
Apatite Biominerals

1
CIRIMAT, Université de Toulouse, CNRS, INPT, UPS, ENSIACET, 4 allée Emile Monso, CS 44362, 31030 Toulouse cedex 4, France
2
CIRIMAT, Université de Toulouse, CNRS, INPT, UPS, Université Paul Sabatier, Faculté de Pharmacie, 35 Chemin des Maraichers, 31062 Toulouse cedex 9, France
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Minerals 2016, 6(2), 34; https://doi.org/10.3390/min6020034
Submission received: 30 December 2015
/
Revised: 20 March 2016
/
Accepted: 21 March 2016
/
Published: 5 April 2016
(This article belongs to the Special Issue Biomineralization: Towards a Unification of Concepts in Chemistry, Physics, Earth Sciences and Biology)
Abstract
:Calcium phosphate apatites offer outstanding biological adaptability that can be attributed to their specific physico-chemical and structural properties. The aim of this review is to summarize and discuss the specific characteristics of calcium phosphate apatite biominerals in vertebrate hard tissues (bone, dentine and enamel). Firstly, the structural, elemental and chemical compositions of apatite biominerals will be summarized, followed by the presentation of the actual conception of the fine structure of synthetic and biological apatites, which is essentially based on the existence of a hydrated layer at the surface of the nanocrystals. The conditions of the formation of these biominerals and the hypothesis of the existence of apatite precursors will be discussed. Then, we will examine the evolution of apatite biominerals, especially during bone and enamel aging and also focus on the adaptability of apatite biominerals to the biological function of their related hard tissues. Finally, the diagenetic evolution of apatite fossils will be analyzed.
1. Introduction
The existence of calcium phosphate biominerals has been reported in living creatures from unicellular organisms to vertebrates [1]. Most of these calcium phosphate biominerals exist as amorphous phase in primitive organisms. However, in evolved organisms and especially in vertebrates, they exist mainly as apatite structures, although a variety of other crystallized calcium phosphate phases (whitlockite, brushite, and octacalcium phosphate) may form in uncontrolled pathologic calcifications [2,3]. Compared to other crystalline biominerals such as calcium carbonates, calcium phosphate apatites exhibit undeniably larger biological adaptability. Such adaptability is notable in specific physico-chemical and structural properties, rendering them useful for a large variety of biological uses such as in the protection of internal organs (shells, scales, and flat bones), internal skeleton (bones), sensors (bones of internal ears, rostrum of whales, otolithes in some species [4] or tympanic bullae [5]) and in the organs of attack and defense (antlers and tusks). In addition, other functional roles have been attributed to apatite biominerals like homeostasis or the inactivation of toxic elements. Several reviews have been published on this subject with different approaches focusing on crystal structures, and calcium phosphate precursors [6], the role of OH-channels [7], the role of unstable amorphous precursors [8], the role of polyphosphates [9], general view including the organic matrices [10], apatite biominerals and biomimetic processing and materials [11], and mineralogical oriented reviews [12]. This review aims to present and discuss the specific characteristics of biomineral apatites and illustrate how these characteristics and the resulting mineral properties have been employed in the adaptation for specific biological functions, in the case of vertebrate hard tissues (bone, dentine and enamel). To start, the structure and composition of biomineral apatites will be reviewed, followed by a description of their biological formation, evolution and maturation particularities and finally their influence on the properties of tissues and their biological behavior will be summarized.
2. Structure and Chemical Formulas
The first structural identifications of calcium phosphate biominerals in vertebrates using X-ray diffraction were obtained by de Jong [13], who established that they corresponded to an apatite structure. Excellent reviews on the apatite structure have been published and interested readers may report to these for more in-depth information [14,15]. Since then, stoichiometric hydroxyapatite (HA):
has been generally used as a model for bone mineral and tooth enamel. However, unlike pure stoichiometric HA which crystallizes in the monoclinic P21/b space group, biological apatites are generally indexed in the hexagonal P63/m space group [15]. The main difference between the HA model and biological apatites is the presence of significant amounts of carbonate ions in all mineralized biological tissues, including pathological apatite biominerals. Early studies on synthetic carbonated apatites established that carbonate ions could in fact be part of the apatite structure [16,17]. Detailed studies indicated that carbonate ions could be located in the two anionic sites of the apatite structure: in the PO43− sites (type B carbonated apatite) and the OH− sites (type A carbonated apatite). Another important characteristic of biological apatites is their non-stoichiometry, often referred to as calcium deficiency, although it appears more complex. Even if it has been the subject of much controversy, the use of non-stoichiometric carbonated apatite as a model for the biological calcification of vertebrate hard tissues is accepted nowadays, with some alterations taking into account the multiplicity of carbonate sites in apatites related to coupled substitutions and interactions [15,17,18,19,20].
Ca10·(PO4)6·(OH)2
Another specificity of biological apatites, which was more recently established, is the presence of hydrogen phosphate (HPO42−) ions in PO43− sites [21,22,23]. These two types of bivalent ions substituting for PO43− (type B CO32− and HPO42−) have been shown to correspond to the formation of calcium deficient apatites, the chemical formulas of which have been the subject of several works, essentially based on the composition of model minerals proposed by mineralogists or synthetic analogues. A general chemical formula proposed by Winand for HPO42−-containing apatites was [24]:
and a similar one by Labarthe et al. for carbonate-containing apatites [16]:
Ca10−x·(PO4)6−x·(HPO4)x·(OH)2−x with 0 ≤ x ≤ 2
Ca10−x·(PO4)6−x·(CO3)x·(OH)2−x with 0 ≤ x ≤ 2
These formulas exhibit similar behavior for the bivalent ion substitution of trivalent phosphates and the necessary maintenance of the structural neutral electrical charge, i.e., the loss of a negative charge due to these substitutions is compensated by the creation of a cationic vacancy and an anionic vacancy in monovalent sites. These chemical formulas are consistent with the limit composition observed (x = 2) and the decrease of the OH− content when the amount of carbonate and/or HPO42− in the apatite increases [25]. Other chemical formulas have been proposed to take into account the existence of other charge compensation mechanisms related to the condition of formation of these non-stoichiometric apatites, such as an excess of calcium (u), usual in carbonate apatites obtained in alkaline media [16] or an intracrystalline hydrolysis of phosphate groups (y). One of the most general one proposed by Rey et al. [26] is however of little relevance for biological apatites:
Ca10−x+u·(PO4)6−x−y·(HPO42− or CO32−)x+y·(OH)2−x+2u+y with 0 ≤ x ≤ 2 and 0 ≤ 2u + y ≤ x
Biological apatites are best approximated by the simple combination of the two previous chemical formulas (2) and (3), taking into account the possible existence of type A carbonates:
Ca10−x·(PO4)6−x·(HPO4 or CO3)x·(OH or 1/2 CO3)2−x with 0 ≤ x ≤ 2
Many ionic substitutions are possible in apatites, involving for example, trivalent cations (e.g., rare earth elements, actinides) or monovalent cations (especially Na+) or other bivalent cations for Ca2+ or tetravalent or bivalent ions replacing PO43− in addition to trivalent ones, and bivalent or monovalent ions replacing OH−. Several charge compensation mechanisms have been proposed. The composition of biological apatites will be developed in Section 4.
3. Non-Apatitic Environments and the Hydrated Layer
More recently, these models, which are based on well-crystallized apatites, were re-examined due to the discovery, using mostly spectroscopic techniques (Fourier transform infrared (FTIR), Raman and solid-state nuclear magnetic resonance (NMR) spectroscopies), of the existence of specific spectral lines in the spectra of biological nanocrystalline apatites which do not appear for well-crystallized apatites and which have been designated as “non-apatitic environments” of the mineral ions [22,23,27,28]. These “non-apatitic” phosphate and carbonate environments have been shown to appear more clearly in the ν4 PO4 and ν2 CO3 domains of FTIR spectra. Synthetic models of nanocrystalline apatites mimicking the main characteristics of biological apatites have been prepared and studied [29,30,31]. These “non-apatitic” phosphate and carbonate environments have been shown, using ion exchange experiments, to share the same surface domain corresponding to a structured hydrated layer on apatite nanocrystals [32]. The structure of the hydrated layer seems very sensitive to its ion content and state of hydration: a loss of the original fine structural details revealed by spectroscopic techniques, in wet state, is observed on drying or when specific ions like magnesium are present, leading to line broadening and amorphization [33,34]. Several rapid and reversible ion exchange reactions have been reported [35,36,37,38] and it has been shown that the adsorption of several ionized organic molecules corresponded to ion exchanges with mineral ions of the hydrated layer [39,40,41]. Consequently, the global chemical formulas of apatites reported in the previous paragraph, which do not take into account the existence of hydrated surface domains, should be re-examined. A definitive formula still seems out of reach due to uncertainties regarding the composition of the hydrated layer and the apatite core.
Based on complementary investigations, using spectroscopic techniques and analytical chemistry, several features of biological apatites and their synthetic analogues have been identified:
- (1)
- Apatite nanocrystals contain non-apatitic anionic and cationic chemical environments,
- (2)
- These environments strongly interact with hydrated domains,
- (3)
- Immature samples (freshly precipitated nanocrystalline apatites) show FTIR fine-band substructure changes upon drying without leading to long-range order modifications,
- (4)
These features allowed for the proposition of a model in which apatite nanocrystals are covered with a rather labile but structured hydrated surface layer, containing relatively mobile ions (mainly bivalent anions and cations: Ca2+, HPO42−, CO32−) in “non-apatitic” environments [43]. However, the exact structure and composition of this hydrated layer is still under investigation. A schematic representation of this hydrated surface layer model of apatite nanocrystals in aqueous medium is given in Figure 1.
As for any other kind of nanomaterial, nanocrystalline apatites, whether biological or synthetic, exhibit a high surface to volume ratio leading to consider experimental results (spectra, physico-chemical or biological properties) as a combination of bulk and surface contributions. The latter is of particular importance, since numerous functions of the bone mineral involve processes or phenomena at the interface between bone apatite nanocrystals and their surroundings. However the role of the apatite core seems determining in the manifestation of some properties. Studies of synthetic analogues of biological nanocrystalline apatites reveal that they exhibit strong reactivity related to this structure, especially a specific ageing process, frequently called maturation in reference to bone. The driving force of this process is the relative instability of the hydrated layer compared to that of apatite domains. Thus, the apatite domains develop slowly at the expense of the hydrated layer. Two main routes of progression have been recognized, depending on the composition of the solution (Figure 2) [30].
When the solution contains carbonate ions, the initial precipitates at time zero contain only a few carbonate ions, mainly in the hydrated layer, and a large amount of HPO42− ions. On ageing, the amount of carbonate ions increases in the hydrated layer, as the amount of labile HPO42− decreases, leaving the Ca/(P + C) atomic ratio almost unchanged. This change in the hydrated layer is accompanied by an increase of carbonate ions in the growing apatite domains, in both type A and type B sites, at an apparent constant ratio during ageing at physiological pH.
When the solution does not contain carbonate ions a different maturation process is observed. Although the hydrated layer converts progressively into apatite on ageing, the Ca/P ratio increases and higher amounts of OH− ions are incorporated in the growing apatite domains, whereas the total amount of HPO42− decreases. This evolution corresponds to a formation of hydroxyapatites closer to stoichiometry.
In both cases, the growing apatite domains show a different composition to that of the hydrated layer and their growth is associated with a release of protons and/or carbonate [44].
4. Composition of the Main Mineralized Tissues
Biomineral apatites always exist in association with organic matrices, and even if the elemental composition of whole hard tissues can be determined it is often difficult to know the precise composition of each component, especially regarding trace elements. The mean elemental composition of the three main human hard tissues (bone, dentine, enamel) is given in Table 1. Conventionally, three types of elements are distinguished by biologists: main, minor and trace. These elements can be part of very different molecular or crystalline structures [45,46,47,48,49].
4.1. Main Elements
Nitrogen is considered to belong mainly to proteins and is often considered to represent the organic matrix, although non-protein organic molecules may also constitute a fraction of the organic matrix. Calcium is essentially located in the apatite structure and is considered to represent the mineral content.
Different mineral-organic associations result in tissues with very different characteristics: tooth enamel with a high content of mineral is the hardest tissue of vertebrates, with good resistance to compression and wear, but it is also rather brittle. Dentine, found underneath the thin enamel layer in teeth, appears much less mineralized than enamel and due to its collagen matrix offers both high compressive and tensile strengths. Bone seems generally less mineralized than dentine, although it is quite similar in composition, with excellent compressive and tensile strengths and a high adaptability to mechanical stress. The mineralization ratio in bone can vary considerably. In some intramuscular fish bones like those found in herrings, for example, mineralization is very low and progresses very slowly within the bone organ, in relation to the growth of the animal [54]. This is also the case for turkey tendons, which transform into bone very slowly and offer, like herring intramuscular bone, a possibility to follow a mineralization process in slow motion. Rather strong variations of mineralization ratios are also found in humans depending on the age and type of bone (trabecular or cortical bone). In infants, for example, bones are much less mineralized than in adults. The mineralization ratio of the tissue should not be confused with the bone mass. In elderly people, the global mass of the skeleton decreases and partly the mineralization ratio of the bone tissue [55]. More recently, a strong adaptation of bone tissue to mechanical stresses has been highlighted during space travels, which can have a strong influence on the bone mass [56]. Hypermineralized bones have been described such as in the rostrum of some whales (up to 85%–90% mineral), where the collagen matrix is progressively lysed and replaced by apatite [57].
Carbon can be found in the organic matrices and also in apatite minerals in the carbonate anions. Oxygen and hydrogen can be found in the organic components and in minerals as components of carbonates, phosphates, hydrogen-phosphate and hydroxide ions, as well as water molecules associated to the organic matrices and minerals. One of the main characteristics of all apatite biominerals is the presence of significant amounts of carbonate species and several authors have suggested banning the term hydroxyapatite in reference to carbonate-apatites, in the case of dentine and bone. The carbonate ions in biological apatites have been located in the apatite structure in both trivalent anionic sites and monovalent sites [17,58,59]. A third location of carbonate ions in the hydrated surface layer of biological nanocrystals has also been identified [58]. These carbonate species present different spectroscopic characteristics in Raman, FTIR or solid-state NMR spectroscopies [27,58,60,61].
