Pd0-Catalyzed Methyl Transfer on Nucleosides and Oligonucleotides, Envisaged as a PET Tracer

Abstract
:1. Introduction

2. Results and Discussion



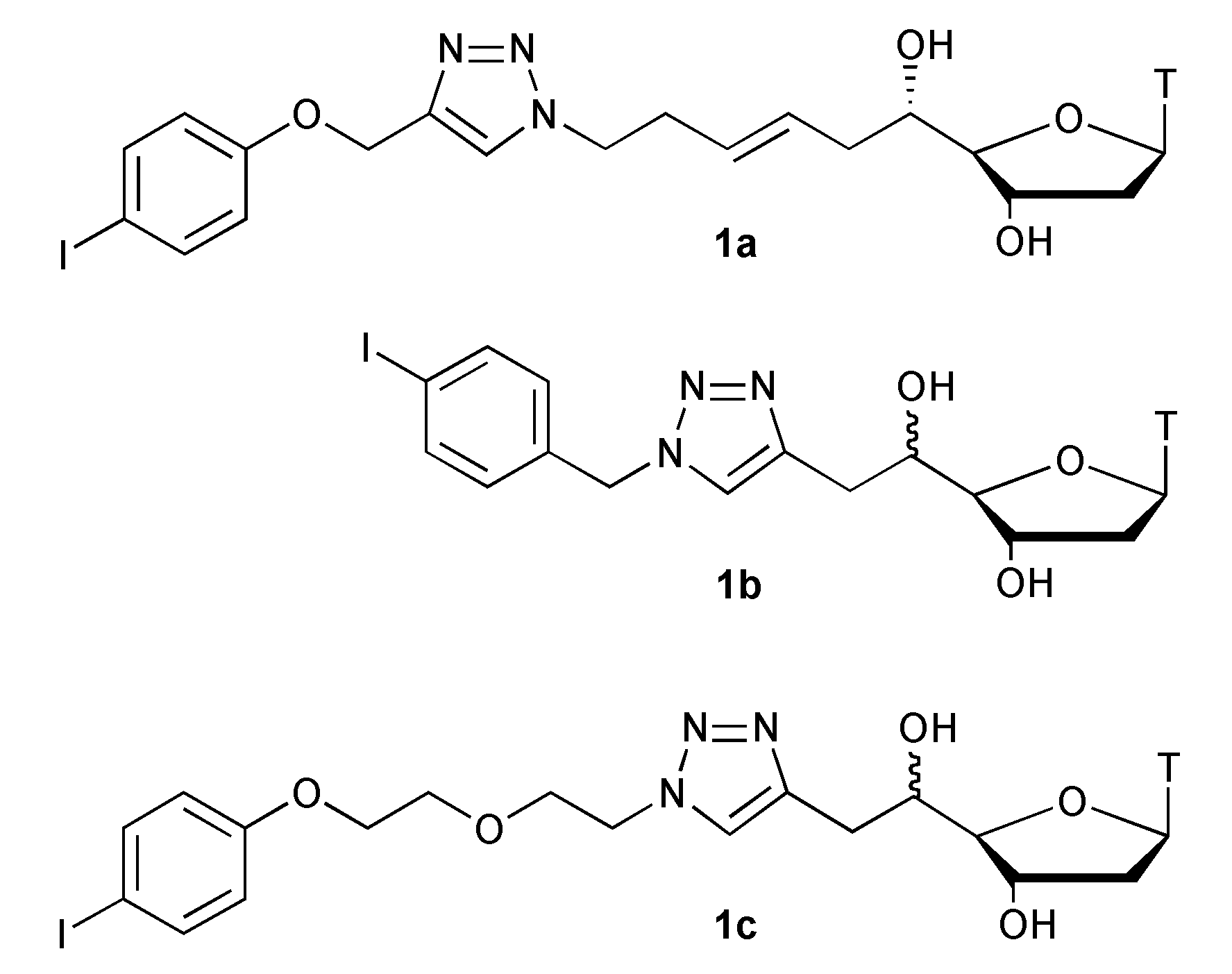{kind=link}
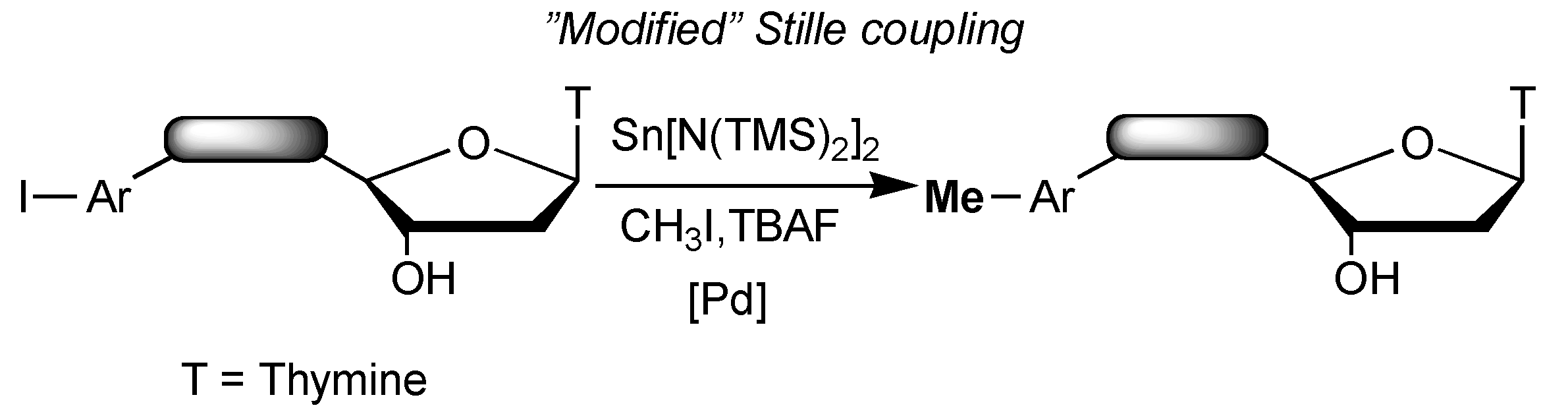{kind=link}
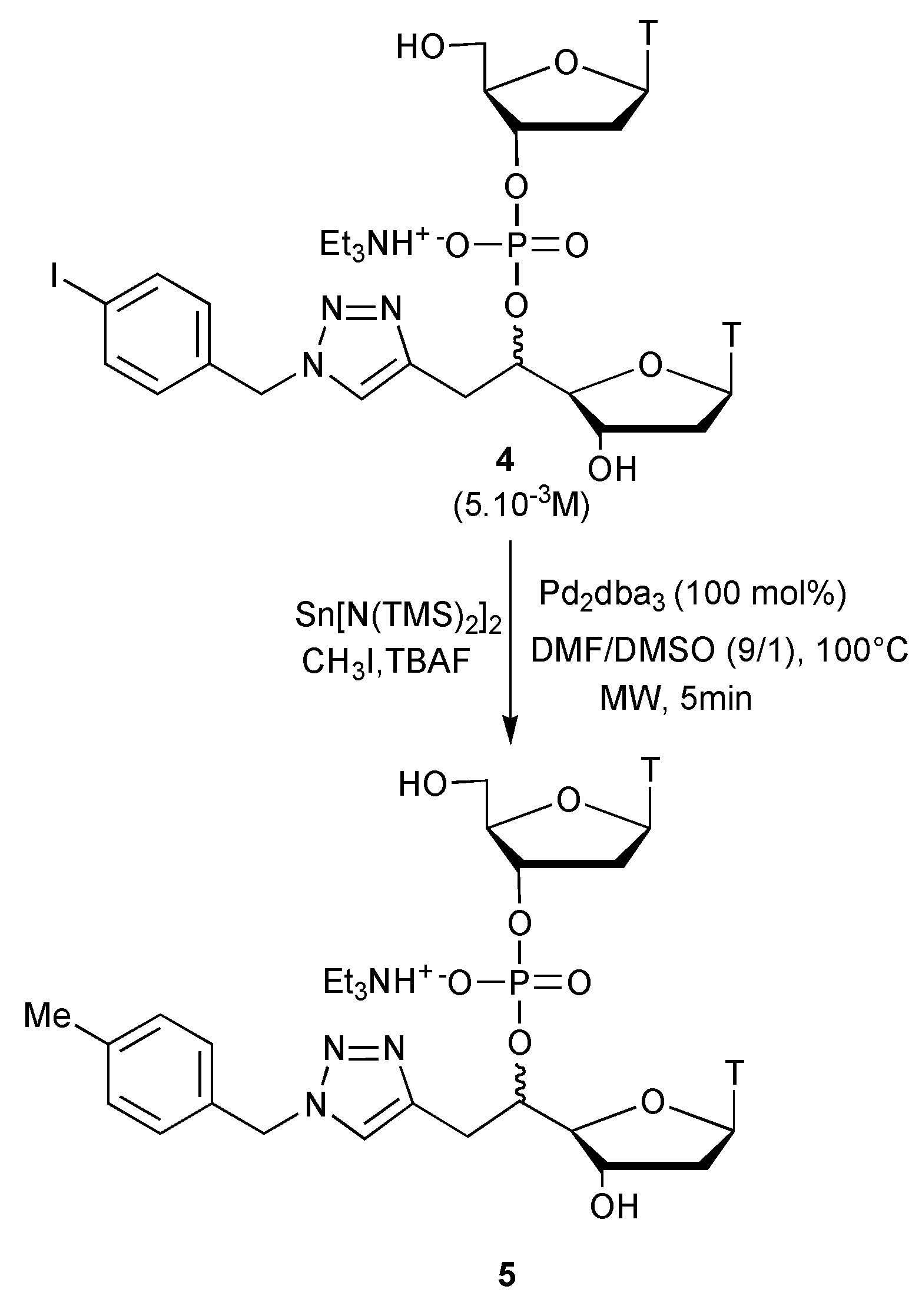{kind=link}
| Entry | Compd.[mol.L−1] | CuI [mol%] | Time [a] [min] | 2/3 [b] |
|---|---|---|---|---|
| 1 | 1a (0.2) | 0 | 5 | 2a/3a: 72/28 [c] |
| 2 | 1a (0.2) | 20 | 50 | 2a/3a: 63/37 |
| 3 | 1a (0.5) | 20 | 20 | 2a/3a: 97/3 |
| 4 | 1a (0.5) | 40 | 5 | 2a/3a: 92/8 |
| 5 | 1b (0.5) | 40 | 5 | 2b/3b: 80/20 |
| 6 | 1c (0.5) | 40 | 50 | 2c/3c: 100/0 |
| 7 | 1c (0.5) | 140 | 5 | 2c/3c: 100/0 |
| Entry | Solvent | CuI [mol%] | MW | Time [min] [a] | 2/3 [b] |
|---|---|---|---|---|---|
| 1 | DMF | 400 | no | 60 | 81/19 [c] |
| 2 | DMF | 2000 | no | - | - |
| 3 | DMF | 0 | no | 5 | 90/10 |
| 4 | DMF | 0 | yes | 5 | 89/11 |
| 5 | DMF/DMSO 9/1 | 0 | no | 5 | 66/34 [c] |
| 6 | DMF/DMSO 9/1 | 0 | yes | 5 | 96/4 |
| 7 | DMF/DMSO 3/1 | 0 | yes | 5 | 91/9 |
| 8 | DMF/H2O 9.5/0.5 | 0 | no | 40 | 54/46 [c] |
| 9 | DMF/H2O 9.5/0.5 | 0 | yes | 5 | 85/15 |
| 10 | DMF/H2O 9/1 | 0 | yes | 5 | 60/40 |

3. Experimental
3.1. General
3.2. Preparation of 4
3.2.1. Coupling (4DCT)
3.2.2. Cleavage of DMTr Group (4CT)
3.2.3. Cleavage of Cyanoethyl Group (4T)
3.2.4. Cleavage of TBDPS p: Preparation of Compound 4
3.3. General Procedure for Methylation (0.2 and 0.5 M)
