 11 | 11.4 | 0.91 ± 0.29 | 37 ± 3 | 9.2 |
| Guanosine | 1.8 ± 0.3 | 251 ± 75 | 12.2 | >500 | >500 | |
| Inosine | 0.44 ± 0.10 | 387 ± 30 | 16.8 | >500 | >500 | |
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.3. Anti-Trypanosome Activity
3.4. Transport Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tseng, C.K.; Marquez, V.E.; Fuller, R.W.; Goldstein, B.M.; Haines, D.R.; McPherson, H.; Parsons, J.L.; Shannon, W.M.; Arnett, G.; Hollingshead, M.; et al. Synthesis of 3-deazaneplanocin A, a powerful inhibitor of S-adenosylhomocysteine hydrolase with potent and selective in vitro and in vivo antiviral activities. J. Med. Chem. 1989, 32, 1442–1446. [Google Scholar] [CrossRef]
- Cantoni, G. The centrality of S-adenosylhomocysteinase in the regulation of the biological utilization of S-adenosylmethionine. In Biological Methylation and Drug Design; Borchardt, R.T., Creveling, C.R., Ueland, P.M., Eds.; Humana Press: Clifton, NJ, USA, 1986; pp. 227–238. [Google Scholar]
- Cantoni, G.L.; Scarano, E. The formation of S-adenosylhomocysteine in enzymatic transmethylation reactions. J. Am. Chem. Soc. 1954, 76, 4744. [Google Scholar] [CrossRef]
- Borchardt, R.T.; Keller, B.T.; Patel-Thombre, U.; Neplanocin, A. A potent inhibitor of S-adenosylhomocysteine hydrolase and of vaccinia virus multiplication in mouse L929 cells. J. Biol. Chem. 1984, 259, 4353–4358. [Google Scholar] [PubMed]
- Bujnicki, J.M.; Prigge, S.T.; Caridha, D.; Chiang, P.K. Structure, evolution, and inhibitor interaction of S-adenosyl-l-homocysteine hydrolase from Plasmodium falciparum. Proteins 2003, 52, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.K. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 1998, 77, 115–134. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. John Montgomery’s legacy: Carbocyclic adenosine analogues as SAH hydrolase inhibitors with broad-spectrum antiviral activity. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1395–1415. [Google Scholar]
- Marquez, V.E. Carbocyclic nucleosides. Adv. Antivir. Drug Des. 1996, 2, 89–146. [Google Scholar]
- Borchardt, R.T.; Wu, Y.-S. S-Aristeromycinyl-l-homocysteine, a potent inhibitor of S-adenosylmethionine-dependent transmethylations. J. Med. Chem. 1976, 19, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Guranowski, A.; Montgomery, J.A.; Cantoni, G.L.; Chiang, P.K. Adenosine analogues as substrates and inhibitors of S-adenosylhomocysteine hydrolase. Biochemistry 1981, 20, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Van Brummelen, A.C.; Olszewski, K.L.; Wilinski, D.; Llinas, M.; Louw, A.I.; Birkholtz, L.M. Co-inhibition of Plasmodium falciparum S-adenosylmethionine decarboxylase/ornithine decarboxylase reveals perturbation-specific compensatory mechanisms by transcriptome, proteome, and metabolome analyses. J. Biol. Chem. 2009, 284, 4635–4646. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Borchardt, R.T. S-adenosyl-l-homocysteine hydrolase as a target for antiviral chemotherapy. J. Med. Chem. 1991, 34, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Hasobe, M.; Liang, H.; Ault-Riche, D.B.; Borcherding, D.R.; Wolfe, M.S.; Borchardt, R.T. (1'R,2'S,3'R)-9-(2',3'-Dihydroxycyclopentan-1'-yl)-adenine and -3-deaza-adenine: Analogs of aristeromycin which exhibit potent antiviral activity with reduced cytotoxicity. Antivir. Chem. Chemother. 1993, 4, 245–248. [Google Scholar]