Phosphorus essentially exists in bones as orthophosphate ions associated with the apatite crystals. Two main species have been identified: PO43− and HPO42−. These species exist on trivalent anionic sites and, as in the case of carbonate, a surface location has also been identified. Hydrogen-phosphate ions have been detected using FTIR and solid-state NMR spectroscopies [22,29,62,63,64]. The bivalent anion content, carbonate and hydrogen phosphate, of apatite is associated with a calcium and hydroxide ion deficiency. The amount of bivalent species varies with the tissue.
Average compositions have been derived for biological apatites neglecting the existence of the hydrated layer and its mineral content. These average compositions of mineral in human bone, dentine and enamel are based on the simplified model of calcium deficient apatites [21] presented above, which do not account for the sodium content, the presence of type A carbonate replacing OH− ions and all minor and trace elements except Mg:
Human bone apatite: Ca8.1·Mg0.2·(PO4)4.3·(HPO4)0.5·(CO3)1.2·(OH)0.3
Human dentine apatite: Ca8.0·Mg0.4·(PO4)4.4·(HPO4)0.7·(CO3)0.9·(OH)0.4
Human enamel apatite: Ca8.8·Mg0.1·(PO4)4.9·(HPO4)0.6·(CO3)0.5·(OH)0.9
This presentation highlights the differences in bone and dentine apatite vacancies content compared to enamel: in humans but also in animals, the amount of vacancies in bone apatites is close to the highest amount of vacancies possible in the apatite structure. In enamel, on the contrary, the amount of vacancies is much lower.
One of the most controversial differences between biological apatite compositions is the hydroxide ion content. In tooth enamel the OH− ions can be directly observed by spectroscopic techniques (FTIR, Raman scattering and solid-state NMR) and the denomination of hydroxyapatite is justified for this biomineral. However, OH− lines in bone or dentine, when they are detected, show very weak intensity, and the reported estimation of the hydroxide content varies considerably, depending on the study and on the bone samples used. Taylor et al. [65] report 50% OH− content of stoichiometric hydroxyapatite (Ca10·(PO4)6 (OH)2) in ox bone, Cho and Ackerman found 20% in chicken bone using solid-state NMR [66]. It should be noted that these values appear in large excess considering the average bone composition of chemical formula (6) and others failed to detect any OH− by Raman or FTIR spectroscopies [67,68]. Recent estimations on different kinds of fresh and freeze-dried bones suggest a null or extremely low OH− content (a few percent) [41]. These discrepancies have been attributed to a possible internal hydrolysis process between water molecules trapped in the apatite lattice and PO43− groups:
Such reactions have been observed in synthetic apatite nanocrystals heated at moderate temperatures (<200 °C) [69]. At higher temperatures an increase of OH− content in calcined bone is always observed. It can be assigned to the decomposition of carbonate species and the hydration of the resulting oxide ions:
H2O + PO43− → HPO42− + OH−
CO32−→ CO2 + O2−
O2− + H2O → 2 OH−
O2− + H2O → 2 OH−
Concerning the OH− content, Pasteris et al., reported a decrease of OH− Raman line intensities in spectra of ground stoichiometric hydroxyapatite with increased crystals strains and decreased crystal sizes [67]. The apatite crystal size or the level of strain does not however necessarily alter the observation of OH− lines, considering that nanocrystalline apatites with clear measurable OH− lines can be prepared [69].
On ageing, several alterations of the bone composition have been reported as will be discussed. In the very early stages, in embryonic bones, the amount of HPO42− is rather high and the carbonate content is very low, leading to calcium deficient apatites with low Ca/P ratios [32,70]. On ageing however, the Ca/P ratio and the carbonate content increases. In fact, these events are linked: as the counter ion of CO32− ions is calcium, the increase in carbonate leads inevitably to an increase of the related calcium ions and of the Ca/P ratio. However, this does not mean that there is an evolution of bone apatite composition towards stoichiometry as is reported sometimes in publications. The Ca/(P + C) atomic ratio, where C stands for carbonate species, remains remarkably constant at about 1.3–1.4, regardless of the age or the animal species [21,70]. This consistency of the Ca/(P + C) ratio simply reveals that the amount of bivalent species (HPO42− or CO32−) is close to constant in bone and that carbonate ions replace hydrogen-phosphate ones upon ageing. On the contrary, in teeth enamel, the carbonate content remains at a low level and during its formation an increase of the Ca/P and Ca/(P + C) ratios is observed, which is related to the presence of hydroxide ions and type A carbonate [30].
4.2. Minor Elements
Although sulfur may enter in the apatite structure this element, present in most biological tissues, is essentially associated with polysaccharides moieties. Strong variations of potassium levels have been found in bone, which could possibly be related to the release of internal cell fluid and can depend on preparation methods. However, the potassium content of enamel appears higher than that of bone and it is possible that this element may be trapped in the enamel apatite mineral. Variations in the chlorine and sodium content of bone and probably in dentine can be attributed in part to the associated biological tissue fluids and may also be related to the tissue preparation methods employed. However, the sodium/chlorine ratio in these tissues appears much higher than in biological extracellular fluid, suggesting that sodium is being incorporated into the mineral crystals. Sodium incorporation could be related to that of carbonate as hypothesized by different researchers [71]. Solid-state NMR spectroscopy analysis confirms that sodium belongs essentially to the mineral phase [72]. A higher content of chlorine in enamel compared to dentine and bone is observed. The incorporation of chlorine as chloride ions in enamel is supported by the occurrence of Cl–OH hydrogen bonding which can be clearly discerned in FTIR and Raman spectra (shift of the OH− line towards lower wavenumbers: 3500 cm−1 instead of 3570 cm−1 in hydroxyapatite) [73]. The reason for the incorporation of chlorine as a substitute for hydroxide ions in enamel apatite, compared to bone or dentine apatite is most probably related to the relatively low level of bivalent ions in enamel apatite and the higher amount of occupied monovalent ionic sites in accordance with the proposed chemical formulas (6)–(8). The magnesium content of bone decreases with increasing age. Although this element can enter into the calcium phosphate apatites as a substitute for calcium, it has also been reported to attach to the surface of the crystals [74].
4.3. Trace Elements
Numerous trace elements are found in hard tissues that are generally divided as essential elements, (i.e., elements necessary for the living organism) such as As, F, Mn, Cu, Zn, and Sr; toxic elements (Al and Pb); and elements without known biological effects. These categories are, however, not clearly separated and several elements may be essential at low concentrations and become toxic at higher ones. In some cases, Al, As, F, Pb and Sr for example, the trace element concentration is related to diet intake, and some of these elements, Al, As, F and Pb for example, are considered to show additive accumulation. The identification of locations of trace elements in hard tissues is a complex task and methods involving the selective destruction of the organic part (hydrazine or hypochlorite treatments) or the mineral component (acid or Ethylenediaminetetraacetic acid (EDTA) dissolutions) do not prevent parallel or post treatment reactions, which may distort the validity of the collected data. Only in a few reported cases has the clear location of trace elements been partly determined, often using spectroscopic techniques. Silicon has been located in the organic matrix in the vicinity of the mineralization front [75]. On the contrary, fluorine has been located in the apatite mineral as a fluoride ion [76,77]. It is important to note that these data do not mean that traces of silicon or fluoride do not exist in other compounds, in other forms. Except for a few elements, like fluoride ions, trace elements are not believed to exhibit a significant physico-chemical effect on the mineral, although most of them can show a biological effect depending on concentration.
One of the most emblematic trace elements is fluorine, existing in the form of fluoride ions, and mostly found in the apatite biomineral (99% of the F body burden). The fluoride content varies strongly depending on dietary uptake, though in normal bone it constitutes 0.05% to 0.1%. A high fluoride content (above 0.3%) is indicative of fluorosis disease, which results in an increase in bone mineral density and in cancellous bone volume and enhanced bone fragility [78]. During the formation of teeth, fluorosis disturbs the formation of enamel. In teeth enamel, the fluoride content has been shown to vary strongly, with a higher content on the surface than in the inner enamel. Globally, the content of F in enamel is lower than that in dentine or bone (Table 1). In addition, in these tissues, the distribution of fluoride and other bone seeking elements is not homogeneous and the zones which are the best vascularized (cancellous bone, dentinal pulp) show a higher fluoride content [79]. This inhomogeneous distribution of trace elements is also found for other bone seeking elements like lead, aluminum and strontium [80]. Some elements like Sr, Si, and Zn have shown interesting biological properties.
5. Crystal Physical Characteristics
The mineralization process is controlled and occurs within delimited tissue boundaries with some degree of order. Bone exhibits elongated plate-like nanocrystals (about 50 nm long, 25 nm width and 10 nm thick) [10,81]. The length corresponds to the c axis of the hexagonal unit-cell, which is parallel to the axis of the collagen fibers. The crystals form agglomerates of parallel platelet crystals. This common orientation begins at the very early stages and has been observed, for example, in mineralizing turkey tendons [82]. The orientation of bone apatite crystals along the collagen fibers has long been considered as an indication of strong interactions between the two main constituents of the bone composite, however recent data obtained by rotational-echo double resonance (REDOR) solid-state NMR failed to find proof of any significant and stable chemical bonds between collagen and bone apatite [83]. One of the most important organic constituents bound to bone crystals are citrate ions, which constitute an important fraction of the organic matter and have been shown to adsorb strongly onto apatite crystals [84]. The citrate ions are the only organic molecules that exist in quantities high enough in bone to have a chemical influence on the crystals. A recent report suggests that these ions could be responsible for the inter-crystalline bonding of the bone crystals and their arrangements in bundles of parallel crystals [85]. Such arrangements have however also been described in synthetic preparations without citrate ions and are considered to result in interactions between the hydrated layers of the nanocrystals [64]. These data do not necessarily exclude the existence of inter-crystalline citrate bonding, but this role has to be confirmed and precisely defined [86]. Interestingly, another common organic acid, lactic acid, has been shown to be stored at significant amounts as lactate in bone tissues and shells of turtles under hypoxic conditions [87], although this buffering effect of bone involves probably an alteration of the mineral, its effect is not yet clarified.
The orientation of crystals in the collagen array does not seem related to strong chemical interactions. A templating effect has been suggested, based on the fact that apatite crystal formation occurs preferably when the fibers are organized in their regular “quarter stagger array”. Disruption of the collagen organization due to diseases (osteogenesis imperfecta for example) [88] is often associated with mineralization impairment. The growth of apatite crystals, towards the free available space left vacant by collagen fibers arrangement, appears as a natural process to consider. Several reports mention that the plate-like crystals of bone appear bent or even folded either in native form in calcifying vesicles, for example [89], in bones [90] or when extracted from the organic matrix of calcifying cartilage [91]. These observations could possibly be related to preparation artifacts altering the very thin bone crystals. Recently McNally et al. [91], analyzing dark field images, concluded that these mineral structures are most probably polycrystalline associations rather than bent single crystals.
Unlike dentine or bone, enamel is composed of needle-like crystals elongated along the hexagonal c axis of the unit-cell. The crystals appear as irregular hexagons, with different dimensions in different species: 50–60 nm wide and 25–30 nm thick for rats mature enamel [92] and 20–180 nm wide, 10–90 nm thick for humans [93]. The crystal length is difficult to determine with accuracy. The crystals are organized in rods of densely grouped parallel crystals with the needle axis perpendicular to the enamel surface. They are believed to be as long as the enamel thickness. Only very few non-collagenous proteins (enamelins and taftelins) remain in mature enamel (about 1%) [94]. Enamel is an acellular tissue that is subject to abrasion and dissolution on contact with acidic foods or acids generated by oral bacteria.
6. Formation
Apatite calcium phosphates are mainly involved in mineral ions metabolism (Ca, P, but also Mg [81] and possibly Zn [95]) in living organisms through dissolution/precipitation/crystallization processes. The fundamental physico-chemical principles involved in crystal formation from solution are: solution supersaturation, nucleation and crystal growth. In the case of biomineral formation, these steps are not only determined by the local ion concentration in the medium but also by the nature of the interfaces present in the biological environment (mineral-organic matrix and -biological environment). In addition, the mineralization process occurs in partly regulated extracellular space that cannot reach thermodynamic equilibrium due to continuous remodeling and circadian variations of main mineral ions concentrations. In the case of bone mineral formation, the extracellular collagen protein matrix constitutes a supramolecular framework to support, delimit and control apatite formation. The formation of bone mineral crystals has been extensively studied in vivo (in embryos, in bone defects [81], in slow mineralizing systems such as turkey tendons [82] or fish bones [54,96]), or in vitro, in osteoblast cell cultures [97]). In all cases, bone tissue formation begins with the extracellular deposition of a structured collagen matrix that exhibits a specific arrangement of the fibrils resulting in the creation of hole zones between aligned fibrils and interfibrillar space for parallel fibrils [98]. The mineral formation begins in the hole zone of the collagen fibrils and is then progressively extended to the inter- and intrafibrillar space. Vesicular mineralization, reported by several authors, seems to play a very limited role in the bone formation of vertebrates, as discussed in the review of Christoffersen and Landis [99].
The composition of bone extracellular fluid has only been the object of a few studies. Most researchers consider that it is close to blood plasma and some of them use simulated body fluid [100] as a model although it is certainly exaggeratedly oversaturated as this model does not consider mineral ions associations with organic molecules of blood [41]. The evaluation of blood plasma free calcium and phosphate concentrations reported by Eidelman et al. seem more realistic [101] and suggest a concentration of these main ions close to that of the solubility product of octacalcium phosphate. This does not establish however the mineral ions concentrations at the time and place of mineral crystals formation.