3.4. Methylation under Dilute Conditions (5 × 10−3 M)
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Phelps, M.E.; Mazziotta, J.C.; Schelbert, H.R. Positron Emission Tomography and Autoradiography; Raven Press: New York, NY, USA, 1986; Chapter 9–11. [Google Scholar]
- Långström, B.; Dannals, R.F. Principles of Nuclear Medicine; W.B. Saunders: Philadelphia, PA, USA, 1995. [Google Scholar]
- Fowler, J.S.; Wolf, A.P. Working against time: Rapid radiotracer synthesis and imaging the human brain. Acc. Chem. Res. 1997, 30, 181–188. [Google Scholar] [CrossRef]
- Allard, M.; Fouquet, E.; James, D.; Szlosek-Pinaud, M. State of the art in 11C labelled radiotracers synthesis. Curr. Med. Chem. 2008, 15, 235–277. [Google Scholar] [CrossRef]
- Cai, L.; Lu, S.; Pike, V.W. Chemistry with [F-18]fluoride ion. Eur. J. Org. Chem. 2008, 2008, 2853–2873. [Google Scholar] [CrossRef]
- Littich, R.; Scott, P.J.H. Novel Strategies for Fluorine-18 Radiochemistry. Angew. Chem. Int. Ed. 2012, 51, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Pretze, M.; Große-Gehling, P.; Mamat, C. Cross-coupling Reactions as valuable tool for the preparation of PET Radiotracers. Molecules 2011, 16, 1129–1165. [Google Scholar] [CrossRef]
- Langer, O.; Forngren, T.; Sandell, J.; Dollé, F.; Långström, B.; Någren, K.; Halldin, C. Preparation of4-[C-11]methylmetaraminol, a potential PET tracer for assessment of myocardial sympathetic innervation. J. Labelled Compd. Radiopharm. 2003, 46, 55–65. [Google Scholar] [CrossRef]