- Wolfe, M.S.; Lee, Y.; Bartlett, W.J.; Borcherding, D.R.; Borchardt, R.T. 4'-Modified analogs of aristeromycin and neplanocin A: Synthesis and inhibitory activity toward S-adenosyl-l-homocysteine hydrolase. J. Med. Chem. 1992, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Richards, H.H.; Chiang, P.K.; Cantoni, G.L. Adenosylhomocysteine hydrolase. Crystallization of the purified enzyme and its properties. J. Biol. Chem. 1978, 253, 4476–4480. [Google Scholar] [PubMed]
- Hasobe, M.; McKee, J.G.; Borcherding, D.R.; Borchardt, R.T. 9-(trans-2',trans-3'-Dihydroxycyclopent-4'-enyl)-adenine and -3-deazaadenine: Analogs of neplanocin A which retain potent antiviral activity but exhibit reduced cytotoxicity. Antimicrob. Agents Chemother. 1987, 31, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.P.; Shen, S.; Ullu, E.; Tschudi, C. Evidence for a capping enzyme with specificity for the trypanosome spliced leader RNA. Mol. Biochem. Parasitol. 2007, 156, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Mair, G.; Ullu, E.; Tschudi, C. Cotranscriptional Cap 4 Formation on the Trypanosoma brucei spliced leader RNA. J. Biol. Chem. 2000, 275, 28994–28999. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, J.R.; Mittra, B.; Campbell, D.A.; Sturm, N.R. Hypermethylated cap 4 maximizes Trypanosoma brucei translation. Mol. Microbiol. 2009, 72, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Zhang, L.; Hagos, A. “Fleximers”. Design and synthesis of two novel split nucleosides. Org. Lett. 2001, 3, 3209–3210. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Zhang, L.; Hagos, A.; Quirk, S. “Fleximers”. Design and synthesis of a new class of novel shape-modified nucleosides. J. Org. Chem. 2002, 67, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Quirk, S.; Salim, S.; Zhang, L.; Hagos, A. Unexpected inhibition of S-adenosyl-l-homocysteine hydrolase by a guanosine nucleoside. Bioorg. Med. Chem. Lett. 2003, 13, 1985–1988. [Google Scholar] [CrossRef] [PubMed]
- Polak, M.; Seley, K.L.; Plavec, J. Conformational properties of shape modified nucleosides—Fleximers. J. Am. Chem. Soc. 2004, 126, 8159–8166. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Salim, S.; Zhang, L. “Molecular chameleons”. Design and synthesis of C-4-substituted imidazole fleximers. Org. Lett. 2005, 7, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Salim, S.; Zhang, L.; O’Daniel, P.I. “Molecular chameleons”. Design and synthesis of a second series of flexible nucleosides. J. Org. Chem. 2005, 70, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.C.; Sadler, J.M.; Andrei, G.; Snoeck, R.; Balzarini, J.; Seley-Radtke, K.L. Carbocyclic 5'-nor “reverse” fleximers. Design, synthesis, and preliminary biological activity. Med. Chem. Commun. 2011, 2, 650–654. [Google Scholar] [CrossRef]
- Wauchope, O.R.; Velasquez, M.; Seley-Radtke, K. Synthetic routes to a series of proximal and distal 2'-deoxy fleximers. Synthesis (Stuttg.) 2012, 44, 3496–3504. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Sadler, J.M.; O’Daniel, P.I.; Kim, N.T.; Seley-Radtke, K.L. “Reverse” carbocyclic fleximers: Synthesis of a new class of adenosine deaminase inhibitors. Nucleosides Nucleotides Nucleic Acids 2013, 32, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Quirk, S.; Seley, K.L. Identification of catalytic amino acids in the human GTP fucose pyrophosphorylase active site. Biochemistry 2005, 44, 13172–13178. [Google Scholar] [CrossRef] [PubMed]