According to the classical theory of crystallization, nucleation represents an activation barrier to the spontaneous precipitation of a solid phase from a supersaturated solution, which is essentially related to the creation of interfacial energy. Crystallization at surfaces (heterogeneous nucleation and crystal growth) may be induced at supersaturations lower than those required for spontaneous precipitation ranging in the domain of metastability. In the presence of a foreign body or surface, the overall free energy change associated with the formation of a critical nucleus under heterogeneous conditions can be less than the corresponding free energy change associated with homogeneous nucleation. Another parameter to consider is the presence of adsorbed molecules that may change the interfacial energy in solution and stabilize smaller nuclei compared to solutions without adsorbate (Figure 3) [102,103]. The stabilization of smaller nuclei favors their formation. Furthermore, the rapid growth of these nuclei is prevented due to the adsorbed molecules at their surface. As a result, fast multiplication of nuclei is observed until most of the molecules are adsorbed, at this stage, the nuclei continue to grow without inhibition and a burst of crystal growth can be eventually observed, with crystal growth rates several orders of magnitude larger than in the case of adsorbate-free nucleation (Figure 3A) [102,103]. Such a process of nuclei multiplication can theoretically occur with any adsorbing molecules, lowering the interfacial energy (Figure 3B). The advantage, regarding an organic matrix mineralization, is to facilitate the control of crystal size through the multiplication of nuclei and multisite growth and the preservation of mineral ion diffusion capabilities in the matrix toward the inner growth sites by delaying a crystal growth obstruction of the diffusion paths. These very classical effects can be related to the non-classical crystallization concept involving transient phase(s) proposed by several authors (amorphous intermediates, dense liquid calcium phosphate clusters, and polymer-induced liquid-precursor phase) (Figure 4) [104,105,106,107,108] and to the action of some ionic additives such as magnesium.
Other than the classical crystallization theory, as illustrated by pathway (a) in Figure 4, several authors [104,105,106,107,108] have also proposed that biomimetic mineralization could take place following a non-classical theory of crystallization, proceeding via an intermediate self-assembly, pathways (b) and (c), prior to crystal fusion or transformation to a metastable or amorphous precursor phase/particle, (d) pathway. These non-classical crystallization routes could especially apply to systems far from thermodynamic equilibrium for which crystal morphology and size cannot be predicted considering classical thermodynamics.
From a kinetic and thermodynamic point of view, if we consider the non-classical crystallization process following the (d) pathway compared to the classical pathway (a) in Figure 4, several authors propose a precursor multistep pathway ((B) pathway in Figure 5) involving a sequence of formed phases rather than a single step pathway (pathway A in Figure 5) for various biomineralizations such as bone mineral formation [104,105,106,107]. This concept is based on Oswald’s step rule, which empirically predicts that when a solution is supersaturated with respect to more than one phase, the first phase formed is the least stable/most soluble/least dense phase due to the lower energy barrier compared to pathway A. The metastable phase formed could then be successively transformed into a more thermodynamically stable phase down to the ultimate step, leading to a more stable phase (HA in the case of calcium phosphate-based biomineralizations). However, as already discussed in the previous sections of this review, we should bear in mind that biological apatites are far from the stoichiometric HA in terms of composition, defects, solubility and thus thermodynamic properties [109]. Consequently, the position of synthetic and biological non-stoichiometric apatites in a free enthalpy variation diagram (Figure 5) would not be that of HA, but would probably be between that of HA and OCP and probably closer to that of OCP [110]. In addition there are uncertainties of the supersaturation ratio at the time and place of mineral formation.
Based on all of these considerations, several authors suggest that the amorphous precursor pathway could be widespread in the formation of biominerals [106,107,111,112]. The possible presence of transient amorphous mineral phases in mineralized vertebrate tissues has been a subject of debate over recent decades, and it still remains an open question. Furthermore, the introduction of an additional precursor step has been suggested by several authors, first in the case of the calcium carbonate system, which involves the formation of a polymer-induced liquid-precursor (PILP) phase before the amorphous phase formation step in the presence of anionic polymer (Figure 5) [104,107]. Such a phenomenon seems very similar to the adsorption effect schematized in Figure 3B. This PILP phase is considered to be a highly hydrated phase, which is more labile than the amorphous phase and could be formed of stabilized nuclei with subcritical radii. The mechanism proposed by Gower for collagen mineralization during bone formation involves a PILP phase [107], i.e., liquid-like amorphous precursor droplets that are drawn into the gap zones of the collagen fibrils through capillary forces. Then, once these drops have infiltrated the gap zones and interfibrillar space of collagen, the PILP phase is transformed into the amorphous (solid) phase (ACP), which in turn crystallizes into apatite nanocrystals.
Veis and Dorvee [105] further examine the mechanism of bone and dentin mineralization through a two-step mechanism involving: (i) the formation of a dense liquid cluster (a dense liquid phase of first-layer water-bound hydrated calcium and phosphate ions); followed by (ii) the formation of a crystal nucleus inside this dense liquid cluster. They also highlight the role of polyelectrolytes/protein interfaces, which have similar dense, liquid, first-layer, water-bound surfaces that can interact with the dense, liquid calcium phosphate nucleation clusters and control crystallization within the bone and dentin collagen fibril matrix most probably in accordance with the mechanism proposed in Figure 3.
Enamel crystals form in direct contact with ameloblast cells in a row-type arrangement, forming aligned bundles of apatite crystals, which are often referred as prisms of apatite. A small deviation of crystal orientation across each prism can be observed in densely packed enamel apatite prisms. Small disorientation angles have also been identified between enamel and dentin crystals excluding epitactic relationships [113]. Several studies report that the ribbon-like shaped structures formed at the earliest stage of enamel formation include a non-crystalline mineral (ACP) and/or octacalcium phosphate (OCP) crystals, both potent precursors of enamel apatite [114,115]. Several authors have identified an amorphous calcium phosphate phase which transforms with time into the final apatitic crystalline mineral during enamel formation in murine incisor teeth [113,115,116]. Beniash et al. [116] hypothesize that this transformation can be triggered by proteolytic degradation of the enamel matrix proteins and suggest that the shape and organization of the mineral particles is determined very early, implying direct control of mineral shape and organization by the matrix proteins. Bodier-Houllé et al. [113] have also identified the presence of amorphous calcium phosphate at the interface of enamel and dentin (dentino-enamel junction).
7. Maturation and Evolution
Mineral structures are often considered stable and unreactive entities. This is not the case for biomineral apatites, especially nanocrystalline ones, which with their hydrated surface layer, appear highly reactive. This reactivity is illustrated by the maturation and evolution of bone mineral after it has been formed, and that of enamel crystals during teeth formation.
7.1. Bone
The maturation and evolution of bone mineral is dependent on physico-chemical and biological events. The physico-chemical transformations are related to the structure of the bone mineral apatite and exhibits similar characteristics to that of synthetic biomimetic apatites as described in Section 3. On ageing, the following events have been reported [22,23,27,32,117,118]:
- a decrease in the labile environments;
- an increase in CO32− and a decrease in HPO42− with a constant Ca/(P + C) ratio; and
- the development of the apatite domains and an increase in their dimensions and crystallinity.
The most important event to consider in regards to the biological alterations is the remodeling process that renews bone tissues at different rates, depending on the bone type, age and mechanical sollicitations. This biological process produces mature bone with its characteristic osteonal structure and high mechanical strength. This sustained bone formation involves the fresh precipitation of nanocrystalline apatite with a well-developed hydrated layer and a high ability to exchange with the bone extracellular fluid, and by extension with blood, allowing for the maintenance and control of homeostasis. Several processes are involved in homeostasis: hormonal regulation implicating bone mineral dissolution and re-formation, and also fast equilibration with bone mineral crystals believed to involve calcium binding molecules [119]. However, physico-chemical data regarding nanocrystalline apatite suggest a possible control by direct equilibration with the hydrated layer of nanocrystalline apatites related to their specific solubilization mechanism [120] depending on the extend of dissolution and the age of mineral crystals. Considering the high levels of calcium and phosphate in body fluids only crystals with a well-developed hydrated layer can be involved in the homeostasis. Strongly packed old bone crystals would be excluded from this fast equilibration due to the reduced water content and packing limiting diffusion to and out of their crystals surface. It has been shown that when nanocrystals have come close to each other in synthetic biomimetic apatite preparations, surface ion exchanges are significantly limited [121]. Thus, remodeling, considered to occur mainly for preserving mechanical properties of the bone tissues, could also be a necessity for homeostasis function.
The mineralization of new osteons is believed to occur in two stages [122,123,124]: a fast primary mineralization leading to about 75% of the total mineral load achievable in bone tissue, followed by a slow secondary mineralization process lasting for several months and even years, leading to mineralization completion. A possible explanation for this mineralization process is that the crystals involved in the primary mineralization, with their hydrated surface layer and lower mineral density, occupy most of the space within the matrix. Several mineral ions stabilize the hydrated layer and/or slow down the development of the apatite domains (mostly Mg2+, CO32− and P2O74−). With ageing and the growth of the more stable apatite domains with higher density, the initial crystal volume decreases and the inter-crystalline space globally increases, leaving space for new mineral deposition. This could results in a partial reconstruction of the hydrated layer, offering new crystal growth possibilities. The secondary mineralization would then be determined by the growth of the apatite domains associated with the shrinkage of the mineral complex. Although this process leads to bone tissue with increased mineral content and mechanical strength [125], it progressively decreases the pool of exchangeable ions useful for homeostasis. Thus bone remodeling can be seen as a quest for equilibrium between the two main functions of the bone tissues, mechanical strength and homeostasis of essential elements.
An important aspect of remodeling is the partial re-use of mineral ions. Although it is linked with bone turn-over, each element has its own turn-over rate depending on its recycling ability in newly formed bone mineral. This recycling rate is often considered to be related to the effect of the element on apatite solubility. Thus, fluoride ions, which attach very easily to apatite and decrease its solubility, even at low concentrations [126], are very difficult to eliminate when they have entered bone mineral apatite and they have a very long half-life in bones evaluated to about 20 years [78]. As the effect of fluoride ions on apatite solubility has been shown to depend on its concentration [126], the clearance delay of fluoride should also depend on its concentration. As well as affecting apatite solubility, fluoride also has a biological effect on bone cells and remodeling [78]. Another aspect of fluoride incorporation is its effect on the hydrated layer. It has been shown that fluoride favors the development of apatite domains at the expense of the hydrated layer [127] and thus influences the probable biological function of the hydrated layer in calcium homeostasis [128]. This can lead, for moderate fluoride intoxications, to hypocalcaemia [129]. Other ions such as Pb2+ are also retained in bone for long periods, which is possibly related to their effect on apatite solubility [130]. On the contrary, ions like Sr2+, which have been found to increase the solubility of Ca-Sr hydroxyapatites [131], can be easily removed from bone mineral within a few weeks after interruption of intake [132]. The possible location of Sr2+ within the hydrated layer has been hypothesized but has not been proven.
From a general point of view, mineral ions can be incorporated preferentially in the apatite domains or in the hydrated layer, although in most cases the partition coefficients are not known and seem to vary with the maturation stage [35]. For example, carbonate or magnesium ions are first incorporated in the hydrated layer during the precipitation of carbonate or Mg containing apatites [35,127]. However, during the maturation process two types of behavior can be distinguished depending on the nature of the mineral ion: carbonate ions are progressively incorporated into the growing apatite domains in type A and B positions whereas magnesium, although it could also belong to the apatite domains, remains preferentially in the hydrated layer and can be easily removed by ion exchange experiments. Strontium, like carbonate, penetrates progressively into the apatite domains from a surface location [35]. The behavior of most mineral ions during maturation is not known, although their preferential location could help to explain their biological effects. For example, infants are more sensitive to acute lead poisoning than adults and their forming mineralized tissues have been shown to easily incorporate this element; the consideration of the possible role of the non-apatitic hydrated layer in these events could be of interest for the improvement of models describing lead metabolism variations with age [133,134].
7.2. Enamel
During its formation enamel mineral is subject to several changes [18,135,136,137]. As already mentioned, little is known of the composition of the first ribbon-like, very elongated crystals. After this first secretory stage the crystals are analogous to those of embryonic bone or mineralizing cartilage, with a low carbonate and a high HPO42− content and no discernible OH− ions [30]. During its development however, a divergence with bone mineral evolution is clearly observed (Figure 2): although the amount of non-apatitic environments decreases, the Ca/(P + C) ratio begins to increase with increasing OH− ion content. This process is similar to that observed for synthetic biomimetic apatites in carbonate poor solutions (Figure 2). In fact, the amount of bicarbonate ions in enamel fluid is very low (about 10 mM) [18] compared to that of bone fluid and blood (27 mM) [45]. It has been shown that the ribbon-like crystals first deposited grow in width and thickness by alternate accretion and maturation of mineral deposits modulated by ameloblast activity, alternating characteristic ruffle-ended and smooth-ended morphologies. These steps are accompanied by pH fluctuations between slightly acidic (6.1–6.8) and physiological (7.2–7.4) values [135]. The low pH values have been associated with mineral deposition and could be related to the development of a hydrated layer on the surface of existing crystals (appearing amorphous on de-hydration). The maturation period also seems associated to changes in the mineral, especially a loss of labile surface carbonate [18]. This evolution can be compared to that of biomimetic apatite precipitates in solutions with a physico-chemical maturation which is driven by the growth of apatite domains at the expense of the hydrated layer. This phenomenon leads to a loss of protons due to the compositional differences between the apatite domains (mainly PO43− ions) and the hydrated layer (mainly HPO42− ions) and can be related to the loss of labile carbonates. Thus, the rest period in enamel crystal growth corresponds to the development of apatite domains and a considerable decrease of the hydrated surface layer due to the removal of one of its stabilizing ions, carbonate. Other changes which may facilitate the apatite growth during the formation of enamel have also been postulated, such as the removal of adsorbed Mg2+ ions related to calcium transport during the maturation stage, and the effect of fluoride ions favoring the development of apatite domains [18].