- Sandell, J.; Yu, M.; Edmon, P.; Garreau, L.; Chalon, S.; Någren, K.; Guilloteau, D.; Halldin, C. Synthesis, Radiolabelling and preliminary biological evaluation of radiolabeled 5-methyl-6-nitroquipazine, a potential radioligand fort he serotonin transporter. Bioorg. Med. Chem. Lett. 2002, 12, 3611–3613. [Google Scholar] [CrossRef]
- Madsen, J.; Merachtsaki, P.; Davoodpour, P.; Bergström, M.; Andersen, K.; Thomsen, C.; Guilloteau, D.; Halldin, C. Synthesis and biological evaluation of novel carbon-11 labelled analogues of citalopram as potential radioligands fort he serotonin transporter. Bioorg. Med. Chem. 2003, 11, 3447–3456. [Google Scholar] [CrossRef]
- Ferrieri, R.A.; Antoni, G.; Khilberg, T.; Långström, B. Handbook of Radiopharmaceuticals. Radiochemistry and Applications; Welch, M.J., Redvanly, C.S., Eds.; Wiley: Chichester, UK, 2003; pp. 229–282. [Google Scholar]
- Huiban, M.; Huet, A.; Barré, L.; Sobrio, F.; Fouquet, E.; Perrio, C. Methyl transfer reaction from methyliodide through modified tin reagent: A versatile method for labelling with carbon-11. Chem. Commun. 2006, 97–99. [Google Scholar]
- Bourdier, T.; Huiban, M.; Huet, A.; Sobrio, F.; Fouquet, E.; Perrio, C.; Barré, L. Tetra- and monoorganotin reagents in palladium-mediated cross-coupling reactions for the labeling with carbon-11 of PET tracers. Synthesis 2008, 6, 978–984. [Google Scholar]