- Quirk, S.; Seley, K.L. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry 2005, 44, 10854–10863. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Scott-Finnigan, T.J. Trypanocidal activity of antitumor antibiotics and other metabolic inhibitors. Antimicrob. Agents Chemother. 1978, 13, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Sufrin, J.R.; Rattendi, D.; Spiess, A.J.; Lane, S.; Marasco, C.J., Jr.; Bacchi, C.J. Antitrypanosomal activity of purine nucleosides can be enhanced by their conversion to O-acetylated derivatives. Antimicrob. Agents Chemother. 1996, 40, 2567–2572. [Google Scholar] [PubMed]
- Cai, S.; Li, Q.S.; Borchardt, R.T.; Kuczera, K.; Schowen, R.L. The antiviral drug ribavirin is a selective inhibitor of S-adenosyl-l-homocysteine hydrolase from Trypanosoma cruzi. Bioorg. Med. Chem. 2007, 15, 7281–7287. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Schneller, S.W.; Rattendi, D.; Bacchi, C.J. (+)-7-deaza-5'-noraristeromycin as an anti-trypanosomal agent. J. Med. Chem. 1997, 40, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ye, W.; Schneller, S.W. Preparation of carbocyclic S-adenosylazamethionine accompanied by a practical synthesis of (−)-aristeromycin. J. Org. Chem. 2004, 69, 3993–3996. [Google Scholar] [CrossRef] [PubMed]
- Luche, J.-L. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J. Am. Chem. Soc. 1978, 100, 2226–2227. [Google Scholar] [CrossRef]
- Swamy, K.C.; Kumar, N.N.; Balaraman, E.; Kumar, K.V. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Moussa, Z. The Hendrickson ‘POP’ reagent and analogues thereof: Synthesis, structure, and application in organic synthesis. ARKIVOC 2012, 432–490. [Google Scholar]
- Kim, J.-H.; Kim, H.O.; Lee, K.M.; Chun, M.W.; Moon, H.R.; Jeong, L.S. Asymmetric synthesis of homo-apioneplanocin A from d-ribose. Tetrahedron 2006, 62, 6339–6342. [Google Scholar] [CrossRef]
- Hassan, J.; Sevignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002, 102, 1359–1469. [Google Scholar] [CrossRef] [PubMed]
- Vo, G.D.; Hartwig, J.F. Palladium-catalyzed coupling of ammonia with aryl chlorides, bromides, iodides, and sulfonates: A general method for the preparation of primary arylamines. J. Am. Chem. Soc. 2009, 131, 11049–11061. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Buchwald, S.L. New ammonia equivalents for the Pd-catalyzed amination of aryl halides. Org. Lett. 2001, 3, 3417–3419. [Google Scholar] [CrossRef] [PubMed]
- Benkeser, R.A.; Buting, W.E. The preparation of aromatic amines with sodium amide in liquid ammonia. J. Am. Chem. Soc. 1952, 74, 3011–3014. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Q.; Luo, M. Reduction of hydrazines to amines with aqueous solution of titanium(iii) trichloride. Org. Biomol. Chem. 2011, 9, 4977–4982. [Google Scholar] [CrossRef] [PubMed]
- Matovu, E.; Stewart, M.L.; Geiser, F.; Brun, R.; Maser, P.; Wallace, L.J.; Burchmore, R.J.; Enyaru, J.C.; Barrett, M.P.; Kaminsky, R.; et al. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Aguinaga Andres, D.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P.; Jarvis, S.M. Adenosine transporters in bloodstream forms of Trypanosoma brucei brucei: Substrate recognition motifs and affinity for trypanocidal drugs. Mol. Pharmacol. 1999, 56, 1162–1170. [Google Scholar] [PubMed]
- De Koning, H.P.; Bridges, D.J.; Burchmore, R.J.S. Purine and pyrimidine transport in pathogenic protozoa: From biology to therapy. FEMS Microbiol. Rev. 2005, 29, 987–1020. [Google Scholar] [CrossRef] [PubMed]