A last developmental stage of enamel occurs after eruption. This post-eruptive maturation corresponds to the completion of mineral formation from saliva leading to a denser, harder enamel surface [138]. A change in the surface enamel composition is observed with decreasing Ca/P ratio, which can be attributed to a decrease in the surface carbonate content and an increase in the fluoride ion content, which in-turn improves the crystallinity of the apatite. These changes also improve the surface resistance of enamel to caries. The slightly acidic media of the mouth favors the release of carbonate ions, and the natural physico-chemical maturation in solutions poor in carbonate ions leads to an evolution of the apatite deposits towards stoichiometry. A physico-chemical factor rarely mentioned in the literature is the dominant orientation of the crystals in enamel, with the hexagonal unit-cell c axis perpendicular to the surface [15]. The c axis direction, in needle-like apatite crystals, corresponds to the fastest growing (and dissolution) direction. This orientation could favor a dissolution-remineralization processes and eventually, after successive cycles, the improvement of surface enamel crystal acid resistance. However, regarding their composition and possibly structure, the resulting crystals appear as heterogeneous entities, as evidenced by the well-established preferential dissolution of the central core of enamel crystals [139] with high HPO42− and low OH− content.
8. Adaptability of Apatite Biominerals to Biological Function
Calcium phosphate biominerals are involved in different tissues with different biological functions. The example of bone and dental enamel illustrate the levels of adaptation of the mineral crystals to their biological function. Dental enamel is an acellular tissue that is subject to high compressive strains, abrasion and dissolution on contact with acidic foods or acids generated by oral bacteria. Bone is a cellular tissue (except in some fishes) with a self-repairing ability when damaged; it is involved in body architecture, protection of internal organs, attachment of muscle and mobility, and homeostasis. From a physico-chemical point of view, a first level of adaptation is the mineral-organic ratio, which is obviously different in bone and enamel (50%–85% mineral in bone and 97%–99% mineral in enamel), but also between other mineralized tissues, and even in bone of different species [125]. Mechanical properties and hardness are strongly related to this parameter but also to other characteristics of the tissues. The biological apatite crystals: composition, dimensions and surface properties can be distinguished as parameters of the biological adaptation to the function.
Apatite composition allowing the existence of different types of ionic lacunas in bone apatites can considerably alter the cohesion of the crystals [20] and their physico-chemical properties, especially their dissolution properties and solubility [120]. Apatites, which are closer to stoichiometry in enamel, are less soluble than bone apatites, which are more lacunar and far from stoichiometry. Less soluble enamel apatites are thus more resistant to acidic attacks from foods or oral bacteria, whereas more soluble bone apatites help facilitate the ion reservoir function of bone through remodeling. In addition to non-stoichiometry, ion substitutions can also specifically act on the solubility; the apatite stability record is thought to be held by shark teeth consisting of fluorapatite, which is among the less soluble apatites.
The crystal size is another variable of biological adaptability. Enamel is comprised of large apatite crystals with low specific surface area for which the dissolution rate is low. On the other hand, bone mineral is comprised of apatite nanometric platelet crystals with high specific surface areas, which favor homeostasis through dissolution phenomena but also directly through rapid surface equilibration reactions [119,128]. As mentioned earlier the orientation of crystals could also play a role in the biological adaptation. If the orientation of enamel crystals could enable improving partial remineralization from saliva, for dentine or bone platelet crystals, the orientation in parallel arrays along collagen fibers favors crystals packing and junction, and some mechanical properties but prevents surface exchanges and reactions and increases stiffness and fragility. An interesting aspect is that the total fusion of crystals is not observed. The limiting phenomenon could be the impossibility to grow an apatite lattice with the residual ions in the hydrated layer (Mg, carbonate and possibly pyrophosphate ions), the presence of adsorbed molecules like citrate ions, amino-acids (especially phosphorylated ones), polypeptides, and more generally any molecules able to bind on the surface of the platelet crystals.
Due to its ion exchange ability, the hydrated surface layer of bone apatite crystals participates in homeostasis. However with ageing, due to the decrease of the labile ion reservoir, which is related to the evolution of the hydrated surface layer and bone apatite crystal maturation [32], its ability to regulate “chemically” the concentration of mineral ions in blood decreases accordingly. This phenomenon parallels the increase of stiffness and fragility mentioned above and determines a cyclical renewal of bone mineral to preserve the ion reservoir and ion exchange properties of freshly formed apatite nanocrystals as well as the original mechanical properties of the bone tissue. This is especially true for well-vascularized epiphysis of long bone and physico-chemical studies in long, weight bearing bones have shown that bone mineral crystals appeared less mature in trabecular bone of the epiphysis than cortical bone of diaphysis, with larger amounts of non-apatitic environments especially in young animals [140].
9. Diagenetic Evolution of Biological Apatite Fossils
Fossils of hard tissues are used for dating purposes, and can also be used to identify life conditions, dietary habits, climate changes, and environmental pollution. It is quite difficult to evaluate the diagenetic alterations of these fossils and their corrections to garner desired information.
Several stages in the evolution of hard tissues after death can be distinguished (Figure 6). After cell death, the first alteration is related to the destruction of the organic matrix by microorganisms and hydrolysis [141,142,143,144,145]. This process is of course important for tissues with a high content of organic components, such as fish scales and bones, and seems rather limited in the case of mature enamel. This degradation can increase the porosity of the tissues, modify its hardness and favor its ulterior degradation. Several driving forces are involved in the evolution of the apatite mineral. Based on our experience on ageing of synthetic analogues in wet conditions, a fast alteration of the hydrated layer in a few months seems likely: the amount of labile ionic environments decrease [146] and can trap surface pollutants in contact with the tissue. However, this phenomenon does not seem to touch the internal apatite core. A second degradation process sees a change in the chemical composition, which is related to the non-stoichiometry of apatites and their relative solubility in media different from body fluids. Numerous elements can enter the apatite lattice at this stage such as fluoride and heavy elements. Considering that apatites offer large ranges of solid solutions, this re-equilibration process, provided it does not involve acidic media, only concerns the surface of apatite crystals in the first stage. Progressively, however, the more stable equilibrated apatite domains will grow through a generally slow dissolution re-precipitation processes driven by the relative thermodynamic stabilities of the apatite fractions, including foreign (non-biological) elements and Ostwald ripening. The crystals grow bigger and their composition can be strongly altered [147]. Although the ions released by dissolution can certainly be locally reused at the precipitation loci, similar to the in vivo remodeling of bone, the system around the fossil is largely open and several foreign elements stabilizing the apatite structure such as fluorine, silicon, heavy metals (Pb, Ba) and rare earth elements play a determining role in this recrystallization, modify the apatite composition and characteristics [113]. The kinetics of these processes depend on the presence of groundwater, dissolved mineral ions, pH and temperature and also on the fossil apatite characteristics (composition, crystal size) and fossil porosity [141,144,147]. Thus small bones and trabecular bone are more strongly altered than cortical, compact bone. Tooth enamel, due the large size of its apatite crystals and its relatively low porosity, is less affected by diagenetic evolution than dentine, although enrichment by foreign elements can be measured even in the case of enamel [142]. Very often isotopic ratios of different elements in fossils are used for paleodietary or paleoclimatology purposes [143,144]. Globally, the mineral ions in largest quantities in the fossil apatite structures (calcium and phosphate and to a lesser extent carbonate) are probably those which are the least affected by foreign contamination, simply because of their mass effect. Thus determinations made using these ions and their isotopes can probably be trusted whereas the use of isotope ratios of elements showing strong diagenetic variations appears uncertain. Other effects that may disturb the diagenetic evolution of fossils also have to be considered, most notably biological degradation involving microorganisms (yeast, fungi, and bacteria) in search of essential elements [141]. Unlike purely physico-chemical diagenetic evolution, driven by thermodynamics, the dissolution–reprecipitation phenomena driven by microorganisms can lead to unstable and reactive precipitates, which can experience a new cycle of diagenetic evolution. These biological alterations would only be limited by the presence of toxic ions for the living organisms involved. Degradation of apatite fossils by other living organisms such as insects or animals has also been observed [141].
10. Conclusions and Perspectives
Diversity and heterogeneity are the two words that may best qualify apatite biominerals. These might not show the original fine morphological details and lace-like patterns of silica and calcium carbonate biomineralizations, but their chemical richness, with large substitution facilities, acceptance of defects, and strong surface reactivity, have led to sophisticated materials with a large adaptability to different essential biological functions in many different animal species living in different environments. Although from an evolutionary perspective the storage of essential elements and detoxifications can be seen as the starting point of apatite biomineralizations, several other functions have emerged necessitating an improvement and a progressive control of all the possibilities given by the chemistry of apatites. The control of chemical composition and non-stoichiometry associated with a surface hydrated layer, allows fast mobilization of mineral ions and a fine tuning of homeostasis adapted to living environments. The control of crystals size, orientation and packing associated with an elaborated architecture participates to improving mechanical performance and lightweight. From the point of view of material science, such elaborated constructions are difficult to imitate although micro-fabrication processes seem to open perspectives. One of the most difficult problems is to manage the reactivity and ageing phenomena, which affect these structures and eventually change their properties. In vivo, the remedy is remodeling which, in some vertebrates, destroys and reconstructs new tissue with some energetic expenses, but allows a preservation of the bone-body fluids equilibria. In some animals, with no remodeling process, the deposition of new mineral is continuous affording at the very least a control of body fluids mineral content. Regarding biomaterials, the fabrication of bone substitutes analogous to the original has long be considered as an ideal. The way seems difficult and costly, in addition, the objective of a bone substitute is to favor bone tissue reconstruction and thus the conception has evolved towards the realization of scaffolds allowing cells recruitments, attachment, proliferation and differentiation and progressively disappearing as the reconstruction the tissue is advancing. The “chemical” imitation of bone tissue in vitro presents an interest as it allows the parameters involved in the mineralization process and mineral evolution and maturation to be assessed and the relations between mechanical properties and the tissue characteristics to be more precisely determined.
Acknowledgments
The authors thank the french state funds managed by the ANR within the Investissements d’Avenir programme under reference ANR-11-IDEX-0004-02, and more specifically within the framework of the Cluster of Excellence MATISSE (MATériaux, Interfaces, Surfaces, Environment) led by Sorbonne Universités for covering the costs to publish this review paper in open access.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lowenstam, H.A. Minerals formed by organisms. Science 1981, 211, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C. Calcium phosphate biominerals. Rev. Mineral. Geochem. 2002, 48, 427–453. [Google Scholar] [CrossRef]
- Bazin, D.; Daudon, M.; Combes, C.; Rey, C. Characterization and some physicochemical aspects of pathological microcalcifications. Chem. Rev. 2012, 112, 5092–5120. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, D. A crystallographic study of vertebrates otoliths. Biol. Bull. 1963, 125, 441–463. [Google Scholar] [CrossRef]
- Li, Z.; Pasteris, J.D. Tracing the pathway of compositional changes in bone mineral with age: Preliminary study of bioapatite aging in hypermineralized dolphin’s bulla. Biochim. Biophys. Acta 2014, 1840, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Sakae, T.; Nakada, H.; LeGeros, J.P. Historical review of biological apatite crystallography. J. Hard Tissue Biol. 2015, 24, 111–122. [Google Scholar] [CrossRef]
- Uskokovic, V. The role of hydroxyl channel in defining selected physicochemical peculiarities exhibited by hydroxyapatite. RSC Adv. 2015, 5, 36614–36633. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Halter, T.J.; Borah, B.M.; Nancollas, G.H. Tracking amorphous precursor formation and transformation during induction stages of nucleation. Cryst. Growth Des. 2014, 14, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Omelon, S.; Ariganello, M.; Bonucci, E.; Grynpas, M.; Nanci, A. A review of phosphate mineral nucleation in biology and geobiology. Calcif. Tissue Int. 2013, 93, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Mineralization of bones and teeth. Elements 2007, 3, 385–391. [Google Scholar] [CrossRef]