- Herve, A.; Rodriguez, A.L.; Fouquet, E. Stille cross-coupling of activated alkyltin reagents under ligandless conditions. J. Org. Chem. 2005, 70, 1953–1956. [Google Scholar] [CrossRef]
- Hosoya, T.; Wakao, M.; Kondo, Y.; Doi, H.; Suzuki, M. Rapid methylation of terminal acetylenes by the Stille coupling of methyl iodide with alkynyltributylstannanes: A general protocol potentially useful fort he synthesis of short-lived (CH3)-C-11-labeled PET tracers with a 1-propynyl group. Org. Biomol. Chem. 2004, 2, 24–27. [Google Scholar] [CrossRef]
- James, D.; Escudier, J.-M.; Amigues, E.; Schulz, J.; Vitry, C.; Bordenave, T.; Szlosek-Pinaud, M.; Fouquet, E. A ‘click-chemistry’ approach to the efficient synthesis of modified nucleosides and oligonucleotides for PET imaging. Tetrahedron Lett. 2010, 51, 1230–1232. [Google Scholar] [CrossRef]
- Harris, D.H.; Lappert, M.F. Monomeric, volatile bivalent amides of group-IVB elements, M(NR12)2 and M(NR12)2 (M=Ge, Sn or Pb-R1=Me3Si, R2=Me3C). J. Chem. Soc. Chem. Commun. 1974, 895–896. [Google Scholar] [CrossRef]
- Schaeffer, C.D.; Zuckerman, J.J. Tin(II)organosilylamines. J. Am. Chem. Soc. 1974, 96, 7160–7162. [Google Scholar] [CrossRef]
- Banuls, V.; Escudier, J.-M.; Zedde, C.; Claparols, C.; Donnadieu, B.; Plaisancié, H. Stereoselective synthesis of (5'S)-5'C-(5-bromo-2-penten-1-yl)-2'-deoxyribofuranosyl thymine, a new convertible nucleoside. Eur. J. Org. Chem. 2001, 4693–4700. [Google Scholar]