- Al-Salabi, M.I.; Wallace, L.J.; Luscher, A.; Maser, P.; Candlish, D.; Rodenko, B.; Gould, M.K.; Jabeen, I.; Ajith, S.N.; de Koning, H.P. Molecular interactions underlying the unusually high adenosine affinity of a novel Trypanosoma brucei nucleoside transporter. Mol. Pharmacol. 2007, 71, 921–929. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P. Transporters in African trypanosomes: Role in drug action and resistance. Int. J. Parasitol. 2001, 31, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.C.; Rojas Lopez, K.E.; Eze, A.A.; Delespaux, V.; van den Abbeele, J.; Rowan, T.; Barrett, M.P.; Morrison, L.J.; de Koning, H.P. Functional expression of TcoAT1 reveals it to be a P1-type nucleoside transporter with no capacity for diminazene uptake. Int. J. Parasitol. Drugs Drug Resist. 2013, 3, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.J.; Candlish, D.; de Koning, H.P. Different substrate recognition motifs of human and trypanosome nucleobase transporters. Selective uptake of purine antimetabolites. J. Biol. Chem. 2002, 277, 26149–26156. [Google Scholar] [CrossRef] [PubMed]
- Natto, M.J.; Wallace, L.J.; Candlish, D.; Al-Salabi, M.I.; Coutts, S.E.; de Koning, H.P. Trypanosoma brucei: Expression of multiple purine transporters prevents the development of allopurinol resistance. Exp. Parasitol. 2005, 109, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.A.; Creek, D.J.; Burgess, K.; Allison, H.C.; Field, M.C.; Maser, P.; de Koning, H.P. Pyrimidine salvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines. Mol. Pharmacol. 2013, 83, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.B.; Yang, X.; Hanke, J.; Mason, K.A.; Schowen, R.L.; Borchardt, R.T.; Yin, D.H. Trypanosoma cruzi: Molecular cloning and characterization of the S-adenosylhomocysteine hydrolase. Exp. Parasitol. 2003, 105, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Geiser, F.; Luscher, A.; de Koning, H.P.; Seebeck, T.; Maser, P. Molecular pharmacology of adenosine transport in Trypanosoma brucei: P1/P2 revisited. Mol. Pharmacol. 2005, 68, 589–595. [Google Scholar] [PubMed]
- Vodnala, S.K.; Lundbäck, T.; Yeheskieli, E.; Sjöberg, B.; Gustavsson, A.-L.; Svensson, R.; Olivera, G.C.; Eze, A.A.; de Koning, H.P.; Hammarström, L.G.J.; et al. Structure-activity relationships of synthetic cordycepin analogues as experimental therapeutics for African Trypanosomiasis. J. Med. Chem. 2013, 56, 9861–9873. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P.; Al-Salabi, M.I.; Cohen, A.M.; Coombs, G.H.; Wastling, J.M. Identification and characterisation of high affinity nucleoside and nucleobase transporters in Toxoplasma gondii. Int. J. Parasitol. 2003, 33, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Quashie, N.B.; Dorin-Semblat, D.; Bray, P.G.; Biagini, G.A.; Doerig, C.; Ranford-Cartwright, L.C.; de Koning, H.P. A comprehensive model of purine uptake by the malaria parasite Plasmodium falciparum: Identification of four purine transport activities in intraerythrocytic parasites. Biochem. J. 2008, 411, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors at this time.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, S.C.; O'Neill, E.; Ebiloma, G.U.; Wallace, L.J.M.; De Koning, H.P.; Seley-Radtke, K.L. Design and Synthesis of a Series of Truncated Neplanocin Fleximers. Molecules 2014, 19, 21200-21214. https://doi.org/10.3390/molecules191221200
Zimmermann SC, O'Neill E, Ebiloma GU, Wallace LJM, De Koning HP, Seley-Radtke KL. Design and Synthesis of a Series of Truncated Neplanocin Fleximers. Molecules. 2014; 19(12):21200-21214. https://doi.org/10.3390/molecules191221200
Chicago/Turabian StyleZimmermann, Sarah C., Elizaveta O'Neill, Godwin U. Ebiloma, Lynsey J. M. Wallace, Harry P. De Koning, and Katherine L. Seley-Radtke. 2014. "Design and Synthesis of a Series of Truncated Neplanocin Fleximers" Molecules 19, no. 12: 21200-21214. https://doi.org/10.3390/molecules191221200