- Gomez-Morales, J.; Iafisco, M.; Delgado-Lopez, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- De Jong, W.F. La substance minérale dans les os. Recueil des Travaux Chimiques des Pays-Bas 1926, 45, 445–448. (In French) [Google Scholar] [CrossRef]
- White, T.J.; Zhili, D. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. Sect. B Struct. Sci. 2003, B59, 1–16. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Labarthe, J.C.; Bonel, G.; Montel, G. Structure and properties of B-type phosphocalcium carbonate apatites. Ann. Chim. 1973, 8, 289–301. [Google Scholar]
- LeGeros, R.Z. Calcium phosphates in oral biology and medecine. Monogr. Oral Sci. 1991, 15, 1–201. [Google Scholar] [PubMed]
- Aoba, T. Recent observations on enamel crystal formation during mammalian amelogenesis. Anat. Rec. 1996, 245, 208–218. [Google Scholar] [CrossRef]
- Fleet, M.E.; Liu, X. Coupled substitution of type A and B carbonate in sodium-bearing apatite. Biomaterials 2007, 28, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C.; Drouet, C.; Grossin, D. Bioactive ceramics: Physical chemistry. In Comprehensive Biomaterials; Ducheyne, P., Healy, K.E., Hutmacher, D.W., Grainger, D.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 1, pp. 187–221. [Google Scholar]
- Legros, R.; Balmain, N.; Bonel, G. Age-related changes in mineral of rat and bovine cortical bone. Calcif. Tissue Int. 1987, 41, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Glimcher, M.J.; Rey, C.; Ackerman, J.L. A unique protonated phosphate group in bone mineral not present in synthetic calcium phosphates. Identification by phosphorus-31 solid state NMR spectroscopy. J. Mol. Biol. 1994, 244, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Shimizu, M.; Collins, B.; Glimcher, M.J. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ions in the early deposits of a solid phase of calcium phosphate in bone and enamel, and their evolution with age. I: Investigations in the ν4 PO4 domain. Calcif. Tissue Int. 1990, 46, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Winand, L. Etude physico-chimique du phosphate tricalcique hydraté et de l’hydroxyapatite. Ann. Chim. 1961, 6, 951–967. (In French) [Google Scholar]
- Montel, G.; Bonel, G.; Heughebaert, J.-C.; Trombe, J.-C.; Rey, C. New concepts in the composition, crystallization and growth of the mineral components of calcified tissues. J. Cryst. Growth 1981, 53, 74–79. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C.; Drouet, C.; Sfihi, H.; Barroug, A. Physico-chemical properties of nanocrystalline apatites: Implications for biominerals and biomaterials. Mater. Sci. Eng. C 2007, 27, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Rey, C.; Renugopalakrishnan, V.; Shimizu, M.; Collins, B.; Glimcher, M.J. A resolution-enhanced Fourier Transform Infrared spectroscopic study of the environment of the CO32− ion in the mineral phase of enamel during its formation and maturation. Calcif. Tissue Int. 1991, 49, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kourkoumelis, N.; Tzaphlidou, M. Spectroscopic assessment of normal cortical bone: Differences in relation to bone site and sex. Sci. World J. 2010, 10, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Jäger, C.; Welzel, T.; Meyer-Zaika, W.; Epple, M. A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Hina, A.; Tofighi, A.; Glimcher, M.J. Maturation of poorly crystalline apatites: Chemical and structural aspects in vivo and in vitro. Cells Mater. 1995, 5, 345–356. [Google Scholar]
- Nassif, N.; Martineau, F.; Syzgantseva, O.; Gobeaux, F.; Willinger, M.; Coradin, T.; Cassaignon, S.; Azaïs, T.; Giraud-Guille, M.-M. In vivo inspired conditions to synthesize biomimetic hydroxyapatite. Chem. Mater. 2010, 22, 3653–3663. [Google Scholar] [CrossRef]
- Cazalbou, S.; Combes, C.; Eichert, D.; Rey, C.; Glimcher, M.J. Poorly crystalline apatites: Evolution and maturation in vitro and in vivo. J. Bone Miner. Metab. 2004, 22, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Eichert, D.; Sfihi, H.; Combes, C.; Rey, C. Specific characteristics of wet nanocrystalline apatites. consequences on biomaterials and bone tissue. Key Eng. Mater. 2004, 254–256, 927–930. [Google Scholar] [CrossRef]
- Eichert, D.; Combes, C.; Drouet, C.; Rey, C. Formation and evolution of hydrated surface layers of apatites. Key Eng. Mater. 2005, 284–286, 3–6. [Google Scholar] [CrossRef]
- Cazalbou, S. Echanges Cationiques Impliquant des Apatites Nanocristallines Analogues au Minéral Osseux. Ph.D. Thesis, Institut National Polytechnique de Toulouse, Toulouse, France, 2000. [Google Scholar]
- Cazalbou, S.; Eichert, D.; Ranz, X.; Drouet, C.; Combes, C.; Harmand, M.F.; Rey, C. Ion exchanges in apatites for biomedical application. J. Mater. Sci. Mater. Med. 2005, 16, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, C.; Combes, C.; Drouet, C.; Lebugle, A.; Sfihi, H.; Barroug, A. Nanocrystalline apatites in biological systems: Characterisation, structure and properties. Materialwissenschaft und Werkstofftechnik 2007, 38, 996–1002. [Google Scholar] [CrossRef]
- Drouet, C.; Carayon, M.; Combes, C.; Rey, C. Surface enrichment of biomimetic apatites with biologically-active ions Mg2+ and Sr2+: A preamble to the activation of bone repair materials. Mater. Sci. Eng. C 2008, 28, 1544–1550. [Google Scholar] [CrossRef] [Green Version]
- Errassifi, F.; Menbaoui, A.; Autefage, H.; Benaziz, L.; Ouizat, S.; Santran, V.; Sarda, S.; Lebugle, A.; Combes, C.; Barroug, A.; et al. Adsorption onto nanocrystalline apatitic calcium phosphates. Applications to growth factors and drugs delivery. In Advances in Bioceramics and Biotechnologies; Narayan, R., McKittrick, J., Singh, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ouizat, S.; Barroug, A.; Legrouri, A.; Rey, C. Adsorption of bovine serum albumin on poorly crystalline apatite: Influence of maturation. Mater. Res. Bull. 2000, 34, 2279–2289. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C. Physical chemistry of biological apatites. In Biomineralization and Biomaterials, Fundamentals and Applications; Aparicio, C., Pau Ginebra, M., Eds.; Woodhead publishing: Swanston, UK, 2015; pp. 95–128. [Google Scholar]
- Brown, W.E. Crystal structure of octacalcium phosphate. Nature 1962, 196, 1048–1050. [Google Scholar] [CrossRef]
- Eichert, D.; Drouet, C.; Sfihi, H.; Rey, C.; Combes, C. Nanocrystalline Apatite Based Biomaterials: Synthesis, Processing and Characterization; Eichert, D., Drouet, C., Sfihi, H., Rey, C., Combes, C., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2009. [Google Scholar]
- Rey, C.; Combes, C.; Drouet, C.; Cazalbou, S.; Grossin, D.; Brouillet, F.; Sarda, S. Surface properties of biomimetic nanocrystalline apatites; applications in biomaterials. Prog. Cryst. Growth Charact. Mater. 2014, 60, 63–73. [Google Scholar] [CrossRef]
- Driessens, F.C.M.; Verbeeck, R.M.H. Biominerals; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Iyengar, G.V.; Tandon, L. Minor and Trace Elements in Human Bones and Teeth; International Atomic Energy Agency: Vienna, Austria, 1999. [Google Scholar]
- Teruel, J.D.D.; Alcolea, A.; Hernández, A.; Ruiz, A.J.O. Comparison of chemical composition of enamel and dentine in human, bovine, porcine and ovine teeth. Arch. Oral Biol. 2015, 60, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.A.B.; Adachi, E.M.; Saiki, M. INAA of enamel and dentine samples of a group of children and adults: A comparative study. J. Radioanal. Nucl. Chem. 2007, 276, 49–52. [Google Scholar] [CrossRef]
- Zenóbio, M.A.F.; Tavares, M.S.N.; Zenóbio, E.G.; Silva, T.A. Elemental composition of dental biologic tissues: Study by means of different analytical techniques. J. Radioanal. Nucl. Chem. 2011, 289, 161–166. [Google Scholar] [CrossRef]
- Ishiguro, K.; Nakagaki, H.; Takeuchi, K.; Mukai, M.; Yoshika, I.; Miyauchi, K.; Robinson, C.; Weatherell, J.A. Distribution of fluoride in the dental tissues and their supporting mandibular bone from the same individual. Arch. Oral Biol. 1994, 39, 535–537. [Google Scholar] [CrossRef]
- Lakomaa, E.L.; Rytömaa, I. Mineral composition of enamel and dentin of primary and permanent teeth in Finland. Scand. J. Dent. Res. 1977, 85, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Adachi, L.K.; Adachi, E.M. Elemental comparison in sound and carious human teeth by instrumental neutron activation analysis. Eur. Radiol. 2014, 24, 29–32. [Google Scholar] [CrossRef]
- Hendricks, S.B.; Hill, W.L. The nature of bone and phosphate rocks. Proc. Natl. Acad. Sci. USA 1950, 36, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Glimcher, M.J. Three-dimensional spatial relationship between the collagen fibrils and the inorganic calcium phosphate crystals of pickerel (Americanus americanus) and herring (Clupea harengus) bone. J. Mol. Biol. 1991, 217, 487–501. [Google Scholar] [CrossRef]
- Bala, Y.; Farlay, D.; Boivin, G. Bone mineralization: From tissue to crystal in normal and pathological contexts. Osteoporos. Int. 2013, 24, 2153–2166. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Abrams, S.A.; David-Street, J.E.; Heer, M.; O’Brien, K.O.; Wastney, M.E.; Zwart, S.R. Fifty years of human space travel: Implications for bone and calcium research. Annu. Rev. Nutr. 2014, 34, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, L.; Traub, W.; de Buffrenil, V.; Allizard, F.; Arad, T.; Weiner, S. Rostrum of a toothed whale: Ultrastructural study of a very dense bone. Bone 1998, 23, 241–247. [Google Scholar] [CrossRef]
- Rey, C.; Collins, B.; Goehl, T.; Dickson, I.R.; Glimcher, M.J. The carbonate environment in bone mineral: A resolution-enhanced fourier transform infrared spectroscopy study. Calcif. Tissue Int. 1989, 45, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C.; Holcom, D.W.; Young, R.A. Infrared determination of the degree of substitution of hydroxyl by carbonate ion in human enamel. Calcif. Tissue Int. 1985, 37, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Penel, G.; Leroy, G.; Rey, C.; Bres, E. Micro-raman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif. Tissue Int. 1998, 63, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Beshah, K.; Rey, C.; Glimcher, M.J.; Shimizu, M.; Griffin, R.G. Solid state carbon-13 and proton NMR studies of carbonate-containing calcium phosphates and enamel. J. Solid State Chem. 1990, 84, 71–81. [Google Scholar] [CrossRef]
- Combes, C.; Rey, C.; Mounic, S. Identification and evaluation of HPO4 ions in biomimetic poorly crystalline apatites and bone mineral. Key Eng. Mater. 2001, 192–195, 143–146. [Google Scholar] [CrossRef]
- Bohic, S.; Rey, C.; Legrand, A.; Sfihi, H.; Rohanizadeh, R.; Martel, C.; Barbier, A.; Daculsi, G. Characterization of the trabecular rat bone mineral: Effect of ovariectomy and bisphosphonate treatment. Bone 2000, 26, 341–348. [Google Scholar] [CrossRef]
- Wang, Y.; Von Euw, S.; Fernandes, F.M.; Cassaignon, S.; Selmane, M.; Laurent, G.; Pehau-Arnaudet, G.; Coelho, C.; Bonhomme-Coury, L.; Giraud-Guille, M.-M.; et al. Water-mediated structuring of bone apatite. Nat. Mater. 2013, 12, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.G.; Parker, S.F.; Mitchell, P.C.H. A study by high energy transfer inelastic neutron scattering spectroscopy of the mineral fraction of ox femur bone. J. Mol. Struct. 2003, 651–653, 123–126. [Google Scholar] [CrossRef]
- Cho, G.; Wu, Y.; Ackerman, J.L. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science 2003, 300, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Pasteris, J.D.; Wopenka, B.; Freeman, J.J.; Rogers, K.; Valsami-Jones, E.; van der Houwen, J.; Silva, M.J. Lack of OH in nanocrystalline apatite as a function of degree of atomic order: Implications for bone and biomaterials. Biomaterials 2004, 25, 229–238. [Google Scholar] [CrossRef]
- Rey, C.; Miquel, J.L.; Facchini, L.; Legrand, A.P.; Glimcher, M.J. Hydroxyl groups in bone mineral. Bone 1995, 16, 583–586. [Google Scholar] [CrossRef]
- Grossin, D.; Rollin-Martinet, S.; Estournès, C.; Rossignol, F.; Champion, E.; Combes, C.; Rey, C.; Geoffroy, C.; Drouet, C. Biomimetic apatite sintered at very low temperature by spark plasma sintering: Physico-chemistry and microstructure aspects. Acta Biomater. 2010, 6, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, C.; Renugopalakrishnan, V.; Collins, B.; Glimcher, M.J. Fourier transform infrared spectroscopic study of the carbonate ions in bone mineral during aging. Calcif. Tissue Int. 1991, 49, 251–258. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.; Trautz, O.R.; LeGeros, J.P.; Klein, E. Carbonate substitution in the apatite structure. Bull. Soc. Chim. Fr. 1968, 1712–1718. [Google Scholar]
- Laurencin, D.; Wong, A.; Chrzanowski, W.; Knowles, J.C.; Qiu, D.; Pickup, D.M.; Newport, R.J.; Gan, Z.; Duer, M.J.; Smith, M.E. Probing the calcium and sodium local environment in bones and teeth using multinuclear solid state NMR and X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Dykes, E.; Elliott, J.C. The occurrence of chloride ions in the apatite lattice of Holly Springs hydroxyapatite and dental enamel. Calcif. Tissue Res. 1971, 7, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Burnell, J.M.; Teubner, E.J.; Miller, A.G. Normal maturational changes in bone matrix, mineral, and crystal size in the rat. Calcif. Tissue Int. 1980, 31, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J.; Lee, D.D.; Brenna, J.T.; Chandra, S.; Morrison, G.H. Detection and localization of silicon and associated elements in vertebrate bone tissue by imaging ion microscopy. Calcif. Tissue Int. 1986, 38, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Code, R.F.; Gelman, R.L.; Armstrong, R.S.; Hallsworth, C.; Lemaire, C.; Cheng, P.T. Field dependence of 19F NMR in rat bone powders. Phys. Med. Biol. 1990, 35, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Grynpas, M.; Rey, C. The effect of fluoride treatment on bone mineral crystals in the rat. Bone 1992, 13, 423–429. [Google Scholar] [CrossRef]
- Boivin, G.; Meunier, P.J. Fluoride and bone: Toxic effects and therapeutic role. In Therapeutic Uses of Trace Elements; Nève, J., Chappuis, P., Lamand, M., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 1996; pp. 283–295. [Google Scholar]
- Nakagaki, H.; Koyama, Y.; Sakakibara, Y.; Weatherell, J.A.; Robinson, C. Distribution of fluoride across human dental enamel, dentine and cementum. Arch. Oral Biol. 1987, 32, 651–654. [Google Scholar] [CrossRef]
- Cazalbou, S.; Hina, A.; Rey, C. Interactions between trace elements and bone mineral matrix. In New Aspects of Trace Element Research; Abdulla, M., Bost, M., Gamon, S., Arnaud, P., Chazot, G., Eds.; Smith-Gordon: London, UK, 1999; pp. 58–62. [Google Scholar]
- Glimcher, M.J. Bone: Nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Rev. Mineral. Geochem. 2006, 64, 223–282. [Google Scholar] [CrossRef]