- Escudier, J.-M.; Dupouy, C.; Fountain, M.A.; del Mundo, I.M.A.; Jacklin, E.M.; Morrow, J.R. Synthesis and luminescence properties of a trinucleotide-europium(III) complex conjugate. Org. Biomol. Chem. 2009, 7, 3251–3257. [Google Scholar] [CrossRef]
- Omuni, A.; Beach, D.G.; Baker, M.; Gabryelski, W.; Manderville, R.A. Postsynthetic guanine arylation of DNA by Suzuki-Miyaura cross-coupling. J. Am. Chem. Soc. 2011, 133, 42–50. [Google Scholar] [CrossRef]
- Wicke, L.; Engels, J.W. Postsynthetic on column RNA labelling via Stille coupling. Bioconjugate Chem. 2012, 23, 627–642. [Google Scholar] [CrossRef]
- Sample Availability: Samples of materials, analytical procedures, characterization data of 2a–c, 3a–b, 4 and 5 are available from the author.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
James, D.; Escudier, J.-M.; Szlosek-Pinaud, M.; Fouquet, E. Pd0-Catalyzed Methyl Transfer on Nucleosides and Oligonucleotides, Envisaged as a PET Tracer. Molecules 2013, 18, 13654-13665. https://doi.org/10.3390/molecules181113654
James D, Escudier J-M, Szlosek-Pinaud M, Fouquet E. Pd0-Catalyzed Methyl Transfer on Nucleosides and Oligonucleotides, Envisaged as a PET Tracer. Molecules. 2013; 18(11):13654-13665. https://doi.org/10.3390/molecules181113654
Chicago/Turabian StyleJames, Damien, Jean-Marc Escudier, Magali Szlosek-Pinaud, and Eric Fouquet. 2013. "Pd0-Catalyzed Methyl Transfer on Nucleosides and Oligonucleotides, Envisaged as a PET Tracer" Molecules 18, no. 11: 13654-13665. https://doi.org/10.3390/molecules181113654