- Landis, W.J.; Song, M.J.; Leith, A.; McEwen, L.; McEwen, B. Mineral and organic matrix interaction in normally calcifying tendon visualized in 3 dimensions by high-voltage electron-microscopic tomography and graphic image-reconstruction. J. Struct. Biol. 1993, 110, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Nikel, O.; Laurencin, D.; Bonhomme, C.; Sroga, G.E.; Besdo, S.; Lorenz, A.; Vashishth, D. Solid state NMR investigation of intact human bone quality: Balancing issues and insight into the structure at the organic-mineral interface. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 6320–6331. [Google Scholar] [CrossRef] [PubMed]
- Skwarek, E.; Janusz, W.; Sternik, D. Adsorption of citrate ions on hydroxyapatite synthetized by various methods. J. Radioanal. Nucl. Chem. 2013, 299, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; Müller, K.H.; Wong, W.C.; Pickard, C.J.; Reid, D.G.; Skepper, J.N.; Duer, M.J. Citrate bridges between mineral platelets in bone. Proc. Natl. Acad. Sci. USA 2014, 111, E1354–E1363. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C. What bridges mineral platelets of bone? BoneKEy Rep. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.C.; Jackson, D.C. Lactate uptake by skeletal bone in anoxic turtles, trachemys scripta. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 146, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J. The strength of a calcified tissue depends in part on the molecular structure and organization of its constituent mineral crystals in their organic matrix. Bone 1995, 16, 533–544. [Google Scholar] [CrossRef]
- Morris, D.C.; Vaananen, H.K.; Anderson, H.C. Matrix vesicle calcification in rat epiphyseal growth palte cartilage prepared anhydrously for electron microscopy. Metab. Bone Dis. Relat. Res. 1983, 5, 131–137. [Google Scholar] [CrossRef]
- Kim, H.-M.; Rey, C.; Glimcher, M.J. X-ray diffraction, electron microscopy, and fourier transform infrared spectroscopy of apatite crystals isolated from chicken and bovine cartilage. Calcif. Tissue Int. 1996, 59, 58–63. [Google Scholar]
- McNally, E.A.; Schwarcz, H.P.; Botton, G.A.; Arsenault, L.A. A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Nylen, M.U.; Eanes, E.D.; Omnell, K.A. Crystal growth in rat enamel. J. Cell Biol. 1963, 18, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Frazier, P.D. Adult human enamel—An SEM study of crystallite size and morphology. J. Ultrastruct. Res. 1968, 22, 1–11. [Google Scholar] [CrossRef]
- Santo, A.R.E.; Line, S.R.P. The enamel organic matrix: Structure and function. Braz. J. Oral Sci. 2005, 4, 716–724. [Google Scholar]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc and health: Current status and future directions zinc homeostasis in humans 1. J. Nutr. 2000, 130, 1360S–1366S. [Google Scholar] [PubMed]
- Burger, C.; Zhou, H.W.; Wang, H.; Sics, I.; Hsiao, B.S.; Chu, B.; Graham, L.; Glimcher, M.J. Lateral packing of mineral crystals in bone collagen fibrils. Biophys. J. 2008, 95, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Kim, H.M.; Gerstenfeld, L.; Glimcher, M.J. Characterization of the apatite crystals of bone and their maturation in osteoclast cell culture: Comparison with native bone crystals. Connect. Tissue Res. 1996, 35, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.J. Molecular models illustrating the possible distribution of holes in simple systematically staggered arrays of type I collagen molecules in native-type fibrils. Connect. Tissue Res. 1989, 21, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, J.; Landis, W.J. A contribution with review to the description of mineralization of bone and other calcified tissues in vivo. Anat. Rec. 1991, 230, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Eidelman, N.; Chow, L.C.; Brown, W.E. Calcium phosphate saturation levels in ultrafiltered serum. Calcif. Tissue Int. 1987, 40, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Combes, C.; Rey, C. Adsorption of proteins and calcium phosphate materials bioactivity. Biomaterials 2002, 23, 2817–2823. [Google Scholar] [CrossRef]
- Combes, C.; Rey, C.; Frèche, M. In vitro crystallization of octacalcium phosphate on type I collagen: Influence of serum albumin. J Mater. Sci. Mater. Med. 1999, 10, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.-Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. R Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Veis, A.; Dorvee, J.R. Biomineralization mechanisms: A new paradigm for crystal nucleation in organic matrices. Calcif. Tissue Int. 2013, 93, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C.; Cölfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Colfen, H.; Antonietti, M. Mesocrystals and Non Classical Crystallization; John Wiley & Sons Ltd.: Chichester, UK, 2008. [Google Scholar]
- Drouet, C. A comprehensive guide to experimental and predicted thermodynamic properties of phosphate apatite minerals in view of applicative purposes. J. Chem. Thermodyn. 2015, 81, 143–159. [Google Scholar] [CrossRef]
- Rollin-Martinet, S.; Navrotsky, A.; Champion, E.; Grossin, D.; Drouet, C. Thermodynamic basis for evolution of apatite in calcified tissues. Am. Mineral. 2013, 98, 2037–2045. [Google Scholar] [CrossRef]
- Weiner, S. Transient precursor strategy in mineral formation of bone. Bone 2006, 39, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; de With, G.; Sommerdijk, N.A.J.M. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010, 9, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Bodier-Houllé, P.; Steuer, P.; Meyer, J.M.; Bigeard, L.; Cuisinier, F.J. High-resolution electron-microscopic study of the relationship between human enamel and dentin crystals at the dentinoenamel junction. Cell Tissue Res. 2000, 301, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Moradian-oldak, J. Control of apatite crystal growth in a fluoride containing amelogenin-rich matrix. Biomaterials 2005, 26, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hanaizumi, Y.; Ohshima, H. Occurrence of amorphous and crystalline mineral deposits at the epithelial-mesenchymal interface of incisors in the calcium-loaded rat: Implication of novel calcium binding domains. Anat. Rec. 1996, 245, 174–185. [Google Scholar] [CrossRef]
- Beniash, E.; Metzler, R.A.; Lam, R.S.K.; Gilbert, P.U.P.A. Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 2009, 166, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Spevak, L.; Flach, C.R.; Hunter, T.; Mendelsohn, R.; Boskey, A. Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif. Tissue Int. 2013, 92, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Bonar, L.C.; Roufosse, A.H.; Sabine, W.K.; Grynpas, M.D.; Glimcher, M.J. X-ray diffraction studies of the crystallinity of bone mineral in newly synthesized and density fractionated bone. Calcif. Tissue Int. 1983, 35, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P. How does bone support calcium homeostasis? Bone 2003, 33, 264–268. [Google Scholar] [CrossRef]
- Baig, A.A.; Fox, J.L.; Wang, Z.; Higuchi, W.I.; Miller, S.C.; Barry, A.M.; Otsuka, M. Metastable equilibrium solubility behavior of bone mineral. Calcif. Tissue Int. 1999, 64, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Eichert, D. Etude de la Réactivité de Surface D’apatites de Synthèse Nanocristallines. Ph.D’s Thesis, Institut National Polytechnique de Toulouse, Toulouse, France, 2001. [Google Scholar]
- Bala, Y.; Farlay, D.; Delmas, P.D.; Meunier, P.J.; Boivin, G. Time sequence of secondary mineralization and microhardness in cortical and cancellous bone from ewes. Bone 2010, 46, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Busa, B.; Miller, L.M.; Rubin, C.T.; Qin, Y.-X.; Judex, S. Rapid establishment of chemical and mechanical properties during lamellar bone formation. Calcif. Tissue Int. 2005, 77, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Meunier, P.J. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif. Tissue Int. 2002, 70, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D. The design of mineralised hard tissues for their mechanical functions. J. Exp. Biol. 1999, 202, 3285–3294. [Google Scholar] [PubMed]
- Moreno, E.; Kresak, M.; Zahradnik, R.T. Fluoridated hydroxyapatite solubility and caries formation. Nature 1974, 247, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Hina, A. Etude de la Réactivité en Milieu Aqueux, D’apatites Phosphocalciques D’intérêt Biologique. Ph.D. Thesis, Institut National Poytechnique de Toulouse, Toulouse, France, 1996. [Google Scholar]
- Talmage, R.V.; Talmage, D.W. Calcium homeostasis: Solving the solubility problem. J. Musculoskelet. Neuronal Interact. 2006, 6, 402–407. [Google Scholar] [PubMed]
- Shashi, A.; Singla, S. Parathyroid function in osteofluorosis. World J. Med. Sci. 2013, 8, 67–73. [Google Scholar]
- Zhu, Y.; Zhu, Z.; Zhao, X.; Liang, Y.; Huang, Y. Characterization, dissolution and solubility of lead hydroxypyromorphite [Pb5(PO4)3OH] at 25–45 °C. J. Chem. 2015, 2015. [Google Scholar] [CrossRef]
- Christoffersen, J.; Christoffersen, M.R.; Kolthoff, N.; Bärenholdt, O. Effects of strontium ions on growth and dissolution of hydroxyapatite and on bone mineral detection. Bone 1997, 20, 47–54. [Google Scholar] [CrossRef]
- Farlay, D.; Boivin, G.; Panczer, G.; Lalande, A.; Meunier, P.J. Long-term strontium ranelate administration in monkeys preserves characteristics of bone mineral crystals and degree of mineralization of bone. J. Bone Miner. Res. 2005, 20, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.W. An age-specific kinetic model of lead metabolism in humans. Eviron. Heal. Perspect. 1993, 101, 598–616. [Google Scholar] [CrossRef]
- Flaherty, E.J.O. A physiologically based kinetic model for lead in children and adults. Environ. Health Perspect. 1998, 106, 1495–1503. [Google Scholar] [CrossRef]
- Simmer, J.P.; Hu, J.C.-C. Dental enamel formation and its impact on clinical dentistry. J. Dent. Educ. 2001, 65, 896–905. [Google Scholar] [PubMed]
- Smith, C.E. Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 1998, 9, 128–161. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Kirkham, J.; Brookes, S.J.; Bonass, W.A.; Shore, R.C. The chemistry of enamel development. Int. J. Dev. Biol. 1995, 39, 145–152. [Google Scholar] [PubMed]
- Lynch, R.J.M. The primary and mixed dentition, post-eruptive enamel maturation and dental caries: A review. Int. Dent. J. 2013, 63, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Arends, J. Mechanism of enamel dissolution and its prevention. J. Biol. Buccale 1977, 5, 219–237. [Google Scholar] [PubMed]
- Kuhn, L.T.; Grynpas, M.D.; Rey, C.C.; Wu, Y.; Ackerman, J.L.; Glimcher, M.J. A comparison of the physical and chemical differences between cancellous and cortical bovine bone mineral at two ages. Calcif. Tissue Int. 2008, 83, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Jalvo, Y.; Sanchez-Chillon, B.; Andrews, P.; Fernandez-Lopez, S.; Alcala Martinez, I. Morphological taphonomic transformations of fossil bones in continental environments, and repercussions on their chemical composition. Archaeometry 2002, 44, 353–361. [Google Scholar] [CrossRef]
- Kohn, M.J.; Schoeninger, M.; Barker, W.W. Altered states: Effects of diagenesis on fossil tooth chemistry. Geochim. Cosmochim. Acta 1999, 63, 2737–2747. [Google Scholar] [CrossRef]
- Clementz, M.T. New insight from old bones: Stable isotope analysis of fossil mammals. J. Mammal. 2012, 93, 368–380. [Google Scholar] [CrossRef]
- Kohn, M.J.; Law, J.M. Stable isotope chemistry of fossil bone as a new paleoclimate indicator. Geochim. Cosmochim. Acta 2006, 70, 931–946. [Google Scholar] [CrossRef]
- Cazalbou, S.; Eichert, D.; Drouet, C.; Combes, C.; Rey, C. Minéralisations biologiques à base de phosphate de calcium. Comptes Rendus Palevol 2004, 3, 563–572. (In French) [Google Scholar] [CrossRef]
- Grunenwald, A.; Keyser, C.; Sautereau, A.M.; Crubézy, E.; Ludes, B.; Drouet, C. Novel contribution on the diagenetic physicochemical features of bone and teeth minerals, as substrates for ancient DNA typing. Anal. Bioanal. Chem. 2014, 406, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, L.; Trueman, C.N.; Palmer, M.R. Protracted diagenetic alteration of REE contents in fossil bioapatites: Direct evidence from Lu-Hf isotope systematics. Geochim. Cosmochim. Acta 2010, 74, 6077–6092. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the surface hydrated layer model for poorly crystalline apatite nanocrystals (Reprinted from Nanocrystalline apatite based biomaterials: synthesis, processing and characterization; Copyright (2009), Eichert D., Drouet C., Sfihi H., Rey C. and Combes C. [43] with permission from Nova Science Publishers Inc.).
Figure 1.
Schematic representation of the surface hydrated layer model for poorly crystalline apatite nanocrystals (Reprinted from Nanocrystalline apatite based biomaterials: synthesis, processing and characterization; Copyright (2009), Eichert D., Drouet C., Sfihi H., Rey C. and Combes C. [43] with permission from Nova Science Publishers Inc.).

Figure 2.
Maturation pathways of nanocrystalline apatites depending on the composition of the medium (the arrows, after the ions, represent an increase (or decrease) of the species considered during maturation).
Figure 2.
Maturation pathways of nanocrystalline apatites depending on the composition of the medium (the arrows, after the ions, represent an increase (or decrease) of the species considered during maturation).

Figure 3.
Example of the effect of adsorbed molecules on crystal growth [102,103]. (A) Effect of bovine serum albumin (BSA) on octacalcium phosphate crystal growth: kinetics results (relative rate of crystal growth of octacalcium phosphate (OCP) in %) and SEM micrographs of OCP crystal growth on collagen in the presence of various concentrations of bovine serum albumin (BSA) using the constant composition crystal growth technique (adapted from [102,103]). At low albumin concentrations, when the solution reaches depletion due to nucleation and adsorption, a burst rate of crystal growth occurs exceeding that observed in the absence of albumin. At high albumin concentrations, however, there is never depletion of free BSA molecules in solution and only the well-known crystal growth inhibitory effect of the molecule on OCP crystals is evidenced. SEM micrographs showed smaller but more numerous OCP crystals in the presence of BSA. (B) Schematization of the effect of nucleation and growth without (a) or with (b) the presence of adsorbing molecules/ions on crystal growth depending on their solution concentration. Adsorbing species may have positive effect on crystal nucleation by stabilizing nuclei with a critical radius smaller than the critical radius in the absence of adsorbing species, due to a decrease of interfacial energy. As a result a multiplication of nuclei is observed in the presence of such adsorbing species. Nuclei grow together slowly and new nuclei form until depletion is reached, a ‘‘burst’’ of growth is then observed when a multitude of nuclei have formed and no crystal growth inhibitory species remains in solution: this leads to small but numerous crystals and provides a relatively homogeneous population of crystals. With larger amounts of adsorbing species, only an inhibiting crystal growth effect dominates.
Figure 3.
Example of the effect of adsorbed molecules on crystal growth [102,103]. (A) Effect of bovine serum albumin (BSA) on octacalcium phosphate crystal growth: kinetics results (relative rate of crystal growth of octacalcium phosphate (OCP) in %) and SEM micrographs of OCP crystal growth on collagen in the presence of various concentrations of bovine serum albumin (BSA) using the constant composition crystal growth technique (adapted from [102,103]). At low albumin concentrations, when the solution reaches depletion due to nucleation and adsorption, a burst rate of crystal growth occurs exceeding that observed in the absence of albumin. At high albumin concentrations, however, there is never depletion of free BSA molecules in solution and only the well-known crystal growth inhibitory effect of the molecule on OCP crystals is evidenced. SEM micrographs showed smaller but more numerous OCP crystals in the presence of BSA. (B) Schematization of the effect of nucleation and growth without (a) or with (b) the presence of adsorbing molecules/ions on crystal growth depending on their solution concentration. Adsorbing species may have positive effect on crystal nucleation by stabilizing nuclei with a critical radius smaller than the critical radius in the absence of adsorbing species, due to a decrease of interfacial energy. As a result a multiplication of nuclei is observed in the presence of such adsorbing species. Nuclei grow together slowly and new nuclei form until depletion is reached, a ‘‘burst’’ of growth is then observed when a multitude of nuclei have formed and no crystal growth inhibitory species remains in solution: this leads to small but numerous crystals and provides a relatively homogeneous population of crystals. With larger amounts of adsorbing species, only an inhibiting crystal growth effect dominates.

Figure 4.
Schematic representation of classical and non-classical theory of crystallization: classical nucleation and crystal growth pathway (a); iso-oriented crystal pathway (b); mesoscale assembly pathway in the presence of polymer or additive (c); and transformation of amorphous precursor particles pathway (d) (from [108]. Copyright 2008 John Wiley & Sons. Reproduced with permission).
Figure 4.
Schematic representation of classical and non-classical theory of crystallization: classical nucleation and crystal growth pathway (a); iso-oriented crystal pathway (b); mesoscale assembly pathway in the presence of polymer or additive (c); and transformation of amorphous precursor particles pathway (d) (from [108]. Copyright 2008 John Wiley & Sons. Reproduced with permission).

Figure 5.
Gibbs free energy of activation associated with nucleation (n), growth (g) and phase transformation (T) through two main pathways: with (B) and without (A) intermediate phase(s) (adapted from Gower’s review paper for the calcium phosphate system [107]).
Figure 5.
Gibbs free energy of activation associated with nucleation (n), growth (g) and phase transformation (T) through two main pathways: with (B) and without (A) intermediate phase(s) (adapted from Gower’s review paper for the calcium phosphate system [107]).

Figure 6.
The various phenomena involved in the diagenetic evolution of biological apatites (adapted from [145]).
Figure 6.
The various phenomena involved in the diagenetic evolution of biological apatites (adapted from [145]).


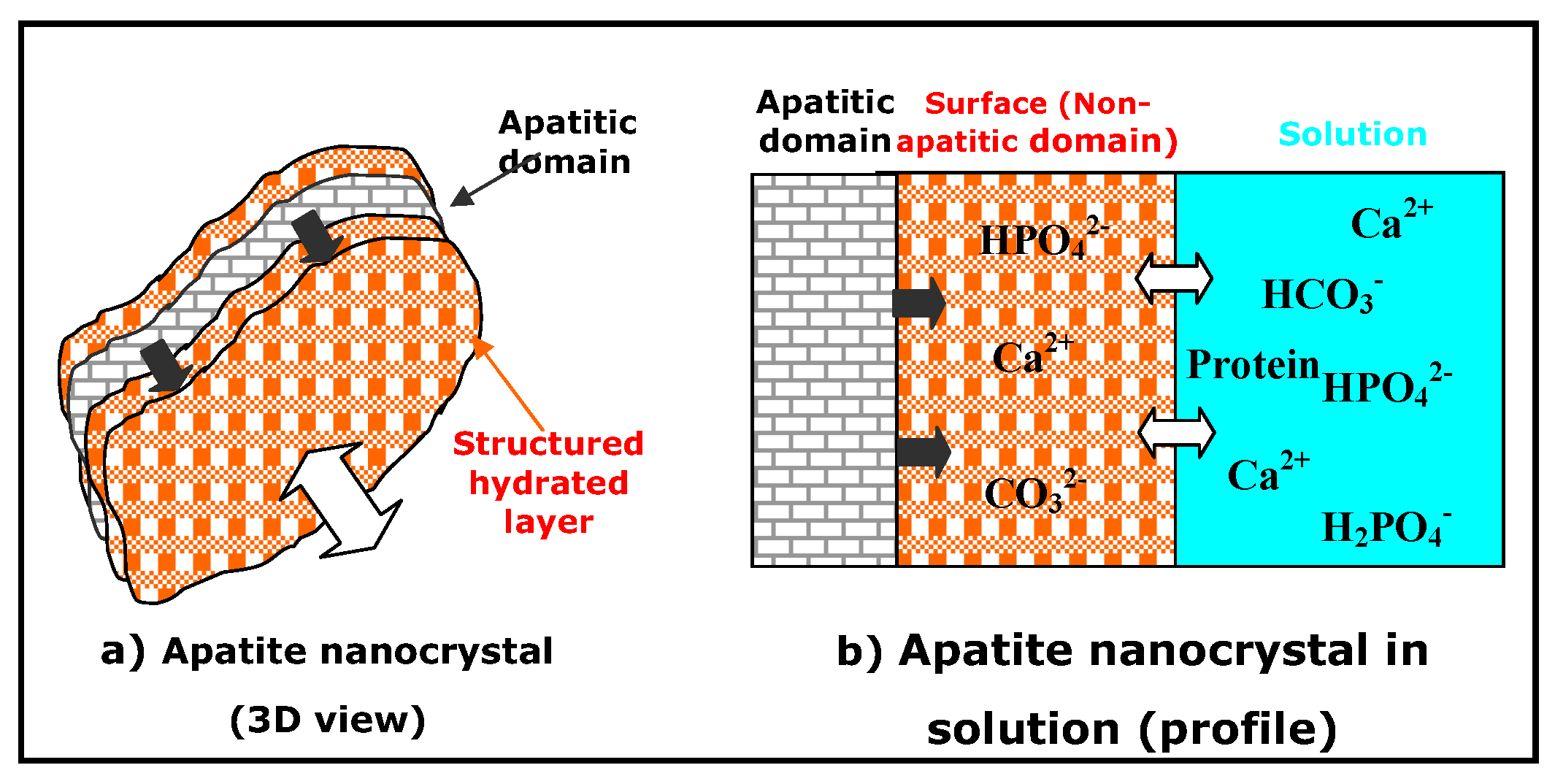{kind=link}
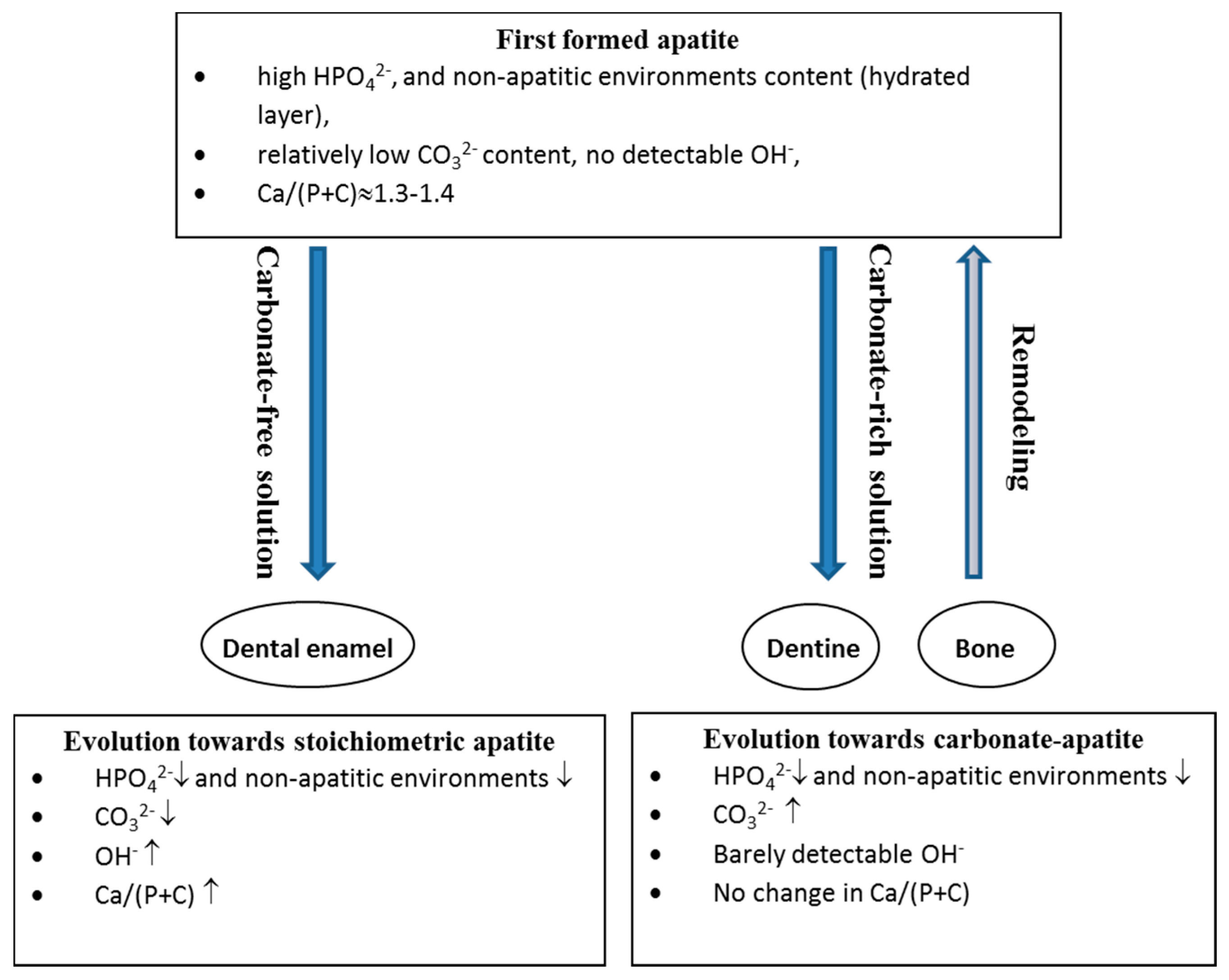{kind=link}
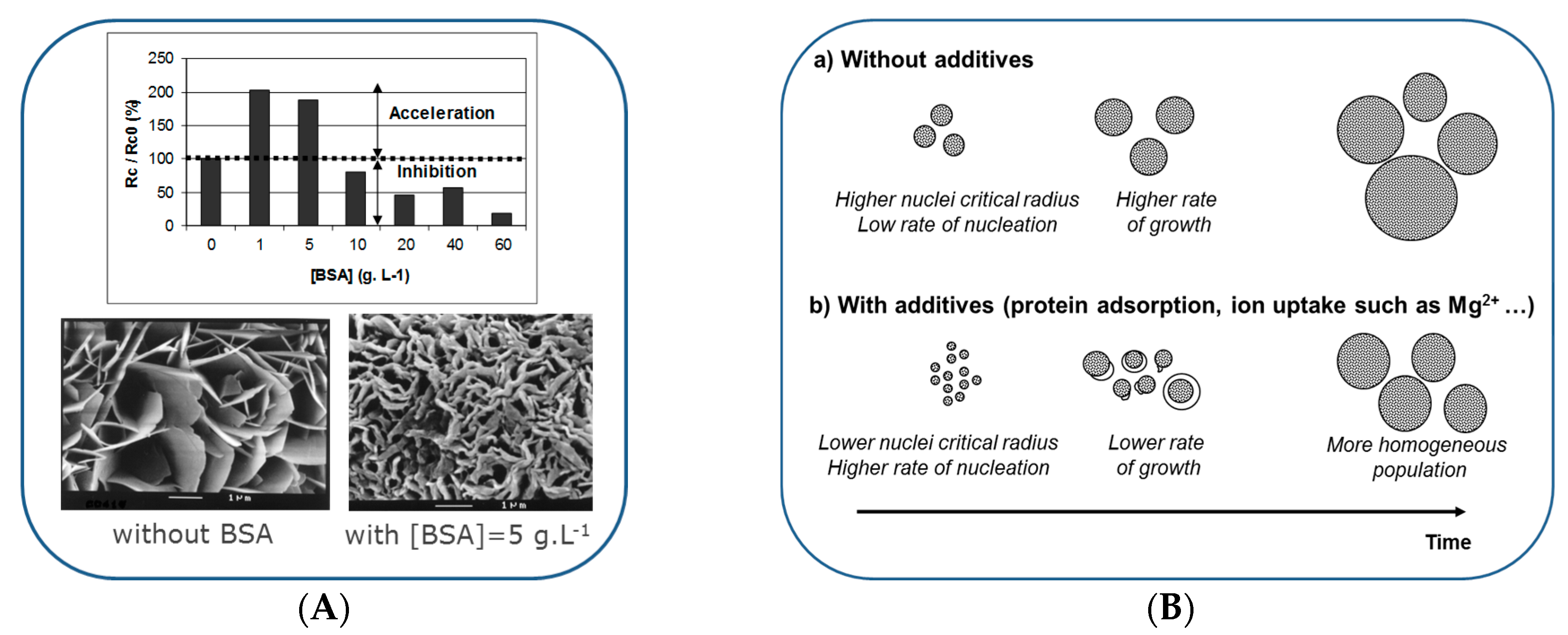{kind=link}
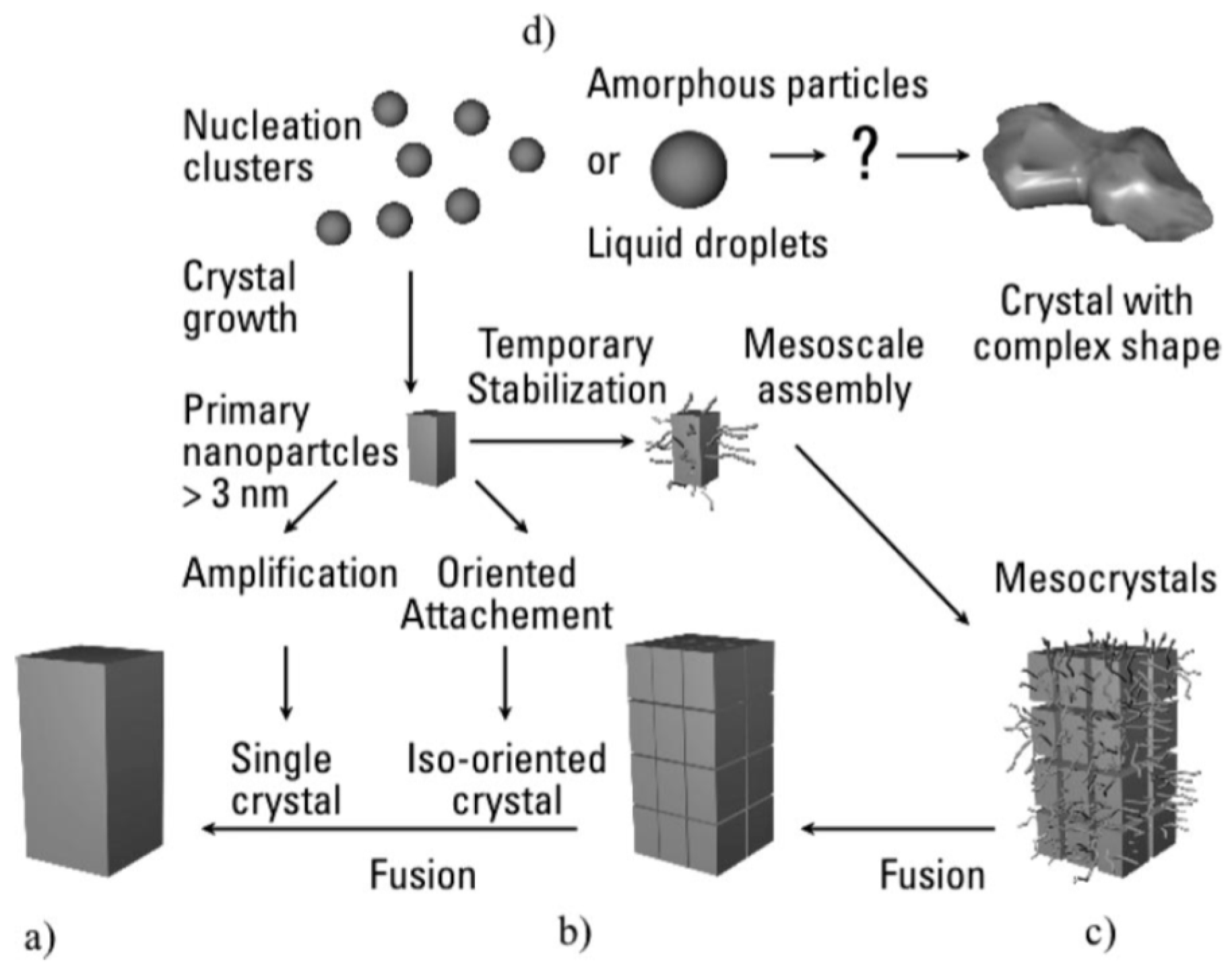{kind=link}
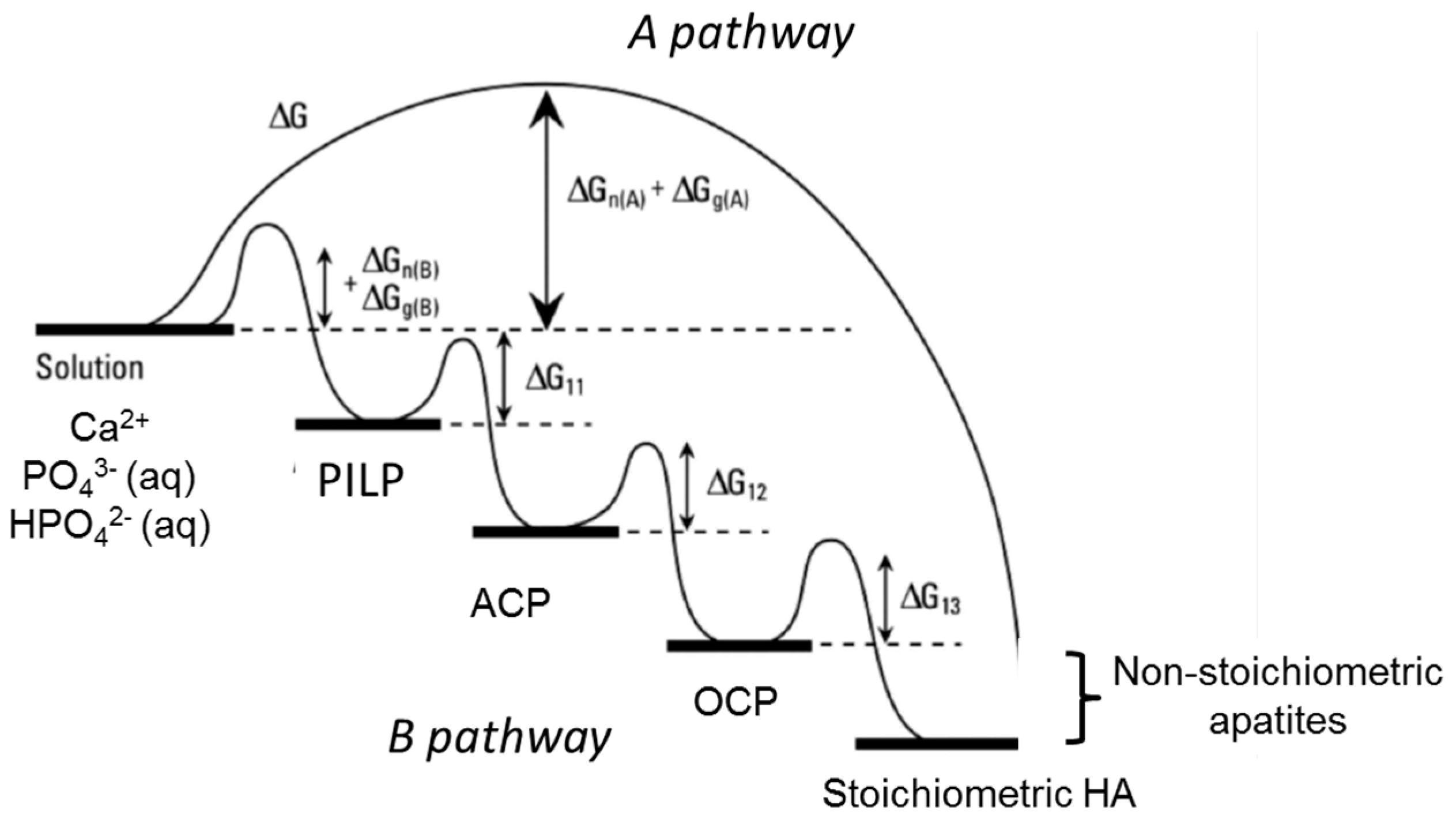{kind=link}
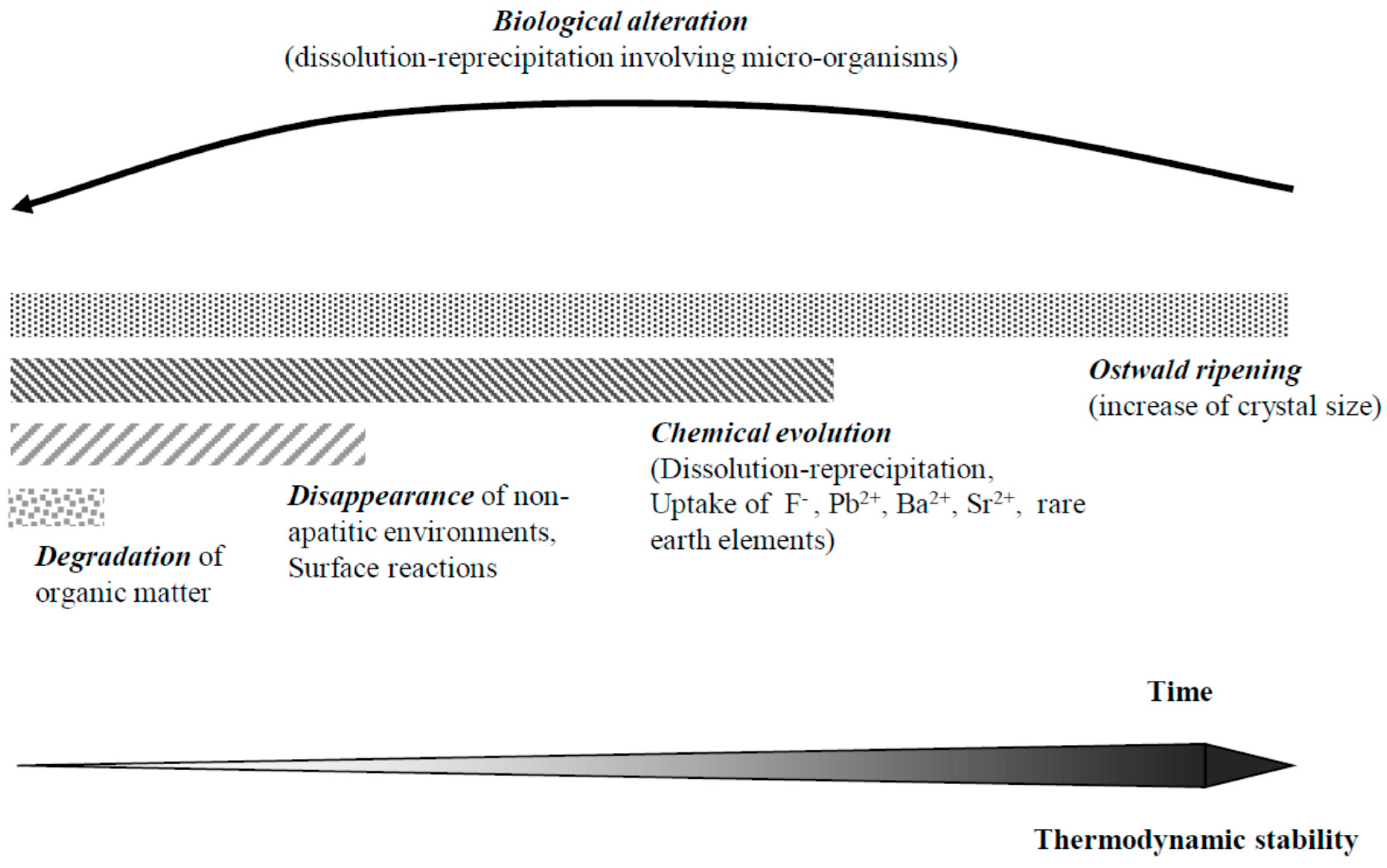{kind=link}
| Elements or Ions | Bone | Dentine | Dental Enamel | References |
|---|---|---|---|---|
| Major elements (wt %) | ||||
| C (total) | 16.7 | 11.8 | 1.4 | bone [41]; dentine and enamel [49] |
| CO32− | 5.6 | 4.6 | 3.2 | bone [53]; dentine and enamel [45] |
| N | 4.9 | 4.0 | 0.32 | dentine and enamel [49] |
| Ca | 25.4 | 26.9 | 36.6 | dentine and enamel [45] |
| P | 11.6 | 13.2 | 17.7 | dentine and enamel [45] |
| Minor elements (wt %) | ||||
| Cl | 0.13 | 0.065 | 0.37 | dentine and enamel [46,47,48,49,51,52] |
| K | 0.0047 | 0.024 | 0.070 | dentine and enamel [46,47,51] |
| Mg | 0.27 | 0.74 | 0.29 | dentine and enamel [46,47,48,51,52] |
| Na | 0.53 | 0.76 | 0.77 | dentine and enamel [47,48,49,51,52] |
| S | 0.08 | 0.070 | 0.021 | dentine and enamel [47] |
| Main trace elements (ppm) | ||||
| Al | 29 | 210 | 55 | dentine and enamel [46,49,51] |
| B | 22 | - | 11 | enamel [45] |
| F | 400 | 215 | 50 | all values from Ishiguro et al. [50] |
| Fe | 76 | 44 | 34 | dentine and enamel [46,47,51,52] |
| Pb | 4.4 | 15 | 17 | all values computed from [46] |
| Sr | 70 | 145 | 173 | dentine and enamel [46,47,48,49,52] |
| Zn | 205 | 148 | 170 | dentine and enamel [46,47,48,49,51,52] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Combes, C.; Cazalbou, S.; Rey, C. Apatite Biominerals. Minerals 2016, 6, 34. https://doi.org/10.3390/min6020034
AMA Style
Combes C, Cazalbou S, Rey C. Apatite Biominerals. Minerals. 2016; 6(2):34. https://doi.org/10.3390/min6020034
Chicago/Turabian StyleCombes, Christèle, Sophie Cazalbou, and Christian Rey. 2016. "Apatite Biominerals" Minerals 6, no. 2: 34. https://doi.org/10.3390/min6020034
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.